Steroid Receptor Signallings as Targets for Resveratrol Actions in Breast and Prostate Cancer

 ,
, 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. ER Signalling in Breast Cancer
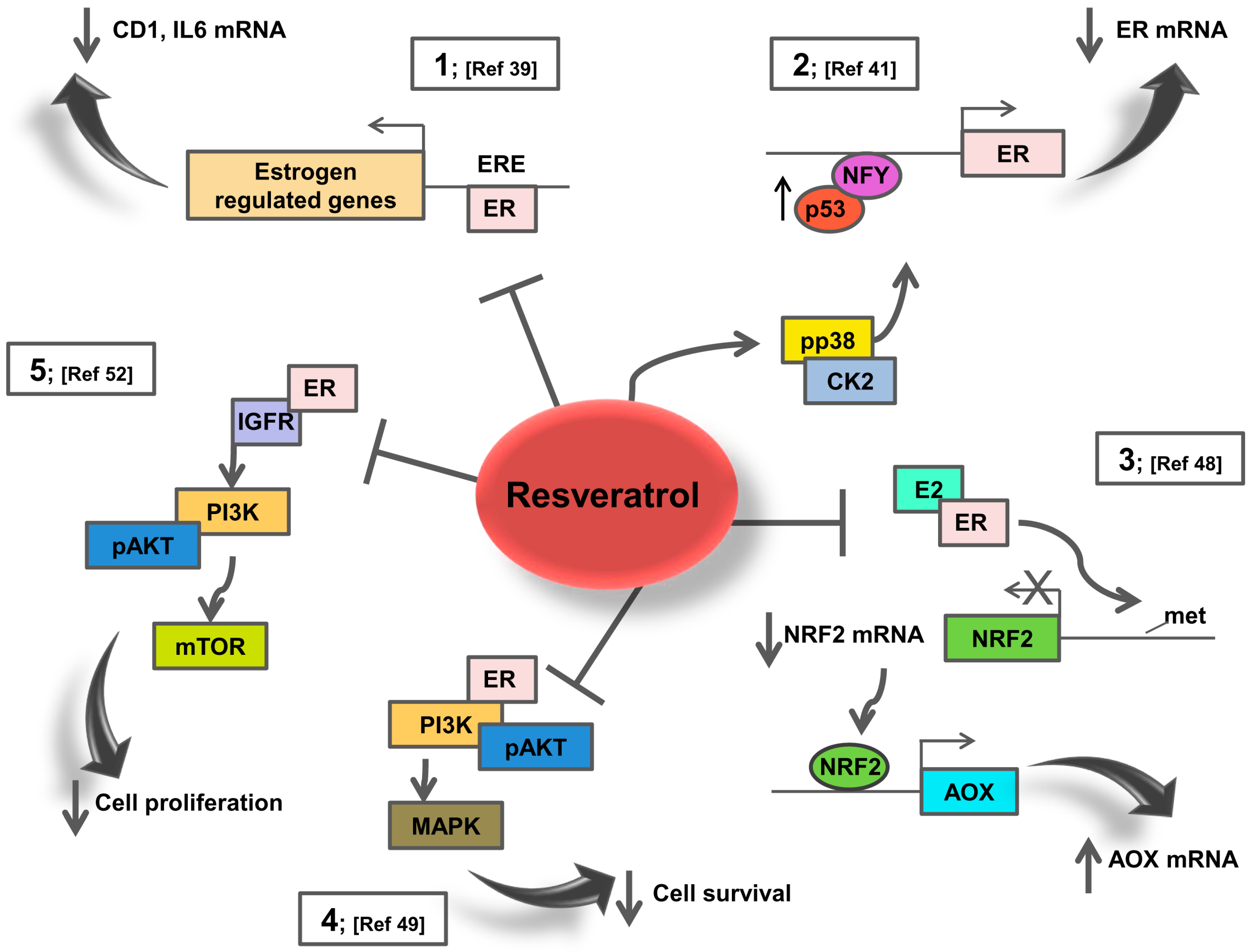
3. Resveratrol Effects on ER Signalling: Potential Action in Breast Cancer Patients
4. AR Signalling in Prostate Cancer
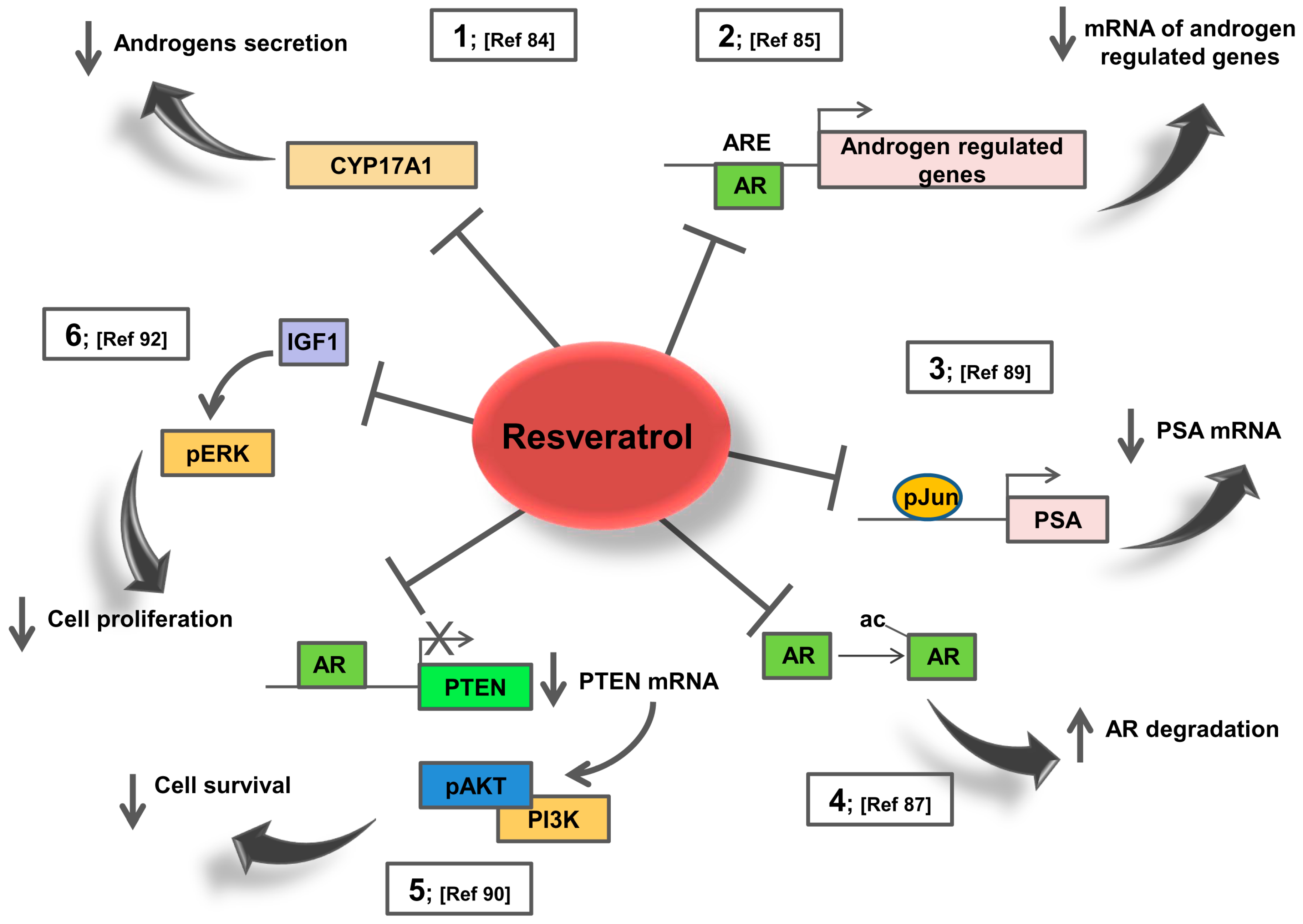
5. Resveratrol Effects on AR Signalling: Potential Action in Prostate Cancer Patients
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Louie, M.C.; Sevigny, M.B. Steroid hormone receptors as prognostic markers in breast cancer. Am. J. Cancer Res. 2017, 7, 1617–1636. [Google Scholar] [PubMed]
- Fujita, K.; Nonomura, N. Role of Androgen Receptor in Prostate Cancer: A Review. World J. Men’s Health 2018. [Google Scholar] [CrossRef] [PubMed]
- Groner, A.C.; Brown, M. Role of steroid receptor and coregulator mutations in hormone-dependent cancers. J. Clin. Investig. 2017, 127, 1126–1135. [Google Scholar] [CrossRef] [PubMed]
- Ulm, M.; Ramesh, A.V.; McNamara, K.M.; Ponnusamy, S.; Sasano, H.; Narayanan, R. Therapeutic advances in hormone-dependent cancers: Focus on prostate, breast and ovarian cancers. Endocr. Connect. 2019, 8, R10–R26. [Google Scholar] [CrossRef] [PubMed]
- Simons, S.S.; Edwards, D.P.; Kumar, R. Minireview: Dynamic Structures of Nuclear Hormone Receptors: New Promises and Challenges. Mol. Endocrinol. 2014, 28, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Kumar, S.; Narasimhan, B. Estrogen alpha receptor antagonists for the treatment of breast cancer: A review. Chem. Cent. J. 2018, 12, 107. [Google Scholar] [CrossRef] [PubMed]
- Gucalp, A.; Traina, T.A. The Androgen Receptor: Is It a Promising Target? Ann. Surg. Oncol. 2017, 24, 2876–2880. [Google Scholar] [CrossRef] [PubMed]
- Nevedomskaya, E.; Baumgart, S.J.; Haendler, B. Recent Advances in Prostate Cancer Treatment and Drug Discovery. Int. J. Mol. Sci. 2018, 19, 1359. [Google Scholar] [CrossRef] [PubMed]
- Tong, C.W.S.; Wu, M.X.; Cho, W.C.S.; To, K.K.W. Recent Advances in the Treatment of Breast Cancer. Front. Oncol. 2018, 8, 227. [Google Scholar] [CrossRef] [PubMed]
- Shoag, J.; Barbieri, C.E. Clinical variability and molecular heterogeneity in prostate cancer. Asian J. Androl. 2016, 18, 543–548. [Google Scholar] [PubMed]
- Lousberg, L.; Collignon, J.; Jerusalem, G. Resistance to therapy in estrogen receptor positive and human epidermal growth factor 2 positive breast cancers: Progress with latest therapeutic strategies. Adv. Med. Oncol. 2016, 8, 429–449. [Google Scholar] [CrossRef] [PubMed]
- Ciccarese, C.; Massari, F.; Iacovelli, R.; Fiorentino, M.; Montironi, R.; Di Nunno, V.; Giunchi, F.; Brunelli, M.; Tortora, G. Prostate cancer heterogeneity: Discovering novel molecular targets for therapy. Cancer Treat. Rev. 2017, 54, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Thakur, R.S.; Ahirwar, B. Natural Compounds A Weapon to Ameliorate Breast Cancer Cells: A Review. Anti-Cancer Agent Med. Chem. 2017, 17, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Bonofiglio, D.; Giordano, C.; De Amicis, F.; Lanzino, M.; Ando, S. Natural Products as Promising Antitumoral Agents in Breast Cancer: Mechanisms of Action and Molecular Targets. Mini-Rev. Med. Chem. 2016, 16, 596–604. [Google Scholar] [CrossRef]
- Rauf, A.; Imran, M.; Butt, M.S.; Nadeem, M.; Peters, D.G.; Mubarak, M.S. Resveratrol as an anti-cancer agent: A review. Crit. Rev. Food Sci. 2018, 58, 1428–1447. [Google Scholar] [CrossRef] [PubMed]
- Conte, A.; Kisslinger, A.; Procaccini, C.; Paladino, S.; Oliviero, O.; de Amicis, F.; Faicchia, D.; Fasano, D.; Caputo, M.; Matarese, G.; et al. Convergent Effects of Resveratrol and PYK2 on Prostate Cells. Int. J. Mol. Sci. 2016, 17, 1542. [Google Scholar] [CrossRef] [PubMed]
- Conte, A.; Procaccini, C.; Iannelli, P.; Kisslinger, A.; De Amicis, F.; Pierantoni, G.M.; Mancini, F.P.; Matarese, G.; Tramontano, D. Effects of Resveratrol on P66shc Phosphorylation in Cultured Prostate Cells. Transl. Med. Unisa 2015, 13, 47–58. [Google Scholar] [PubMed]
- Lecomte, S.; Lelong, M.; Bourgine, G.; Efstathiou, T.; Saligaut, C.; Pakdel, F. Assessment of the potential activity of major dietary compounds as selective estrogen receptor modulators in two distinct cell models for proliferation and differentiation. Toxicol. Appl. Pharmacol. 2017, 325, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Kumar, A.; Butt, N.A.; Zhang, L.F.; Williams, R.; Rimando, A.M.; Biswas, P.K.; Levenson, A.S. Molecular insight into the differential anti-androgenic activity of resveratrol and its natural analogs: In silico approach to understand biological actions. Mol. Biosyst. 2016, 12, 1702–1709. [Google Scholar] [CrossRef] [PubMed]
- Maggi, A.; Villa, A. In vivo dynamics of estrogen receptor activity: The ERE-Luc model. J. Steroid Biochem. Mol. Biol. 2014, 139, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Singhal, H.; Greene, M.E.; Tarulli, G.; Zarnke, A.L.; Bourgo, R.J.; Laine, M.; Chang, Y.F.; Ma, S.H.; Dembo, A.G.; Raj, G.V.; et al. Genomic agonism and phenotypic antagonism between estrogen and progesterone receptors in breast cancer. Sci. Adv. 2016, 2, e1501924. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, G.; Miano, V.; Beccuti, M.; Balbo, G.; De Bortoli, M.; Cordero, F. Dissecting the genomic activity of a transcriptional regulator by the integrative analysis of omics data. Sci. Rep. 2017, 7, 8564. [Google Scholar] [CrossRef] [PubMed]
- Yi, P.; Wang, Z.; Feng, Q.; Chou, C.K.; Pintilie, G.D.; Shen, H.; Foulds, C.E.; Fan, G.Z.; Serysheva, I.; Ludtke, S.J.; et al. Structural and Functional Impacts of ER Coactivator Sequential Recruitment. Mol. Cell 2017, 67, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Rothenberger, N.J.; Somasundaram, A.; Stabile, L.P. The Role of the Estrogen Pathway in the Tumor Microenvironment. Int. J. Mol. Sci. 2018, 19, 611. [Google Scholar] [CrossRef] [PubMed]
- Vanneste, M.; Hanoux, V.; Bouakka, M.; Bonnamy, P.J. Hyaluronate synthase-2 overexpression alters estrogen dependence and induces histone deacetylase inhibitor-like effects on ER-driven genes in MCF7 breast tumor cells. Mol. Cell. Endocrinol. 2017, 444, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Siersbaek, R.; Kumar, S.; Carroll, J.S. Signaling pathways and steroid receptors modulating estrogen receptor a function in breast cancer. Genes Dev. 2018, 32, 1141–1154. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, D.P.; Norris, J.D.; Chang, C.Y. Neomorphic ER alpha Mutations Drive Progression in Breast Cancer and Present a Challenge for New Drug Discovery. Cancer Cell 2018, 33, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Kok, M.; Zwart, W.; Holm, C.; Fles, R.; Hauptmann, M.; Van’t Veer, L.J.; Wessels, L.F.A.; Neefjes, J.; Stal, O.; Linn, S.C.; et al. PKA-induced phosphorylation of ER alpha at serine 305 and high PAK1 levels is associated with sensitivity to tamoxifen in ER-positive breast cancer. Breast Cancer Res. Treat. 2011, 125, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Mayer, J.A.; Mazumdar, A.; Fertuck, K.; Kim, H.; Brown, M.; Brown, P.H. Estrogen Induces c-myc Gene Expression via an Upstream Enhancer Activated by the Estrogen Receptor and the AP-1 Transcription Factor. Mol. Endocrinol. 2011, 25, 1527–1538. [Google Scholar] [CrossRef] [PubMed]
- Merino, D.; Lok, S.W.; Visvader, J.E.; Lindeman, G.J. Targeting BCL-2 to enhance vulnerability to therapy in estrogen receptor-positive breast cancer. Oncogene 2016, 35, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Brechbuhl, H.M.; Finlay-Schultz, J.; Yamamoto, T.M.; Gillen, A.E.; Cittelly, D.M.; Tan, A.C.; Sams, S.B.; Pillai, M.M.; Elias, A.D.; Robinson, W.A.; et al. Fibroblast Subtypes Regulate Responsiveness of Luminal Breast Cancer to Estrogen. Clin. Cancer Res. 2017, 23, 1710–1721. [Google Scholar] [CrossRef] [PubMed]
- Mauro, L.; Pellegrino, M.; Giordano, F.; Ricchio, E.; Rizza, P.; De Amicis, F.; Catalano, S.; Bonofiglio, D.; Panno, M.L.; Ando, S. Estrogen receptor-alpha drives adiponectin effects on cyclin D1 expression in breast cancer cells. FASEB J. 2015, 29, 2150–2160. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.M.; Livingston, M.K.; Warrick, J.W.; Stanek, E.M.; Alarid, E.T.; Beebe, D.J.; Johnson, B.P. Mammary fibroblasts reduce apoptosis and speed estrogen-induced hyperplasia in an organotypic MCF7-derived duct model. Sci. Rep. 2018, 8, 7139. [Google Scholar] [CrossRef] [PubMed]
- Stender, J.D.; Nwachukwu, J.C.; Kastrati, I.; Kim, Y.; Strid, T.; Yakir, M.; Srinivasan, S.; Nowak, J.; Izard, T.; Rangarajan, E.S.; et al. Structural and Molecular Mechanisms of Cytokine-Mediated Endocrine Resistance in Human Breast Cancer Cells. Mol. Cell 2017, 65, 1122–1135. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Kim, J.G.; Kim, N.D.; Yang, K.; Shim, J.W.; Heo, K. Estradiol, TGF-1 and hypoxia promote breast cancer stemness and EMT-mediated breast cancer migration. Oncol. Lett. 2016, 11, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Nagini, S. Breast Cancer: Current Molecular Therapeutic Targets and New Players. Anti-Cancer Agent Med. Chem. 2017, 17, 152–163. [Google Scholar] [CrossRef]
- Chin, Y.T.; Hsieh, M.T.; Yang, S.H.; Tsai, P.W.; Wang, S.H.; Wang, C.C.; Lee, Y.S.; Cheng, G.Y.; HuangFu, W.C.; London, D.; et al. Anti-proliferative and gene expression actions of resveratrol in breast cancer cells in vitro. Oncotarget 2014, 5, 12891–12907. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.E.; Lathe, R. The promiscuous estrogen receptor: Evolution of physiological estrogens and response to phytochemicals and endocrine disruptors. J. Steroid Biochem. Mol. Biol. 2018, 184, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Basly, J.P.; Marre-Fournier, F.; Le Bail, J.C.; Habrioux, G.; Chulia, A.J. Estrogenic/antiestrogenic and scavenging properties of (E)- and (Z)-resveratrol. Life Sci. 2000, 66, 769–777. [Google Scholar] [CrossRef]
- Azios, N.G.; Dharmawardhane, S.F. Resveratrol and estradiol exert disparate effects on cell migration, cell surface actin structures, and focal adhesion assembly in MDA-MB-231 human breast cancer. Neoplasia 2005, 7, 128–140. [Google Scholar] [CrossRef] [PubMed]
- De Amicis, F.; Giordano, F.; Vivacqua, A.; Pellegrino, M.; Panno, M.L.; Tramontano, D.; Fuqua, S.A.W.; Ando, S. Resveratrol, through NF-Y/p53/Sin3/HDAC1 complex phosphorylation, inhibits estrogen receptor alpha gene expression via p38(MAPK)/CK2 signaling in human breast cancer cells. FASEB J. 2011, 25, 3695–3707. [Google Scholar] [CrossRef] [PubMed]
- Kala, R.; Tollefsbol, T.O. A Novel Combinatorial Epigenetic Therapy Using Resveratrol and Pterostilbene for Restoring Estrogen Receptor-alpha (ER alpha) Expression in ER alpha-Negative Breast Cancer Cells. PLoS ONE 2016, 11, e0155057. [Google Scholar] [CrossRef] [PubMed]
- Saluzzo, J.; Hallman, K.M.; Aleck, K.; Dwyer, B.; Quigley, M.; Mladenovik, V.; Siebert, A.E.; Dinda, S. The regulation of tumor suppressor protein, p53, and estrogen receptor (ERalpha) by resveratrol in breast cancer cells. Genes Cancer 2016, 7, 414–425. [Google Scholar] [PubMed]
- Fernandes, G.F.S.; Silva, G.D.B.; Pavan, A.R.; Chiba, D.E.; Chin, C.M.; Dos Santos, J.L. Epigenetic Regulatory Mechanisms Induced by Resveratrol. Nutrients 2017, 9, 1201. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.S.; Chiou, Y.S.; Ho, C.T.; Pan, M.H. Chemoprevention by resveratrol and pterostilbene: Targeting on epigenetic regulation. Biofactors 2018, 44, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Andreani, C.; Bartolacci, C.; Wijnant, K.; Crinelli, R.; Bianchi, M.; Magnani, M.; Hysi, A.; Iezzi, M.; Amici, A.; Marchini, C. Resveratrol fuels HER2 and ER alpha-positive breast cancer behaving as proteasome inhibitor. Aging 2017, 9, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Pichardo, L.; Cubano, L.A.; Dharmawardhane, S. Dietary grape polyphenol resveratrol increases mammary tumor growth and metastasis in immunocompromised mice. BMC Complement. Altern. Med. 2013, 13, 6. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Shoulson, R.; Chatterjee, A.; Ronghe, A.; Bhat, N.K.; Dim, D.C.; Bhat, H.K. Resveratrol inhibits estrogen-induced breast carcinogenesis through induction of NRF2-mediated protective pathways. Carcinogenesis 2014, 35, 1872–1880. [Google Scholar] [CrossRef] [PubMed]
- Pozo-Guisado, E.; Lorenzo-Benayas, M.J.; Fernandez-Salguero, P.M. Resveratrol modulates the phosphoinositide 3-kinase pathway through an estrogen receptor alpha-dependent mechanism: Relevance in cell proliferation. Int. J. Cancer 2004, 109, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Sarkar, N.; Biswas, J.; Bishayee, A. Resveratrol for breast cancer prevention and therapy: Preclinical evidence and molecular mechanisms. Semin. Cancer Biol. 2016, 40–41, 209–232. [Google Scholar] [CrossRef] [PubMed]
- Menazza, S.; Murphy, E. The Expanding Complexity of Estrogen Receptor Signaling in the Cardiovascular System. Circ. Res. 2016, 118, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Kang, N.H.; Hwang, K.A.; Lee, H.R.; Choi, D.W.; Choi, K.C. Resveratrol regulates the cell viability promoted by 17 beta-estradiol or bisphenol A via down-regulation of the cross-talk between estrogen receptor alpha and insulin growth factor-1 receptor in BG-1 ovarian cancer cells. Food Chem. Toxicol. 2013, 59, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.C.; Tsai, S.H.; Chen, L.; Lin-Shiau, S.Y.; Lin, J.K. Resveratrol-induced G(2) arrest through the inhibition of CDK7 and p34(CDC2) kinases in colon carcinoma HT29 cells. Biochem. Pharmacol. 2003, 65, 1053–1060. [Google Scholar] [CrossRef]
- Coutinho, D.D.; Pacheco, M.T.; Frozza, R.L.; Bernardi, A. Anti-Inflammatory Effects of Resveratrol: Mechanistic Insights. Int. J. Mol. Sci. 2018, 19, 1812. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.; Kim, D.H.; Surh, Y.J. Resveratrol suppresses migration, invasion and sternness of human breast cancer cells by interfering with tumor-stromal cross-talk. Arch. Biochem. Biophys. 2018, 643, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Rossi, E.L.; Khatib, S.A.; Doerstling, S.S.; Bowers, L.W.; Pruski, M.; Ford, N.A.; Glickman, R.D.; Niu, M.M.; Yang, P.Y.; Cui, Z.R.; et al. Resveratrol inhibits obesity-associated adipose tissue dysfunction and tumor growth in a mouse model of postmenopausal claudin-low breast cancer. Mol. Carcinog. 2018, 57, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Jernberg, E.; Bergh, A.; Wikstrom, P. Clinical relevance of androgen receptor alterations in prostate cancer. Endocr. Connect. 2017, 6, R146–R161. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, H.; Ju, Q.; Ding, Z.; Ge, X.; Shi, Q.M.; Zhou, J.L.; Zhou, X.L.; Zhang, J.P.; Zhang, M.R.; et al. The co-regulators SRC-1 and SMRT are involved in interleukin-6-induced androgen receptor activation. Eur. Cytokine Netw. 2016, 27, 108–113. [Google Scholar] [PubMed]
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P.; Qiu, W.; Montgomery, D.; Digiammarino, E.L.; Hansen, T.M.; Risi, R.M.; Frey, R.; Manaves, V.; et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550, 128. [Google Scholar] [CrossRef] [PubMed]
- Wadosky, K.M.; Koochekpour, S. Androgen receptor splice variants and prostate cancer: From bench to bedside. Oncotarget 2017, 8, 18550–18576. [Google Scholar] [CrossRef] [PubMed]
- McGuinness, D.; McEwan, I.J. Posttranslational Modifications of Steroid Receptors: Phosphorylation. Methods Mol. Biol. 2016, 1443, 105–117. [Google Scholar] [PubMed]
- Deng, Q.; Zhang, Z.; Wu, Y.; Yu, W.Y.; Zhang, J.W.; Jiang, Z.M.; Zhang, Y.; Liang, H.; Gui, Y.T. Non-Genomic Action of Androgens is Mediated by Rapid Phosphorylation and Regulation of Androgen Receptor Trafficking. Cell. Physiol. Biochem. 2017, 43, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Thoma, C. Prostate cancer: Role for EGFR &HER2 in bone metastasis. Nat. Rev. Urol. 2017, 14, 7. [Google Scholar] [PubMed]
- Chen, J.Q.; Li, L.; Yang, Z.; Luo, J.; Yeh, S.Y.; Chang, C.S. Androgen-deprivation therapy with enzalutamide enhances prostate cancer metastasis via decreasing the EPHB6 suppressor expression. Cancer Lett. 2017, 408, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Galletti, G.; Leach, B.I.; Lam, L.; Tagawa, S.T. Mechanisms of resistance to systemic therapy in metastatic castration-resistant prostate cancer. Cancer Treat. Rev. 2017, 57, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Le, B.; Powers, G.L.; Tam, Y.T.; Schumacher, N.; Malinowski, R.L.; Steinke, L.; Kwon, G.; Marker, P.C. Multi-drug loaded micelles delivering chemotherapy and targeted therapies directed against HSP90 and the PI3K/AKT/mTOR pathway in prostate cancer. PLoS ONE 2017, 12, e0174658. [Google Scholar] [CrossRef] [PubMed]
- De Amicis, F.; Lanzino, M.; Kisslinger, A.; Cali, G.; Chieffi, P.; Ando, S.; Mancini, F.P.; Tramontano, D. Loss of proline-rich tyrosine kinase 2 function induces spreading and motility of epithelial prostate cells. J. Cell. Physiol. 2006, 209, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Brizzolara, A.; Benelli, R.; Vene, R.; Barboro, P.; Poggi, A.; Tosetti, F.; Ferrari, N. The ErbB family and androgen receptor signaling are targets of Celecoxib in prostate cancer. Cancer Lett. 2017, 400, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Patek, S.C.; Willder, J.M.; Heng, J.S.; Taylor, B.; Horgan, P.G.; Leung, H.Y.; Underwood, M.A.; Edwards, J. Androgen receptor phosphorylation status at serine 578 predicts poor outcome in prostate cancer patients. Oncotarget 2017, 8, 4875–4887. [Google Scholar] [PubMed]
- Kang, M.; Lee, K.H.; Lee, H.S.; Park, Y.H.; Jeong, C.W.; Ku, J.H.; Kim, H.H.; Kwak, C. PDLIM2 Suppression Efficiently Reduces Tumor Growth and Invasiveness of Human Castration-Resistant Prostate Cancer-Like Cells. Prostate 2016, 76, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.F.; Louie, M.C.; Desai, S.J.; Yang, J.; Chen, H.W.; Evans, C.P.; Kung, H.J. Interleukin-8 confers androgen-independent growth and migration of LNCaP: Differential effects of tyrosine kinases Src and FAK. Oncogene 2004, 23, 2197–2205. [Google Scholar] [CrossRef] [PubMed]
- Gorowska-Wojtowicz, E.; Hejmej, A.; Kaminska, A.; Pardyak, L.; Kotula-Balak, M.; Dulinska-Litewka, J.; Laidler, P.; Bilinska, B. Anti-androgen 2-hydroxyflutamide modulates cadherin, catenin and androgen receptor phosphorylation in androgen-sensitive LNCaP and androgen-independent PC3 prostate cancer cell lines acting via PI3K/Akt and MAPK/ERK1/2 pathways. Toxicol. In Vitro 2017, 40, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Baron, S.; Manin, M.; Beaudoin, C.; Leotoing, L.; Communal, Y.; Veyssiere, G.; Morel, L. Androgen receptor mediates non-genomic activation of phosphatidylinositol 3-OH kinase in androgen-sensitive epithelial cells. J. Biol. Chem. 2004, 279, 14579–14586. [Google Scholar] [CrossRef] [PubMed]
- Hamzeh, M.; Robaire, B. Androgens activate mitogen-activated protein kinase via epidermal growth factor receptor/insulin-like growth factor 1 receptor in the mouse PC-1 cell line. J. Endocrinol. 2011, 209, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Lai, D.V.; Wu, W.J.; Webb, Z.D.; Yang, Q.; Zhao, L.C.; Yu, Z.X.; Thorpe, J.E.; Disch, B.C.; Ihnat, M.A.; et al. Transition from androgenic to neurosteroidal action of 5 alpha-androstane-3 alpha, 17 beta-diol through the type A gamma-aminobutyric acid receptor in prostate cancer progression. J. Steroid Biochem. Mol. Biol. 2018, 178, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Zarif, J.C.; Lamb, L.E.; Schulz, V.V.; Nollet, E.A.; Miranti, C.K. Androgen receptor non-nuclear regulation of prostate cancer cell invasion mediated by Src and matriptase. Oncotarget 2015, 6, 6862–6876. [Google Scholar] [CrossRef] [PubMed]
- Eiro, N.; Fernandez-Gomez, J.; Sacristan, R.; Fernandez-Garcia, B.; Lobo, B.; Gonzalez-Suarez, J.; Quintas, A.; Escaf, S.; Vizoso, F. Stromal factors involved in human prostate cancer development, progression and castration resistance. J. Cancer Res. Clin. Oncol. 2017, 143, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Palethorpe, H.M.; Leach, D.A.; Need, E.F.; Drew, P.A.; Smith, E. Myofibroblast androgen receptor expression determines cell survival in co-cultures of myofibroblasts and prostate cancer cells in vitro. Oncotarget 2018, 9, 19100–19114. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.A.; Need, E.F.; Trotta, A.P.; Grubisha, M.J.; DeFranco, D.B.; Buchanan, G. Hic-5 influences genomic and non-genomic actions of the androgen receptor in prostate myofibroblasts. Mol. Cell. Endocrinol. 2014, 384, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Goreczny, G.J.; Forsythe, I.J.; Turner, C.E. Hic-5 regulates fibrillar adhesion formation to control tumor extracellular matrix remodeling through interaction with tensin1. Oncogene 2018, 37, 1699–1713. [Google Scholar] [CrossRef] [PubMed]
- Al Aameri, R.F.H.; Sheth, S.; Alanisi, E.M.A.; Borse, V.; Mukherjea, D.; Rybak, L.P.; Ramkumar, V. Tonic suppression of PCAT29 by the IL-6 signaling pathway in prostate cancer: Reversal by resveratrol. PLoS ONE 2017, 12, e0177198. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.H.; Zhu, W.; Young, C.Y.F. Resveratrol inhibits the expression and function of the androgen receptor in LNCaP prostate cancer cells. Cancer Res. 1999, 59, 5892–5895. [Google Scholar] [PubMed]
- Gao, S.; Liu, G.Z.; Wang, Z.X. Modulation of androgen receptor-dependent transcription by resveratrol and genistein in prostate cancer cells. Prostate 2004, 59, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Marti, N.; Bouchoucha, N.; Sauter, K.S.; Fluck, C.E. Resveratrol inhibits androgen production of human adrenocortical H295R cells by lowering CYP17 and CYP21 expression and activities. PLoS ONE 2017, 12, e0174224. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Murata, Y.; Yamaji, R.; Miura, T.; Inui, H.; Nakano, Y. Resveratrol down-regulates the androgen receptor at the post-translational level in prostate cancer cells. J. Nutr. Sci. Vitam. 2007, 53, 556–560. [Google Scholar] [CrossRef]
- Jones, S.B.; DePrimo, S.E.; Whitfield, M.L.; Brooks, J.D. Resveratrol-induced gene expression profiles in human prostate cancer cells. Cancer Epidemiol. Biomark. 2005, 14, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Atarashi, K.; Murata, Y.; Yamaji, R.; Nakano, Y.; Inui, H. Inhibitory mechanisms of the transcriptional activity of androgen receptor by resveratrol: Implication of DNA binding and acetylation of the receptor. J. Steroid Biochem. Mol. Biol. 2011, 123, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.; Cavero, L.; Tong, D.L.; Liu, Q.L.; Geary, K.; Talamonti, N.; Xu, J.; Fu, J.J.; Jiang, J.; Zhang, D.Z. Resveratrol enhances polyubiquitination-mediated ARV7 degradation in prostate cancer cells. Oncotarget 2017, 8, 54683–54693. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.Q.; Pan, Y.Q.; Young, C.Y.F. Overexpression of c-Jun induced by quercetin and resverol inhibits the expression and function of the androgen receptor in human prostate cancer cells. Cancer Lett. 2004, 213, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Romigh, T.; He, X.; Orloff, M.S.; Silverman, R.H.; Heston, W.D.; Eng, C. Resveratrol regulates the PTEN/AKT pathway through androgen receptor-dependent and -independent mechanisms in prostate cancer cell lines. Hum. Mol. Genet. 2010, 19, 4319–4329. [Google Scholar] [CrossRef] [PubMed]
- Benitez, D.A.; Pozo-Guisado, E.; Clementi, M.; Castellon, E.; Fernandez-Salguero, P.M. Non-genomic action of resveratrol on androgen and oestrogen receptors in prostate cancer: Modulation of the phosphoinositide 3-kinase pathway. Br. J. Cancer 2007, 96, 1595–1604. [Google Scholar] [CrossRef] [PubMed]
- Harper, C.E.; Patel, B.B.; Wang, J.; Arabshahi, A.; Eltoum, I.A.; Lamartiniere, C.A. Resveratrol suppresses prostate cancer progression in transgenic mice. Carcinogenesis 2007, 28, 1946–1953. [Google Scholar] [CrossRef] [PubMed]
- Seeni, A.; Takahashi, S.; Takeshita, K.; Tang, M.X.; Sugiura, S.; Sato, S.; Shirai, T. Suppression of Prostate Cancer Growth by Resveratrol in The Transgenic Rat for Adenocarcinoma of Prostate (TRAP) Model. Asian Pac. J. Cancer Prev. 2008, 9, 7–14. [Google Scholar] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Amicis, F.; Chimento, A.; Montalto, F.I.; Casaburi, I.; Sirianni, R.; Pezzi, V. Steroid Receptor Signallings as Targets for Resveratrol Actions in Breast and Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 1087. https://doi.org/10.3390/ijms20051087
De Amicis F, Chimento A, Montalto FI, Casaburi I, Sirianni R, Pezzi V. Steroid Receptor Signallings as Targets for Resveratrol Actions in Breast and Prostate Cancer. International Journal of Molecular Sciences. 2019; 20(5):1087. https://doi.org/10.3390/ijms20051087
Chicago/Turabian StyleDe Amicis, Francesca, Adele Chimento, Francesca Ida Montalto, Ivan Casaburi, Rosa Sirianni, and Vincenzo Pezzi. 2019. "Steroid Receptor Signallings as Targets for Resveratrol Actions in Breast and Prostate Cancer" International Journal of Molecular Sciences 20, no. 5: 1087. https://doi.org/10.3390/ijms20051087
APA StyleDe Amicis, F., Chimento, A., Montalto, F. I., Casaburi, I., Sirianni, R., & Pezzi, V. (2019). Steroid Receptor Signallings as Targets for Resveratrol Actions in Breast and Prostate Cancer. International Journal of Molecular Sciences, 20(5), 1087. https://doi.org/10.3390/ijms20051087