ER Stress is Involved in Epithelial-To-Mesenchymal Transition of Alveolar Epithelial Cells Exposed to a Hypoxic Microenvironment

 , ,
, ,
Abstract
:1. Introduction
2. Results
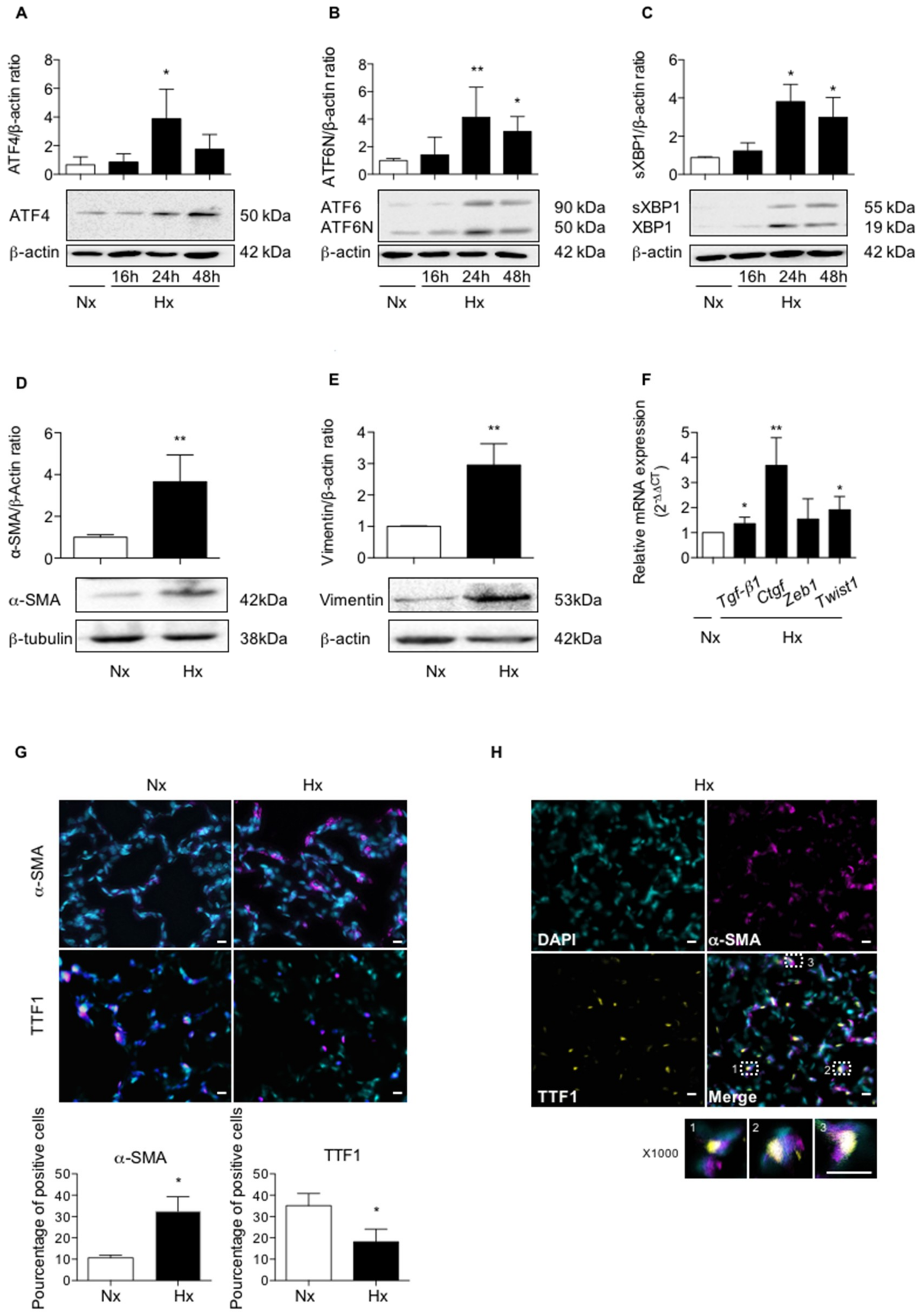
2.1. Acute Hypoxia Upregulates UPR Pathways and Induces Phenotypic Alterations of Alveolar Epithelial Cells in Rat Lungs
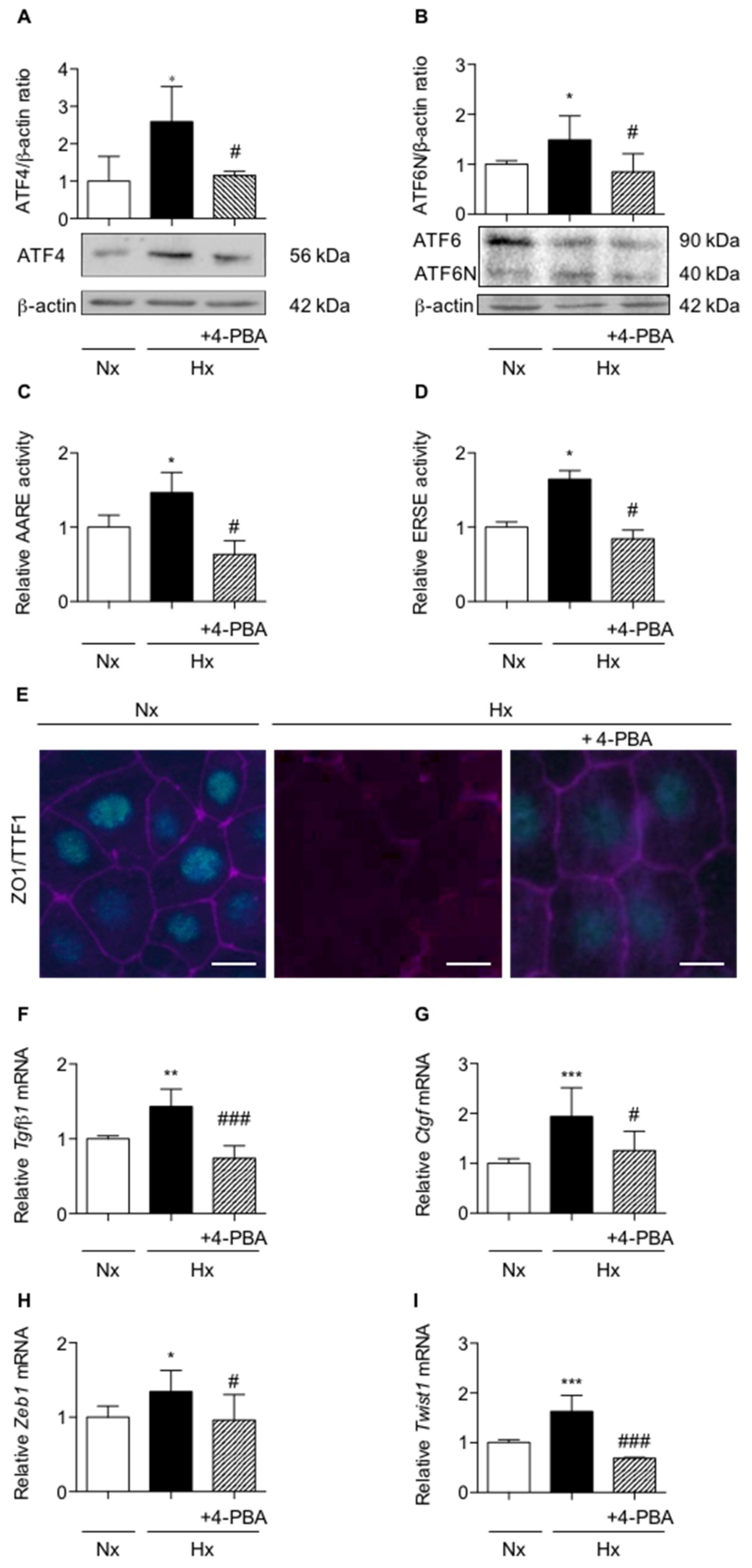
2.2. Activation of UPR Pathways is Involved in the Loss of Alveolar Epithelial Cell Phenotype Induced in Vitro by Hypoxic Exposure
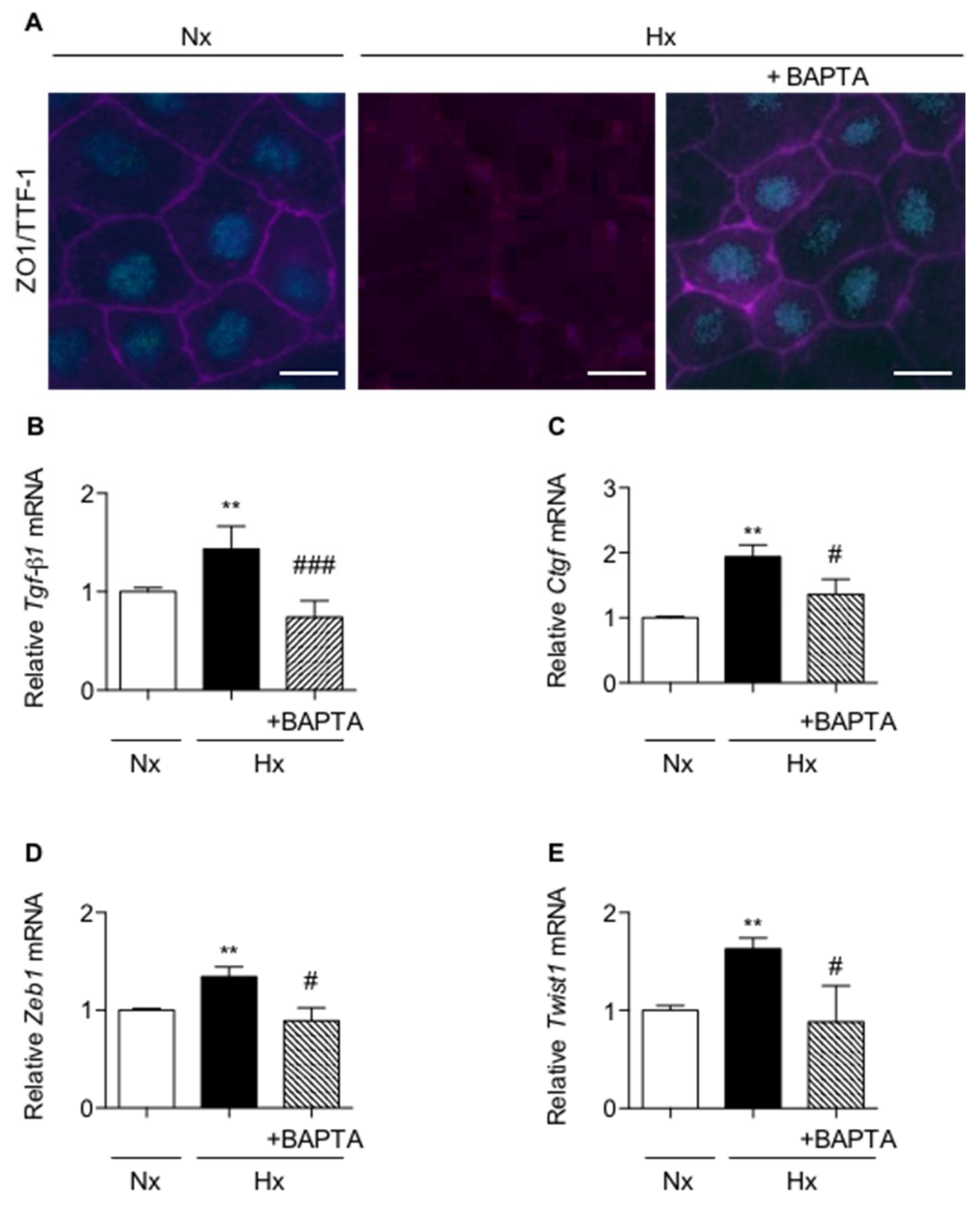
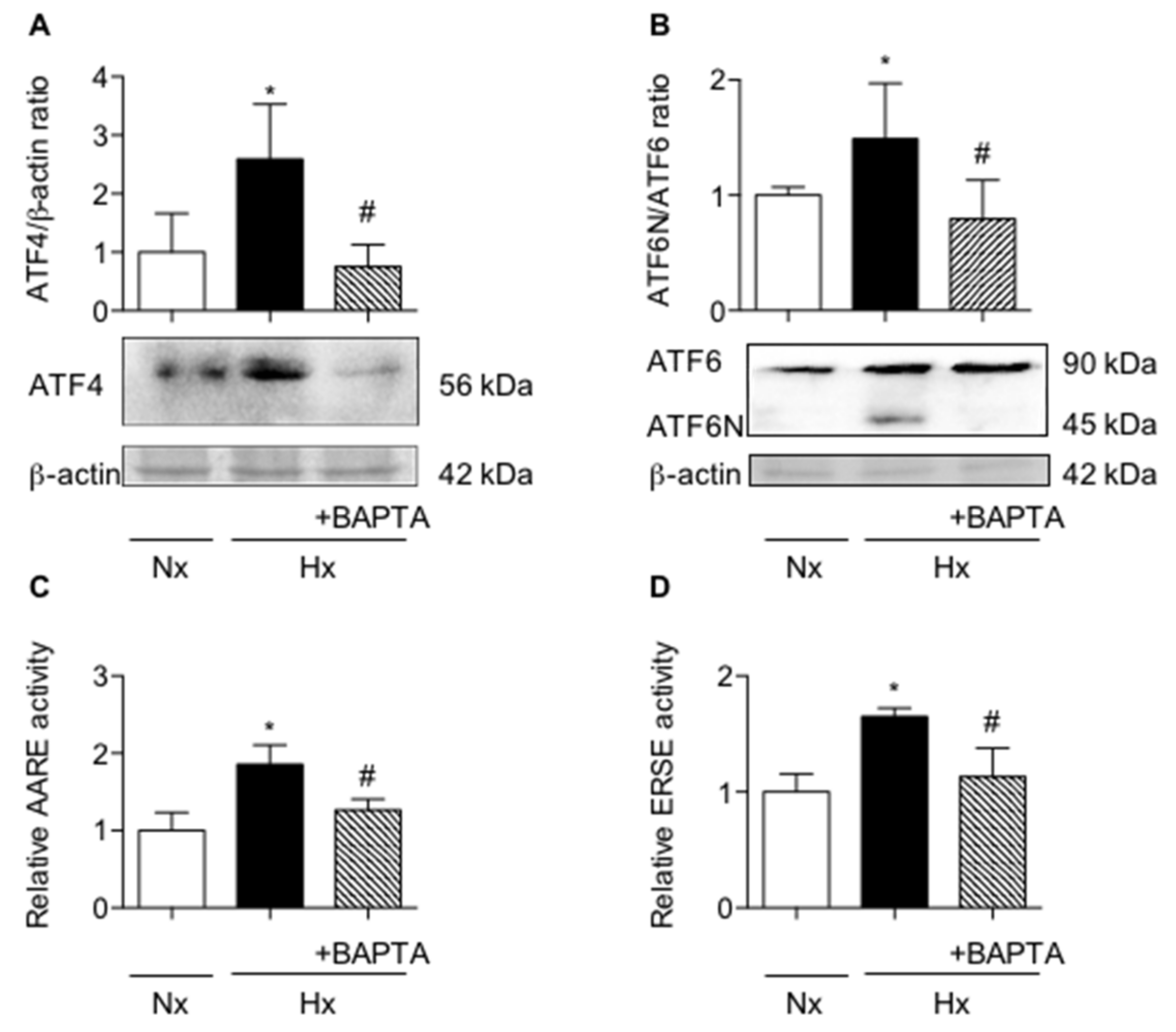
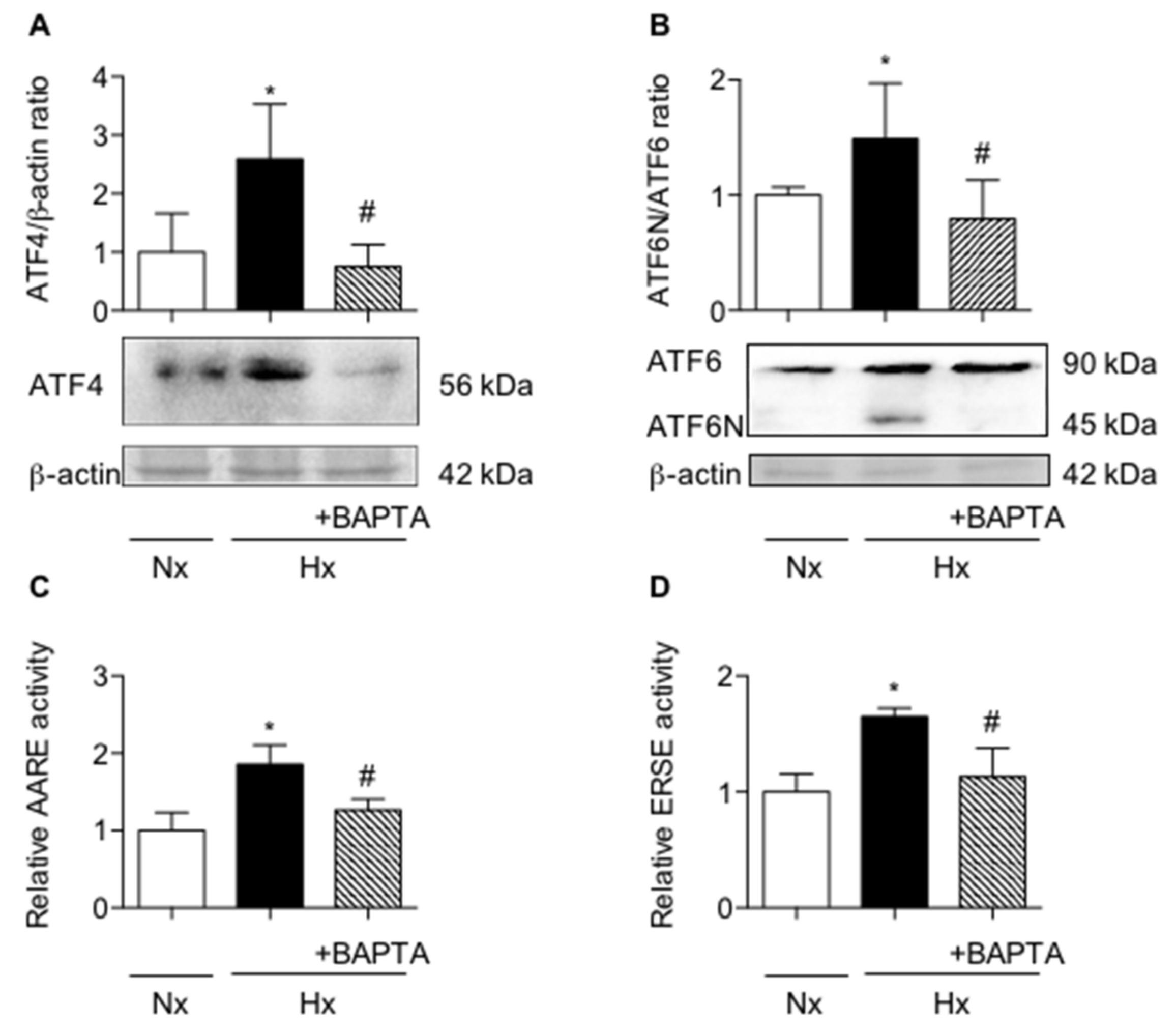
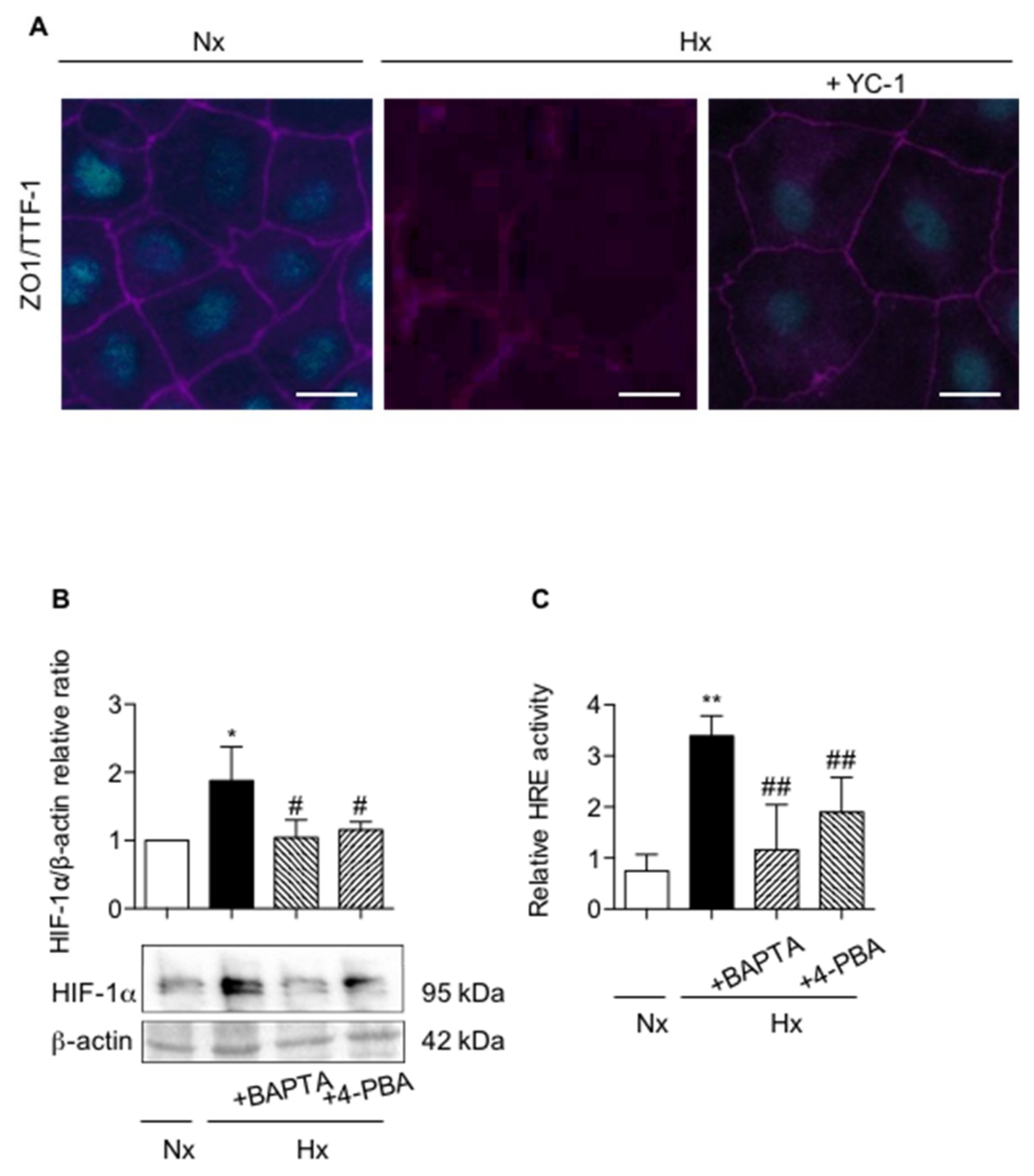
2.3. Calcium Chelation Limits Cell Alterations and UPR Pathways Activation in AECs Exposed to Hypoxia
2.4. Pharmacological Inhibition of UPR Pathways and Chelation of Calcium Modulates HIF-1 Expression and Activity
3. Discussion
4. Materials and Methods
4.1. Rat Model of Acute Hypoxic Exposure
4.2. Alveolar Epithelial Cells Isolation
4.3. Alveolar Epithelial Cells Treatments
4.4. In Vivo and In Vitro Immunofluorescence Stainings
4.5. Transient Transfection of AECs and Luciferase Activity Assay
4.6. Western Blot Analyses
4.7. RNA Extraction and Reverse Transcription-Quantitative Polymerase Chain Reaction
4.8. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-PBA | 4-phenylbutyrate |
| AARE | Amino-acid responsive element |
| AEC | Alveolar epithelial cell |
| ATF | Activating transcription factor |
| BAPTA | 1,2-bis(o-aminophenoxy)ethane-N,N,N,N-tetraacetic acid |
| ER | Endoplasmic reticulum |
| ERSE | ER stress element |
| HIF | Hypoxia inducible factor |
| HRE | Hypoxia responsive element |
| IPF | Idiopathic pulmonary fibrosis |
| IRE-1 | Inositol-requiring endonuclaease-1 |
| mRNA | messenger RNA |
| PERK | PKR-ER like kinase |
| RL | Renilla reniformis luciferase |
| TTF1 | Thyroid transcription factor 1 |
| UPR | Unfolded protein response |
| YC-1 | 5′-hydroxymethyl-2′-furyl)-1-benzyl indazole |
| ZO-1 | Zonula occludens 1 |
References
- Selman, M.; King, T.E.; Pardo, A.; American Thoracic Society; European Respiratory Society; American College of Chest Physicians. Idiopathic Pulmonary Fibrosis: Prevailing and Evolving Hypotheses about Its Pathogenesis and Implications for Therapy. Ann. Intern. Med. 2001, 134, 136–151. [Google Scholar] [CrossRef]
- Goldmann, T.; Zissel, G.; Watz, H.; Drömann, D.; Reck, M.; Kugler, C.; Rabe, K.F.; Marwitz, S. Human Alveolar Epithelial Cells Type II Are Capable of TGFβ-Dependent Epithelial-Mesenchymal-Transition and Collagen-Synthesis. Respir. Res. 2018, 19, 138. [Google Scholar] [CrossRef]
- Kugler, K.; Matthias, C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar Epithelial Cell Mesenchymal Transition Develops in Vivo during Pulmonary Fibrosis and Is Regulated by the Extracellular Matrix. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185. [Google Scholar] [CrossRef]
- Willis, B.C.; Liebler, J.M.; Luby-Phelps, K.; Nicholson, A.G.; Crandall, E.D.; du Bois, R.M.; Borok, Z. Induction of Epithelial-Mesenchymal Transition in Alveolar Epithelial Cells by Transforming Growth Factor-Beta1: Potential Role in Idiopathic Pulmonary Fibrosis. Am. J. Pathol. 2005, 166, 1321–1332. [Google Scholar] [CrossRef]
- Gora, S.; Maouche, S.; Atout, R.; Wanherdrick, K.; Lambeau, G.; Cambien, F.; Ninio, E.; Karabina, S.-A. Phospholipolyzed LDL Induces an Inflammatory Response in Endothelial Cells through Endoplasmic Reticulum Stress Signaling. FASEB J. 2010, 24, 3284–3297. [Google Scholar] [CrossRef]
- Guleria, A.; Singh, V.; Chandna, S. An Attenuated Calcium Signaling and Pre-Emptive Activation of UPR Pathway Together Contribute to ER and Calcium Stress Resilience of Lepidopteran Insect Cells. Biochim. Biophys. Acta (BBA) Gen. Subj. 2017, 1861, 504–521. [Google Scholar] [CrossRef]
- Korfei, M.; Ruppert, C.; Mahavadi, P.; Henneke, I.; Markart, P.; Koch, M.; Lang, G.; Fink, L.; Bohle, R.-M.; Seeger, W.; et al. Epithelial Endoplasmic Reticulum Stress and Apoptosis in Sporadic Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 838–846. [Google Scholar] [CrossRef]
- Lawson, W.E.; Crossno, P.F.; Polosukhin, V.V.; Roldan, J.; Cheng, D.-S.; Lane, K.B.; Blackwell, T.R.; Xu, C.; Markin, C.; Ware, L.B.; et al. Endoplasmic Reticulum Stress in Alveolar Epithelial Cells Is Prominent in IPF: Association with Altered Surfactant Protein Processing and Herpesvirus Infection. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L1119–L1126. [Google Scholar] [CrossRef]
- Burman, A.; Kropski, J.A.; Calvi, C.L.; Serezani, A.P.; Pascoalino, B.D.; Han, W.; Sherrill, T.; Gleaves, L.; Lawson, W.E.; Young, L.R.; et al. Localized Hypoxia Links ER Stress to Lung Fibrosis through Induction of C/EBP Homologous Protein. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Delbrel, E.; Soumare, A.; Naguez, A.; Label, R.; Bernard, O.; Bruhat, A.; Fafournoux, P.; Tremblais, G.; Marchant, D.; Gille, T.; et al. HIF-1α Triggers ER Stress and CHOP-Mediated Apoptosis in Alveolar Epithelial Cells, a Key Event in Pulmonary Fibrosis. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Guo, L.; Xu, J.; Liu, L.; Liu, S.; Zhu, R. Hypoxia-Induced Epithelial-Mesenchymal Transition Is Involved in Bleomycin-Induced Lung Fibrosis. BioMed Res. Int. 2015, 2015, 1–10. [Google Scholar] [CrossRef]
- Tzouvelekis, A.; Harokopos, V.; Paparountas, T.; Oikonomou, N.; Chatziioannou, A.; Vilaras, G.; Tsiambas, E.; Karameris, A.; Bouros, D.; Aidinis, V. Comparative Expression Profiling in Pulmonary Fibrosis Suggests a Role of Hypoxia-Inducible Factor-1α in Disease Pathogenesis. Am. J. Respir. Crit. Care Med. 2007, 176, 1108–1119. [Google Scholar] [CrossRef]
- Zhang, W.; Shi, X.; Peng, Y.; Wu, M.; Zhang, P.; Xie, R.; Wu, Y.; Yan, Q.; Liu, S.; Wang, J. HIF-1α Promotes Epithelial-Mesenchymal Transition and Metastasis through Direct Regulation of ZEB1 in Colorectal Cancer. PLoS ONE 2015, 10, e0129603. [Google Scholar] [CrossRef]
- Mingyuan, X.; Qianqian, P.; Shengquan, X.; Chenyi, Y.; Rui, L.; Yichen, S.; Jinghong, X. Hypoxia-Inducible Factor-1α Activates Transforming Growth Factor-B1/Smad Signaling and Increases Collagen Deposition in Dermal Fibroblasts. Oncotarget 2017, 9, 3188–3197. [Google Scholar] [CrossRef]
- Gusarova, G.A.; Trejo, H.E.; Dada, L.A.; Briva, A.; Welch, L.C.; Hamanaka, R.B.; Mutlu, G.M.; Chandel, N.S.; Prakriya, M.; Sznajder, J.I. Hypoxia Leads to Na,K-ATPase Downregulation via Ca2+ Release-Activated Ca2+ Channels and AMPK Activation. Mol. Cell. Biol. 2011, 31, 3546–3556. [Google Scholar] [CrossRef]
- Davis, F.M.; Azimi, I.; Faville, R.A.; Peters, A.A.; Jalink, K.; Putney, J.W.; Goodhill, G.J.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Induction of Epithelial–Mesenchymal Transition (EMT) in Breast Cancer Cells Is Calcium Signal Dependent. Oncogene 2014, 33, 2307–2316. [Google Scholar] [CrossRef]
- Zhong, Q.; Zhou, B.; Ann, D.K.; Minoo, P.; Liu, Y.; Banfalvi, A.; Krishnaveni, M.S.; Dubourd, M.; Demaio, L.; Willis, B.C.; et al. Role of Endoplasmic Reticulum Stress in Epithelial–Mesenchymal Transition of Alveolar Epithelial Cells: Effects of Misfolded Surfactant Protein. Am. J. Respir. Cell Mol. Biol. 2011, 45, 498–509. [Google Scholar] [CrossRef]
- Uzunhan, Y.; Bernard, O.; Marchant, D.; Dard, N.; Vanneaux, V.; Larghero, J.; Gille, T.; Clerici, C.; Valeyre, D.; Nunes, H.; et al. Mesenchymal Stem Cells Protect from Hypoxia-Induced Alveolar Epithelial-Mesenchymal Transition. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L439–L451. [Google Scholar] [CrossRef]
- Williams, J.A.; Hou, Y.; Ni, H.-M.; Ding, W.-X. Role of Intracellular Calcium in Proteasome Inhibitor-Induced Endoplasmic Reticulum Stress, Autophagy and Cell Death. Pharm. Res. 2013, 30, 2279–2289. [Google Scholar] [CrossRef]
- Planes, C.; Friedlander, G.; Loiseau, A.; Amiel, C.; Clerici, C. Inhibition of Na-K-ATPase Activity after Prolonged Hypoxia in an Alveolar Epithelial Cell Line. Am. J. Physiol. Lung Cell. Mol. Physiol. 1996, 271, L70–L78. [Google Scholar] [CrossRef]
- Zhou, G.; Dada, L.A.; Wu, M.; Kelly, A.; Trejo, H.; Zhou, Q.; Varga, J.; Sznajder, J.I. Hypoxia-Induced Alveolar Epithelial-Mesenchymal Transition Requires Mitochondrial ROS and Hypoxia-Inducible Factor 1. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L1120–L1130. [Google Scholar] [CrossRef]
- Tanjore, H.; Xu, X.C.; Polosukhin, V.V.; Degryse, A.L.; Li, B.; Han, W.; Sherrill, T.P.; Plieth, D.; Neilson, E.G.; Blackwell, T.S.; et al. Contribution of Epithelial-Derived Fibroblasts to Bleomycin-Induced Lung Fibrosis. Am. J. Respir. Crit. Care Med. 2009, 180, 657–665. [Google Scholar] [CrossRef]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 Promotes Triple-Negative Breast Cancer by Controlling the HIF1α Pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef]
- Li, H.; Chen, X.; Gao, Y.; Wu, J.; Zeng, F.; Song, F. XBP1 Induces Snail Expression to Promote Epithelial- to-Mesenchymal Transition and Invasion of Breast Cancer Cells. Cell. Signal. 2015, 27, 82–89. [Google Scholar] [CrossRef]
- Banerjee, A.; Ahmed, H.; Yang, P.; Czinn, S.J.; Blanchard, T.G. Endoplasmic Reticulum Stress and IRE-1 Signaling Cause Apoptosis in Colon Cancer Cells in Response to Andrographolide Treatment. Oncotarget 2016, 7. [Google Scholar] [CrossRef]
- Mo, X.-T.; Zhou, W.-C.; Cui, W.-H.; Li, D.-L.; Li, L.-C.; Xu, L.; Zhao, P.; Gao, J. Inositol-Requiring Protein 1 —X-Box-Binding Protein 1 Pathway Promotes Epithelial–Mesenchymal Transition via Mediating Snail Expression in Pulmonary Fibrosis. Int. J. Biochem. Cell Biol. 2015, 65, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Kumar, J.; Hermanson, K.; Sun, Y.; Qureshi, H.; Perley, D.; Scheidegger, A.; Singh, B.B.; Dhasarathy, A. The Calcium Channel Proteins ORAI3 and STIM1 Mediate TGF-& β2; Induced Snai1 Expression. Oncotarget 2018, 9. [Google Scholar] [CrossRef]
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca2+ Homeostasis and Endoplasmic Reticulum (ER) Stress: An Integrated View of Calcium Signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef]
- Belaidi, E.; Thomas, A.; Bourdier, G.; Moulin, S.; Lemarié, E.; Levy, P.; Pépin, J.-L.; Korichneva, I.; Godin-Ribuot, D.; Arnaud, C. Endoplasmic Reticulum Stress as a Novel Inducer of Hypoxia Inducible Factor-1 Activity: Its Role in the Susceptibility to Myocardial Ischemia-Reperfusion Induced by Chronic Intermittent Hypoxia. Int. J. Cardiol. 2016, 210, 45–53. [Google Scholar] [CrossRef]
- Bernard, O.; Jeny, F.; Uzunhan, Y.; Dondi, E.; Terfous, R.; Label, R.; Sutton, A.; Larghero, J.; Vanneaux, V.; Nunes, H.; et al. Mesenchymal Stem Cells Reduce Hypoxia-Induced Apoptosis in Alveolar Epithelial Cells by Modulating HIF and ROS Hypoxic Signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L360–L371. [Google Scholar] [CrossRef]
- Migneault, F.; Boncoeur, É.; Morneau, F.; Pascariu, M.; Dagenais, A.; Berthiaume, Y. Cycloheximide and Lipopolysaccharide Downregulate ΑENaC MRNA via Different Mechanisms in Alveolar Epithelial Cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L747–L755. [Google Scholar] [CrossRef]
- Chaveroux, C.; Sarcinelli, C.; Barbet, V.; Belfeki, S.; Barthelaix, A.; Ferraro-Peyret, C.; Lebecque, S.; Renno, T.; Bruhat, A.; Fafournoux, P.; et al. Nutrient Shortage Triggers the Hexosamine Biosynthetic Pathway via the GCN2-ATF4 Signalling Pathway. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GENE | Forward Primer | Reverse Primer |
|---|---|---|
| Twist1 (108 bp) | 5′-CTACGCCTTCTCCGTCTGGA-3′ | 5′-CAATGACATCTAGGTCTCCGGC-3′ |
| Ctgf (100 bp) | 5′-CCTAGCTGCCTACCGACTGG-3′ | 5′-CTTAGAACAGGCGCTCCACT-3′ |
| Tgf-β1 (146 bp) | 5′-TGAGTGGCTGTCTTTTGACG-3′ | 5′-TGGGACTGATCCCATTGATT-3′ |
| Zeb1 (90 bp) | 5′-CCGTAAGTTCAAGTGCACCG-3′ | 5′-GTGGGACTGCCACTGTGGAT-3′ |
| β-actin (74 bp) | 5′-ACCGTGAAAAGATGACCCAGA-3′ | 5′-CACAGCCTGGATGGCTACGT-3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delbrel, E.; Uzunhan, Y.; Soumare, A.; Gille, T.; Marchant, D.; Planès, C.; Boncoeur, E. ER Stress is Involved in Epithelial-To-Mesenchymal Transition of Alveolar Epithelial Cells Exposed to a Hypoxic Microenvironment. Int. J. Mol. Sci. 2019, 20, 1299. https://doi.org/10.3390/ijms20061299
Delbrel E, Uzunhan Y, Soumare A, Gille T, Marchant D, Planès C, Boncoeur E. ER Stress is Involved in Epithelial-To-Mesenchymal Transition of Alveolar Epithelial Cells Exposed to a Hypoxic Microenvironment. International Journal of Molecular Sciences. 2019; 20(6):1299. https://doi.org/10.3390/ijms20061299
Chicago/Turabian StyleDelbrel, Eva, Yurdagül Uzunhan, Abdoulaye Soumare, Thomas Gille, Dominique Marchant, Carole Planès, and Emilie Boncoeur. 2019. "ER Stress is Involved in Epithelial-To-Mesenchymal Transition of Alveolar Epithelial Cells Exposed to a Hypoxic Microenvironment" International Journal of Molecular Sciences 20, no. 6: 1299. https://doi.org/10.3390/ijms20061299
APA StyleDelbrel, E., Uzunhan, Y., Soumare, A., Gille, T., Marchant, D., Planès, C., & Boncoeur, E. (2019). ER Stress is Involved in Epithelial-To-Mesenchymal Transition of Alveolar Epithelial Cells Exposed to a Hypoxic Microenvironment. International Journal of Molecular Sciences, 20(6), 1299. https://doi.org/10.3390/ijms20061299