Causes and Mechanisms of Hematopoietic Stem Cell Aging




{kind=link}
{kind=link}
Abstract
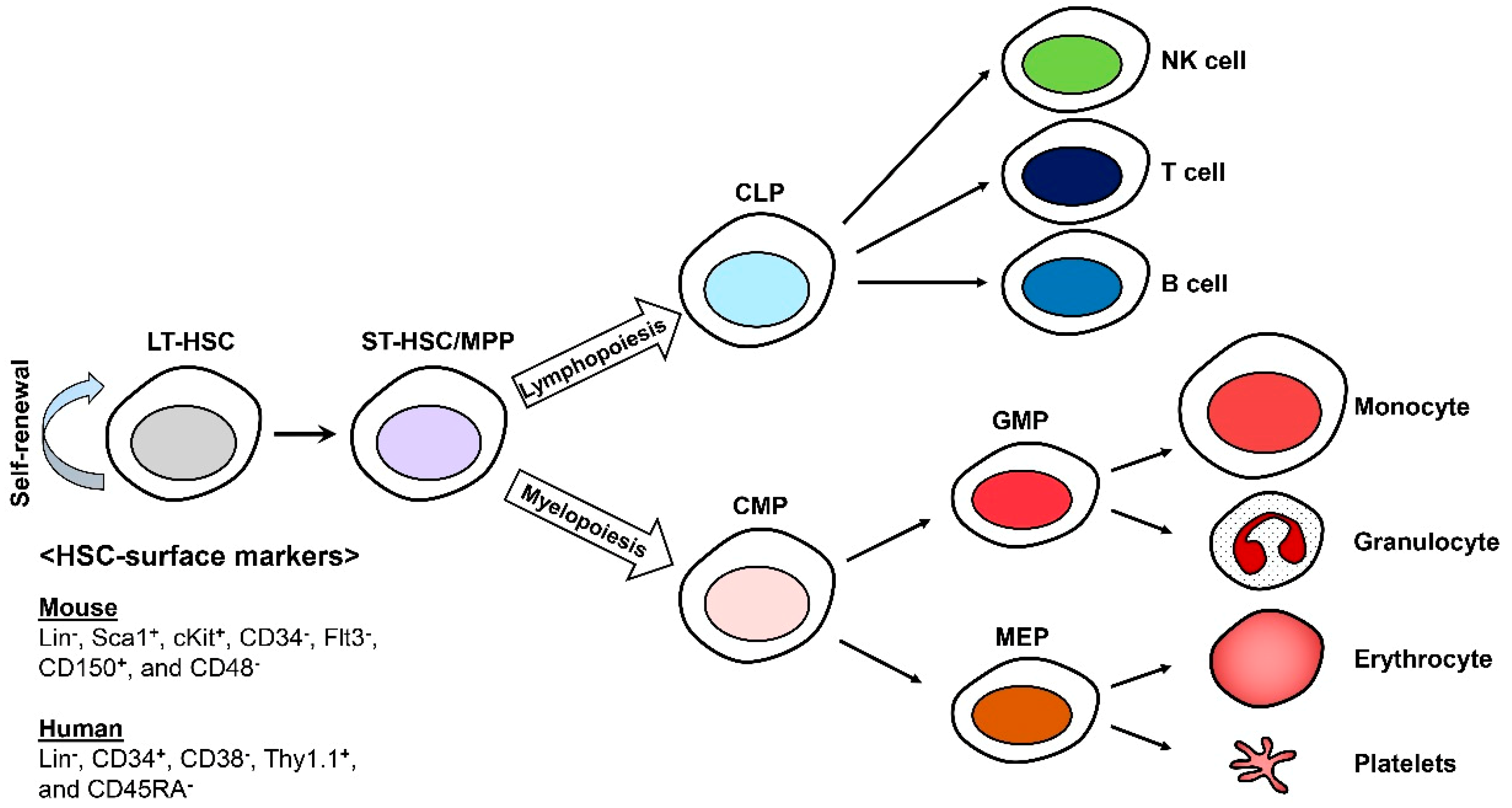
1. Introduction
2. Hallmarks of HSC Aging
2.1. Defect in Repopulating Capacity
2.2. Defect in Homing and Increase in Mobilization
2.3. Lineage Skewing
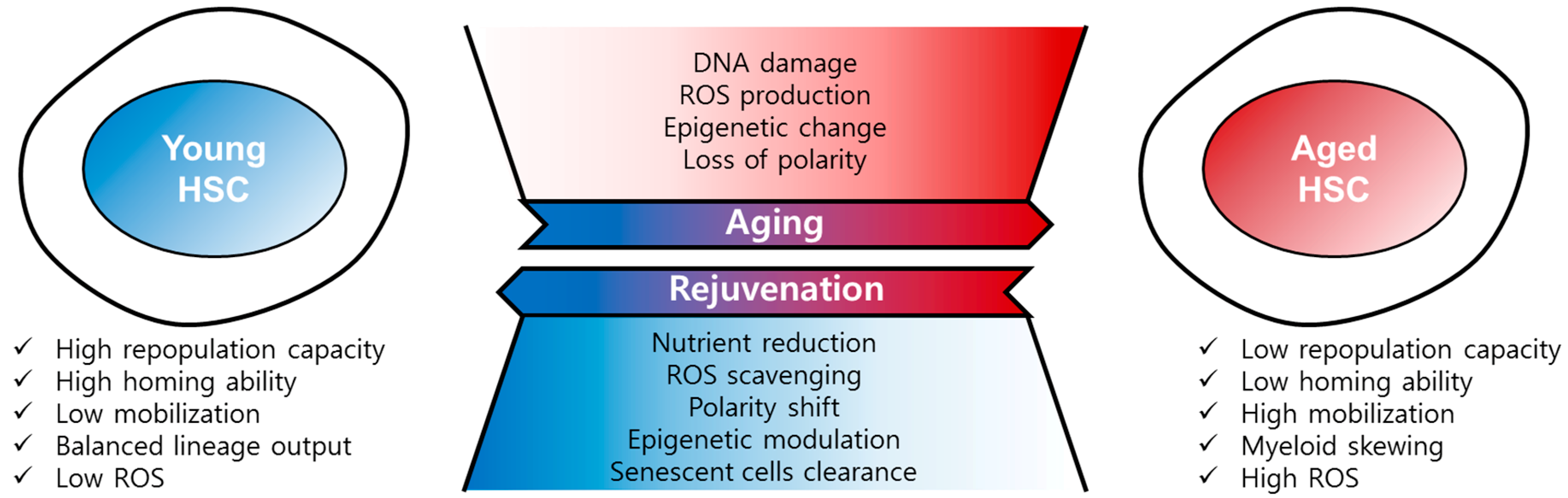
3. Causes and Mechanisms of HSC Aging
3.1. DNA Damage
3.2. ROS (Reactive Oxygen Species)
3.3. Epigenetic Changes
3.4. Polarity Changes
4. Rejuvenation of Aged HSCs
4.1. Reduction of Nutrient Supply
4.2. ROS Scavenging
4.3. Epigenetic Modulation
4.4. Clearance of Senescent Cells
5. Emerging Technologies for HSC Study
5.1. Single-Cell RNA-Sequencing (scRNA-Seq)
5.2. Single-Cell Transplantation
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Akunuru, S.; Geiger, H. Aging, Clonality, and Rejuvenation of Hematopoietic Stem Cells. Trends Mol. Med. 2016, 22, 701–712. [Google Scholar] [CrossRef]
- Moehrle, B.M.; Geiger, H. Aging of hematopoietic stem cells: DNA damage and mutations? Exp. Hematol. 2016, 44, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Ergen, A.V.; Goodell, M.A. Mechanisms of hematopoietic stem cell aging. Exp. Gerontol. 2010, 45, 286–290. [Google Scholar] [CrossRef]
- Kiel, M.J.; Yilmaz, O.H.; Iwashita, T.; Yilmaz, O.H.; Terhorst, C.; Morrison, S.J. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005, 121, 1109–1121. [Google Scholar] [CrossRef]
- Jung, H.; Kim, M.J.; Kim, D.O.; Kim, W.S.; Yoon, S.J.; Park, Y.J.; Yoon, S.R.; Kim, T.D.; Suh, H.W.; Yun, S.; et al. TXNIP maintains the hematopoietic cell pool by switching the function of p53 under oxidative stress. Cell Metab. 2013, 18, 75–85. [Google Scholar] [CrossRef]
- Notta, F.; Doulatov, S.; Laurenti, E.; Poeppl, A.; Jurisica, I.; Dick, J.E. Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science 2011, 333, 218–221. [Google Scholar] [CrossRef]
- Latchney, S.E.; Calvi, L.M. The aging hematopoietic stem cell niche: Phenotypic and functional changes and mechanisms that contribute to hematopoietic aging. Semin. Hematol. 2017, 54, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Pang, W.W.; Schrier, S.L.; Weissman, I.L. Age-associated changes in human hematopoietic stem cells. Semin. Hematol. 2017, 54, 39–42. [Google Scholar] [CrossRef]
- De Haan, G.; Lazare, S.S. Aging of hematopoietic stem cells. Blood 2018, 131, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef]
- Warren, L.A.; Rossi, D.J. Stem Cells and aging in the hematopoietic system. Mech. Ageing Dev. 2009, 130, 46–53. [Google Scholar] [CrossRef]
- Geiger, H.; de Haan, G.; Florian, M.C. The ageing haematopoietic stem cell compartment. Nat. Rev. Immunol. 2013, 13, 376–389. [Google Scholar] [CrossRef]
- Rossi, D.J.; Bryder, D.; Zahn, J.M.; Ahlenius, H.; Sonu, R.; Wagers, A.J.; Weissman, I.L. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc. Natl. Acad. Sci. USA 2005, 102, 9194–9199. [Google Scholar] [CrossRef] [PubMed]
- De Haan, G.; Nijhof, W.; Van Zant, G. Mouse strain-dependent changes in frequency and proliferation of hematopoietic stem cells during aging: Correlation between lifespan and cycling activity. Blood 1997, 89, 1543–1550. [Google Scholar] [PubMed]
- Sudo, K.; Ema, H.; Morita, Y.; Nakauchi, H. Age-associated characteristics of murine hematopoietic stem cells. J. Exp. Med. 2000, 192, 1273–1280. [Google Scholar] [CrossRef]
- Pang, W.W.; Price, E.A.; Sahoo, D.; Beerman, I.; Maloney, W.J.; Rossi, D.J.; Schrier, S.L.; Weissman, I.L. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc. Natl. Acad. Sci. USA 2011, 108, 20012–20017. [Google Scholar] [CrossRef] [PubMed]
- Yahata, T.; Takanashi, T.; Muguruma, Y.; Ibrahim, A.A.; Matsuzawa, H.; Uno, T.; Sheng, Y.; Onizuka, M.; Ito, M.; Kato, S.; et al. Accumulation of oxidative DNA damage restricts the self-renewal capacity of human hematopoietic stem cells. Blood 2011, 118, 2941–2950. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Moon, H.B.; Spangrude, G.J. Major age-related changes of mouse hematopoietic stem/progenitor cells. Ann. N. Y. Acad. Sci. 2003, 996, 195–208. [Google Scholar] [CrossRef]
- Jung, H.; Kim, D.O.; Byun, J.E.; Kim, W.S.; Kim, M.J.; Song, H.Y.; Kim, Y.K.; Kang, D.K.; Park, Y.J.; Kim, T.D.; et al. Thioredoxin-interacting protein regulates haematopoietic stem cell ageing and rejuvenation by inhibiting p38 kinase activity. Nat. Commun. 2016, 7, 13674. [Google Scholar] [CrossRef]
- Chen, J. Hematopoietic stem cell development, aging and functional failure. Int. J. Hematol. 2011, 94, 3–10. [Google Scholar] [CrossRef]
- Spangrude, G.J.; Heimfeld, S.; Weissman, I.L. Purification and characterization of mouse hematopoietic stem cells. Science 1988, 241, 58–62. [Google Scholar] [CrossRef]
- Xing, Z.; Ryan, M.A.; Daria, D.; Nattamai, K.J.; Van Zant, G.; Wang, L.; Zheng, Y.; Geiger, H. Increased hematopoietic stem cell mobilization in aged mice. Blood 2006, 108, 2190–2197. [Google Scholar] [CrossRef]
- Nilsson, S.K.; Dooner, M.S.; Weier, H.U.; Frenkel, B.; Lian, J.B.; Stein, G.S.; Quesenberry, P.J. Cells capable of bone production engraft from whole bone marrow transplants in nonablated mice. J. Exp. Med. 1999, 189, 729–734. [Google Scholar] [CrossRef]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef]
- Zhang, J.; Niu, C.; Ye, L.; Huang, H.; He, X.; Tong, W.G.; Ross, J.; Haug, J.; Johnson, T.; Feng, J.Q.; et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 2003, 425, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Lo Celso, C.; Fleming, H.E.; Wu, J.W.; Zhao, C.X.; Miake-Lye, S.; Fujisaki, J.; Cote, D.; Rowe, D.W.; Lin, C.P.; Scadden, D.T. Live-animal tracking of individual haematopoietic stem/progenitor cells in their niche. Nature 2009, 457, 92–96. [Google Scholar] [CrossRef]
- Xie, Y.; Yin, T.; Wiegraebe, W.; He, X.C.; Miller, D.; Stark, D.; Perko, K.; Alexander, R.; Schwartz, J.; Grindley, J.C.; et al. Detection of functional haematopoietic stem cell niche using real-time imaging. Nature 2009, 457, 97–101. [Google Scholar] [CrossRef]
- Domingues, M.J.; Nilsson, S.K.; Cao, B. New agents in HSC mobilization. Int. J. Hematol. 2017, 105, 141–152. [Google Scholar] [CrossRef]
- Liang, Y.; Van Zant, G.; Szilvassy, S.J. Effects of aging on the homing and engraftment of murine hematopoietic stem and progenitor cells. Blood 2005, 106, 1479–1487. [Google Scholar] [CrossRef]
- Bendall, L.J.; Bradstock, K.F. G-CSF: From granulopoietic stimulant to bone marrow stem cell mobilizing agent. Cytokine Growth Factor Rev. 2014, 25, 355–367. [Google Scholar] [CrossRef]
- Lee, J.; Cho, Y.S.; Jung, H.; Choi, I. Pharmacological Regulation of Oxidative Stress in Stem Cells. Oxid. Med. Cell. Longev. 2018, 2018, 4081890. [Google Scholar] [CrossRef]
- Liu, F.; Poursine-Laurent, J.; Link, D.C. Expression of the G-CSF receptor on hematopoietic progenitor cells is not required for their mobilization by G-CSF. Blood 2000, 95, 3025–3031. [Google Scholar]
- Tesio, M.; Golan, K.; Corso, S.; Giordano, S.; Schajnovitz, A.; Vagima, Y.; Shivtiel, S.; Kalinkovich, A.; Caione, L.; Gammaitoni, L.; et al. Enhanced c-Met activity promotes G-CSF-induced mobilization of hematopoietic progenitor cells via ROS signaling. Blood 2011, 117, 419–428. [Google Scholar] [CrossRef]
- Dorshkind, K.; Montecino-Rodriguez, E.; Signer, R.A. The ageing immune system: Is it ever too old to become young again? Nat. Rev. Immunol. 2009, 9, 57–62. [Google Scholar] [CrossRef]
- Geiger, H.; Rudolph, K.L. Aging in the lympho-hematopoietic stem cell compartment. Trends Immunol. 2009, 30, 360–365. [Google Scholar] [CrossRef]
- Wahlestedt, M.; Erlandsson, E.; Kristiansen, T.; Lu, R.; Brakebusch, C.; Weissman, I.L.; Yuan, J.; Martin-Gonzalez, J.; Bryder, D. Clonal reversal of ageing-associated stem cell lineage bias via a pluripotent intermediate. Nat. Commun. 2017, 8, 14533. [Google Scholar] [CrossRef]
- Beerman, I.; Bhattacharya, D.; Zandi, S.; Sigvardsson, M.; Weissman, I.L.; Bryder, D.; Rossi, D.J. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc. Natl. Acad. Sci. USA 2010, 107, 5465–5470. [Google Scholar] [CrossRef]
- Cho, R.H.; Sieburg, H.B.; Muller-Sieburg, C.E. A new mechanism for the aging of hematopoietic stem cells: Aging changes the clonal composition of the stem cell compartment but not individual stem cells. Blood 2008, 111, 5553–5561. [Google Scholar] [CrossRef]
- Rundberg Nilsson, A.; Soneji, S.; Adolfsson, S.; Bryder, D.; Pronk, C.J. Human and Murine Hematopoietic Stem Cell Aging Is Associated with Functional Impairments and Intrinsic Megakaryocytic/Erythroid Bias. PLoS ONE 2016, 11, e0158369. [Google Scholar] [CrossRef]
- Li, T.; Zhou, Z.W.; Ju, Z.; Wang, Z.Q. DNA Damage Response in Hematopoietic Stem Cell Ageing. Genom. Proteom. Bioinform. 2016, 14, 147–154. [Google Scholar] [CrossRef]
- Park, Y.; Gerson, S.L. DNA repair defects in stem cell function and aging. Annu. Rev. Med. 2005, 56, 495–508. [Google Scholar] [CrossRef]
- Rossi, D.J.; Bryder, D.; Seita, J.; Nussenzweig, A.; Hoeijmakers, J.; Weissman, I.L. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 2007, 447, 725–729. [Google Scholar] [CrossRef]
- Rube, C.E.; Fricke, A.; Widmann, T.A.; Furst, T.; Madry, H.; Pfreundschuh, M.; Rube, C. Accumulation of DNA damage in hematopoietic stem and progenitor cells during human aging. PLoS ONE 2011, 6, e17487. [Google Scholar] [CrossRef]
- Hamilton, M.L.; Van Remmen, H.; Drake, J.A.; Yang, H.; Guo, Z.M.; Kewitt, K.; Walter, C.A.; Richardson, A. Does oxidative damage to DNA increase with age? Proc. Natl. Acad. Sci. USA 2001, 98, 10469–10474. [Google Scholar] [CrossRef]
- Simsek, T.; Kocabas, F.; Zheng, J.; Deberardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef]
- Jang, Y.Y.; Sharkis, S.J. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 2007, 110, 3056–3063. [Google Scholar] [CrossRef]
- Porto, M.L.; Rodrigues, B.P.; Menezes, T.N.; Ceschim, S.L.; Casarini, D.E.; Gava, A.L.; Pereira, T.M.; Vasquez, E.C.; Campagnaro, B.P.; Meyrelles, S.S. Reactive oxygen species contribute to dysfunction of bone marrow hematopoietic stem cells in aged C57BL/6 J mice. J. Biomed. Sci. 2015, 22, 97. [Google Scholar] [CrossRef]
- Urao, N.; Ushio-Fukai, M. Redox regulation of stem/progenitor cells and bone marrow niche. Free Rad. Biol. Med. 2013, 54, 26–39. [Google Scholar] [CrossRef]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C.; et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef]
- Miyamoto, K.; Araki, K.Y.; Naka, K.; Arai, F.; Takubo, K.; Yamazaki, S.; Matsuoka, S.; Miyamoto, T.; Ito, K.; Ohmura, M.; et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 2007, 1, 101–112. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Chandel, N.S. Hypoxia. 2. Hypoxia regulates cellular metabolism. Am. J. Physiol. Cell Physiol. 2011, 300, C385–C393. [Google Scholar] [CrossRef]
- Kramer, A.; Challen, G.A. The epigenetic basis of hematopoietic stem cell aging. Semin. Hematol. 2017, 54, 19–24. [Google Scholar] [CrossRef]
- Sun, D.; Luo, M.; Jeong, M.; Rodriguez, B.; Xia, Z.; Hannah, R.; Wang, H.; Le, T.; Faull, K.F.; Chen, R.; et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell 2014, 14, 673–688. [Google Scholar] [CrossRef]
- Trowbridge, J.J.; Snow, J.W.; Kim, J.; Orkin, S.H. DNA methyltransferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell 2009, 5, 442–449. [Google Scholar] [CrossRef]
- Broske, A.M.; Vockentanz, L.; Kharazi, S.; Huska, M.R.; Mancini, E.; Scheller, M.; Kuhl, C.; Enns, A.; Prinz, M.; Jaenisch, R.; et al. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat. Genet. 2009, 41, 1207–1215. [Google Scholar] [CrossRef]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2011, 44, 23–31. [Google Scholar] [CrossRef]
- Florian, M.C.; Dorr, K.; Niebel, A.; Daria, D.; Schrezenmeier, H.; Rojewski, M.; Filippi, M.D.; Hasenberg, A.; Gunzer, M.; Scharffetter-Kochanek, K.; et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell 2012, 10, 520–530. [Google Scholar] [CrossRef]
- Cau, J.; Hall, A. Cdc42 controls the polarity of the actin and microtubule cytoskeletons through two distinct signal transduction pathways. J. Cell Sci. 2005, 118, 2579–2587. [Google Scholar] [CrossRef]
- Florian, M.C.; Geiger, H. Concise review: Polarity in stem cells, disease, and aging. Stem Cells 2010, 28, 1623–1629. [Google Scholar] [CrossRef]
- Guevara-Aguirre, J.; Balasubramanian, P.; Guevara-Aguirre, M.; Wei, M.; Madia, F.; Cheng, C.W.; Hwang, D.; Martin-Montalvo, A.; Saavedra, J.; Ingles, S.; et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci. Transl. Med. 2011, 3, 70ra13. [Google Scholar] [CrossRef]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span--from yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Geloen, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef]
- Cheng, C.W.; Adams, G.B.; Perin, L.; Wei, M.; Zhou, X.; Lam, B.S.; Da Sacco, S.; Mirisola, M.; Quinn, D.I.; Dorff, T.B.; et al. Prolonged fasting reduces IGF-1/PKA to promote hematopoietic-stem-cell-based regeneration and reverse immunosuppression. Cell Stem Cell 2014, 14, 810–823. [Google Scholar] [CrossRef]
- Shin, J.; Zhang, D.; Chen, D. Reversible acetylation of metabolic enzymes celebration: SIRT2 and p300 join the party. Mol. Cell 2011, 43, 3–5. [Google Scholar] [CrossRef]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef]
- Brown, K.; Xie, S.; Qiu, X.; Mohrin, M.; Shin, J.; Liu, Y.; Zhang, D.; Scadden, D.T.; Chen, D. SIRT3 reverses aging-associated degeneration. Cell Rep. 2013, 3, 319–327. [Google Scholar] [CrossRef]
- Mohrin, M.; Shin, J.; Liu, Y.; Brown, K.; Luo, H.; Xi, Y.; Haynes, C.M.; Chen, D. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 2015, 347, 1374–1377. [Google Scholar] [CrossRef]
- Ito, K.; Hirao, A.; Arai, F.; Matsuoka, S.; Takubo, K.; Hamaguchi, I.; Nomiyama, K.; Hosokawa, K.; Sakurada, K.; Nakagata, N.; et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 2004, 431, 997–1002. [Google Scholar] [CrossRef]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar] [CrossRef]
- Le, Q.; Yao, W.; Chen, Y.; Yan, B.; Liu, C.; Yuan, M.; Zhou, Y.; Ma, L. GRK6 regulates ROS response and maintains hematopoietic stem cell self-renewal. Cell Death Dis. 2016, 7, e2478. [Google Scholar] [CrossRef]
- Satoh, Y.; Yokota, T.; Sudo, T.; Kondo, M.; Lai, A.; Kincade, P.W.; Kouro, T.; Iida, R.; Kokame, K.; Miyata, T.; et al. The Satb1 protein directs hematopoietic stem cell differentiation toward lymphoid lineages. Immunity 2013, 38, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Feng, W.; Li, H.; Gardner, D.; Luo, Y.; Wang, Y.; Liu, L.; Meng, A.; Sharpless, N.E.; Zhou, D. Total body irradiation causes long-term mouse BM injury via induction of HSC premature senescence in an Ink4a- and Arf-independent manner. Blood 2014, 123, 3105–3115. [Google Scholar] [CrossRef] [PubMed]
- Le, O.N.; Rodier, F.; Fontaine, F.; Coppe, J.P.; Campisi, J.; DeGregori, J.; Laverdiere, C.; Kokta, V.; Haddad, E.; Beausejour, C.M. Ionizing radiation-induced long-term expression of senescence markers in mice is independent of p53 and immune status. Aging Cell 2010, 9, 398–409. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Papalexi, E.; Satija, R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol. 2018, 18, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, M.S.; Tirosh, I.; Heckl, D.; Rao, T.N.; Dixit, A.; Haas, B.J.; Schneider, R.K.; Wagers, A.J.; Ebert, B.L.; Regev, A. Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res. 2015, 25, 1860–1872. [Google Scholar] [CrossRef]
- Rodriguez-Fraticelli, A.E.; Wolock, S.L.; Weinreb, C.S.; Panero, R.; Patel, S.H.; Jankovic, M.; Sun, J.; Calogero, R.A.; Klein, A.M.; Camargo, F.D. Clonal analysis of lineage fate in native haematopoiesis. Nature 2018, 553, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Grover, A.; Sanjuan-Pla, A.; Thongjuea, S.; Carrelha, J.; Giustacchini, A.; Gambardella, A.; Macaulay, I.; Mancini, E.; Luis, T.C.; Mead, A.; et al. Single-cell RNA sequencing reveals molecular and functional platelet bias of aged haematopoietic stem cells. Nat. Commun. 2016, 7, 11075. [Google Scholar] [CrossRef] [PubMed]
- Baron, C.S.; Kester, L.; Klaus, A.; Boisset, J.C.; Thambyrajah, R.; Yvernogeau, L.; Kouskoff, V.; Lacaud, G.; van Oudenaarden, A.; Robin, C. Single-cell transcriptomics reveal the dynamic of haematopoietic stem cell production in the aorta. Nat. Commun. 2018, 9, 2517. [Google Scholar] [CrossRef]
- Muller-Sieburg, C.E.; Cho, R.H.; Thoman, M.; Adkins, B.; Sieburg, H.B. Deterministic regulation of hematopoietic stem cell self-renewal and differentiation. Blood 2002, 100, 1302–1309. [Google Scholar] [PubMed]
- Dykstra, B.; Kent, D.; Bowie, M.; McCaffrey, L.; Hamilton, M.; Lyons, K.; Lee, S.J.; Brinkman, R.; Eaves, C. Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell Stem Cell 2007, 1, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, S.E.W.; Nerlov, C. Haematopoiesis in the era of advanced single-cell technologies. Nat. Cell Biol. 2019, 21, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Carrelha, J.; Meng, Y.; Kettyle, L.M.; Luis, T.C.; Norfo, R.; Alcolea, V.; Boukarabila, H.; Grasso, F.; Gambardella, A.; Grover, A.; et al. Hierarchically related lineage-restricted fates of multipotent haematopoietic stem cells. Nature 2018, 554, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, R.; Wilkinson, A.C.; Ooehara, J.; Lan, X.; Lai, C.Y.; Nakauchi, Y.; Pritchard, J.K.; Nakauchi, H. Large-Scale Clonal Analysis Resolves Aging of the Mouse Hematopoietic Stem Cell Compartment. Cell Stem Cell 2018, 22, 600–607e604. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Yoon, S.R.; Choi, I.; Jung, H. Causes and Mechanisms of Hematopoietic Stem Cell Aging. Int. J. Mol. Sci. 2019, 20, 1272. https://doi.org/10.3390/ijms20061272
Lee J, Yoon SR, Choi I, Jung H. Causes and Mechanisms of Hematopoietic Stem Cell Aging. International Journal of Molecular Sciences. 2019; 20(6):1272. https://doi.org/10.3390/ijms20061272
Chicago/Turabian StyleLee, Jungwoon, Suk Ran Yoon, Inpyo Choi, and Haiyoung Jung. 2019. "Causes and Mechanisms of Hematopoietic Stem Cell Aging" International Journal of Molecular Sciences 20, no. 6: 1272. https://doi.org/10.3390/ijms20061272
APA StyleLee, J., Yoon, S. R., Choi, I., & Jung, H. (2019). Causes and Mechanisms of Hematopoietic Stem Cell Aging. International Journal of Molecular Sciences, 20(6), 1272. https://doi.org/10.3390/ijms20061272