The Reactive Plasticity of Hippocampal Ionotropic Glutamate Receptors in Animal Epilepsies

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Types of iGluRs in the Mammalian Brain
2.1. Tissue Localization of iGluRs and Subunits in the Hippocampus
2.1.1. AMPA Receptors and Subunits
2.1.2. Kainate Receptor Subunits
2.1.3. NMDA Receptor Subunits
2.1.4. Rearrangement of iGluR Subunits Following Chronic Deafferentation
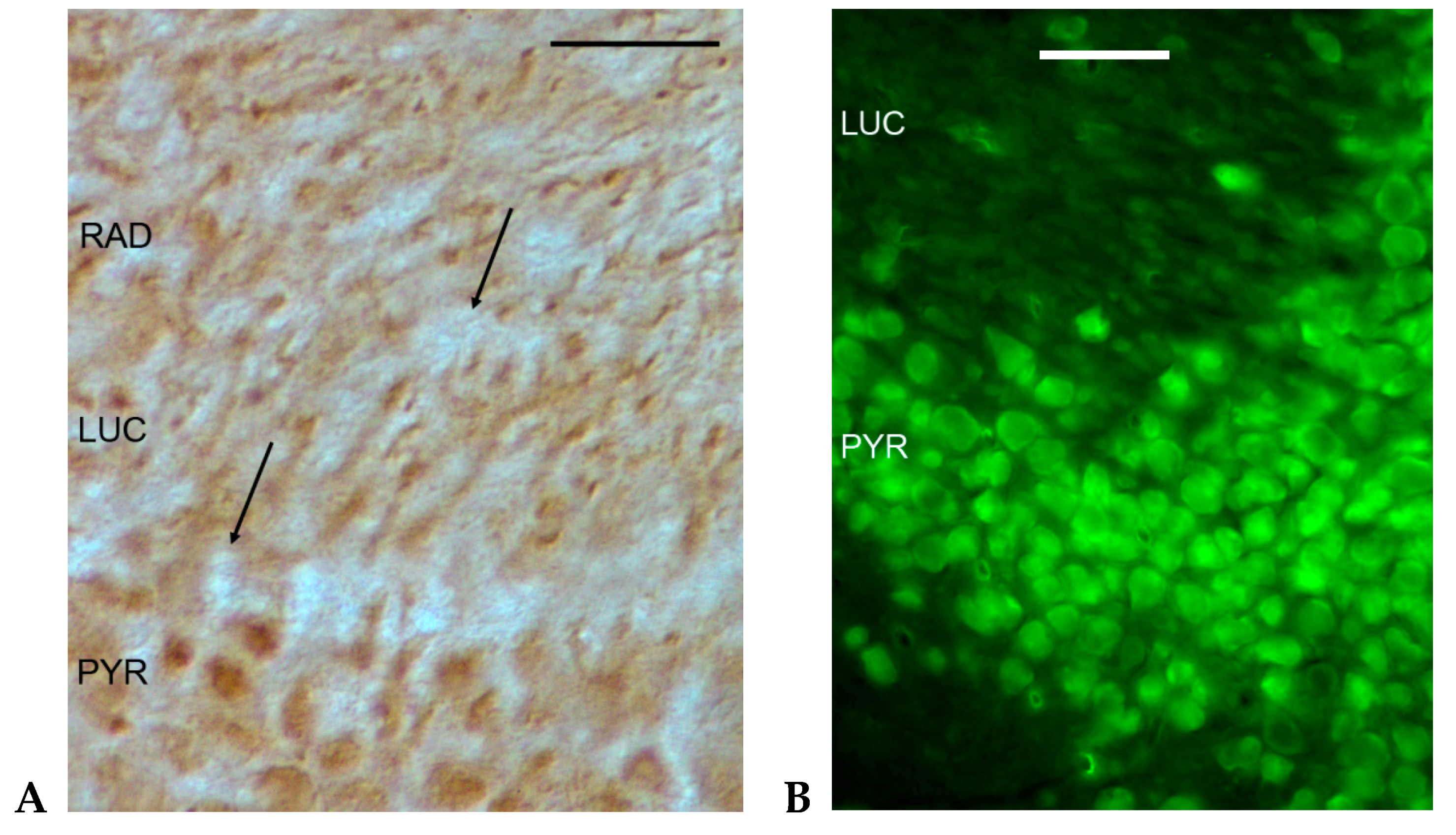
2.2. Electron Microscopic Immunohistochemistry of the iGluR Subunits in Rodent Hippocampus
3. Functional Alterations, Expression, and Distribution of iGluR Subunits Following Seizures
- (1)
- (2)
3.1. Alterations of iGluRs after Repeated, Short Convulsions Caused by 4-AP
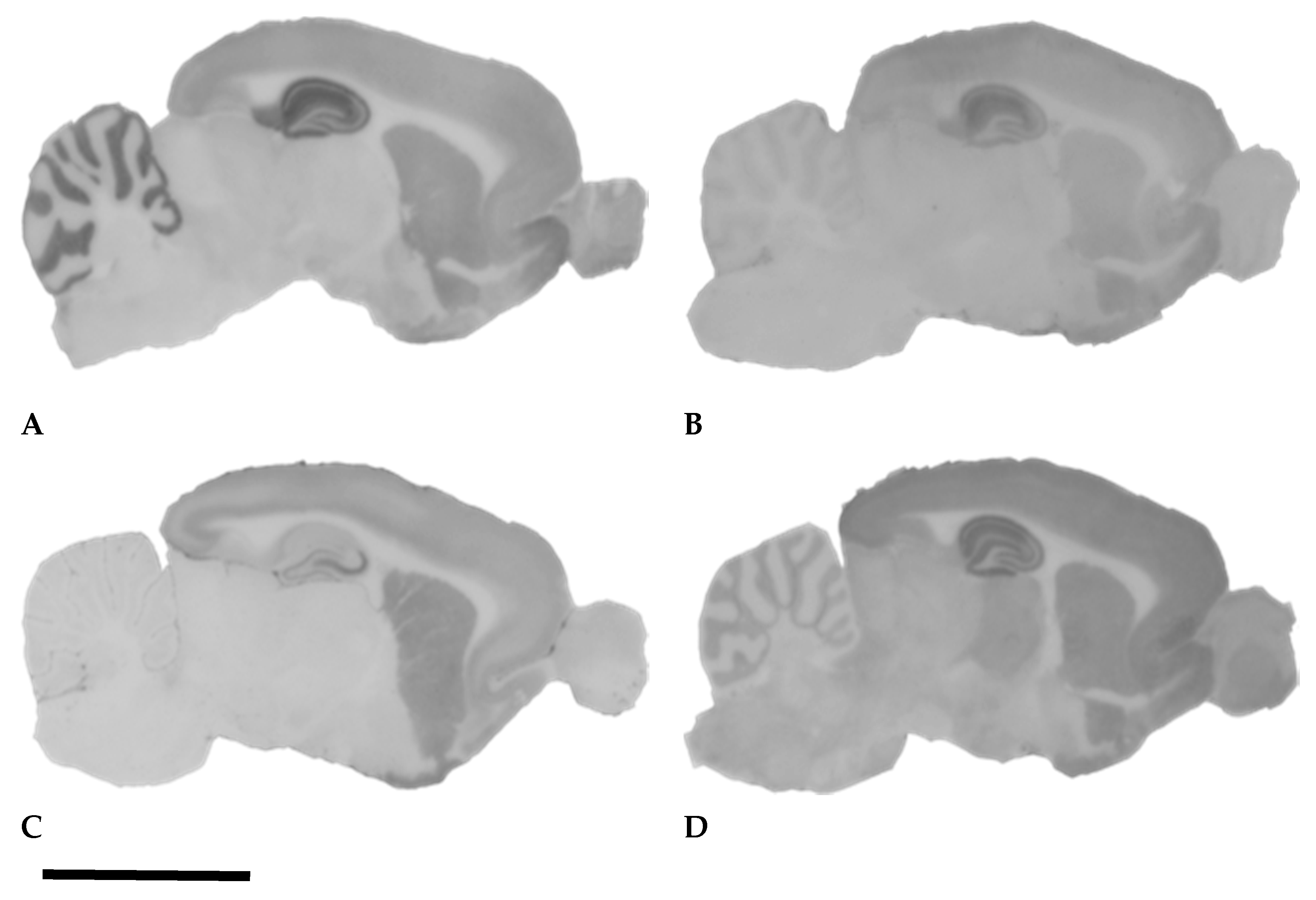
3.2. Alterations of iGluRs after Pilocarpine Seizures and Hippocampal Neuronal Degeneration
4. The Reactive Plasticity of iGluRs in Animal Epilepsies
4.1. AMPA Receptors
4.2. Kainate Receptors
4.2.1. Presynaptic KARs
4.2.2. Postsynaptic KARs
4.3. NMDA Receptors
5. Conclusions
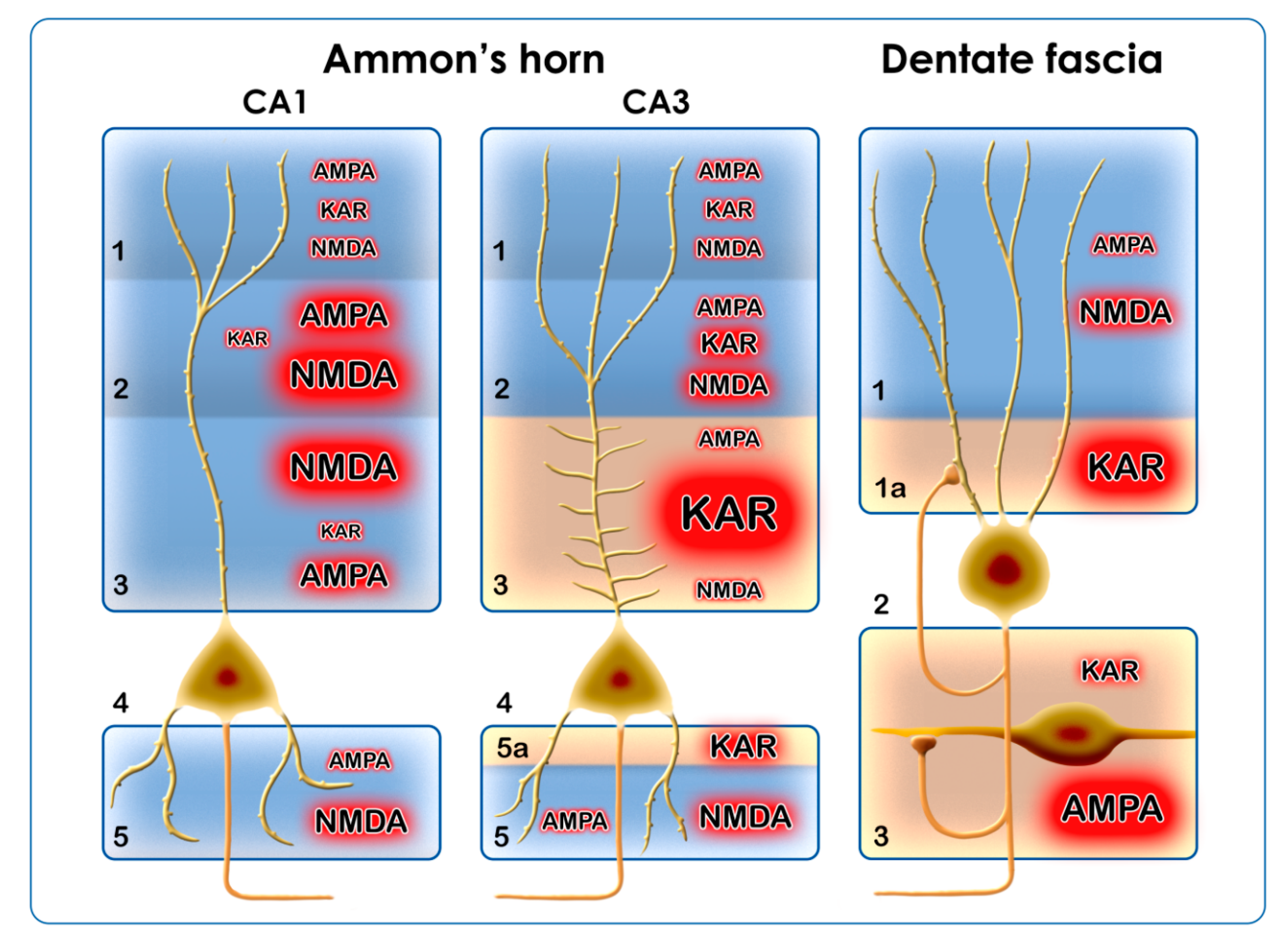
- Summarizing our own results and the literature data, we can conclude that the main alteration of AMPARs during epilepsy is the increase of their Ca2+ permeability. This will be achieved through the downregulation of the GluA2 subunit at the transcriptional, translational, and/or post-translational levels. The phosphorylation/dephosphorylation cycles of the subunits are important too, because phosphorylation alone induces Ca2+ permeability and changes the trafficking of the receptor. AMPARs are localized in every region of the hippocampal formation; therefore, the transformation of AMPARs will affect every region and cell type of the hippocampus (Figure 3).
- The Ca2+ permeability of KARs contributes to epileptogenesis presynaptically by increasing the release of Glu, and postsynaptically by increasing intracytoplasmic Ca2+ concentration and the neurotoxicity of Glu. The KARs have an outstanding role in the DF–CA4–CA3 regions. In chronic pilocarpine epilepsy, there is an extensive axonal sprouting in these regions, which originates from the dentate granule cells. Sprouting mossy fibers in the IML, SLUC, and hilus of the dentate fascia form synapses [97] and probably maintain neurotoxicity through their KAR content (Figure 3).
- The NMDARs display reactive plasticity in epilepsy; their localization and subunit composition are subject to changes from the acute phase until the chronic phase of epilepsy. The alterations of the GluN1 and GluN2 subunits include transcriptional, translational, post-translational, local phosphorylation, and trafficking changes. According to literature data, the GluN2A and GluN2B subunits are the most frequently involved. The NMDARs function mainly in the postsynaptic regions of the CA1 and the ML of the DF, where they may display synaptic and extrasynaptic localizations. In every case, they mediate the toxic effects of Glu (Figure 3).
- The reactive plasticity of the iGluRs in different hippocampal regions accommodates to neuronal types (principal neurons/interneurons) and afferent connections (Figure 3). The inhibitory interneurons of the hippocampus utilize KARs, NMDARs, and Ca2+-permeable AMPA receptors, and these iGluR combinations may be responsible for the ongoing degeneration of hippocampal inhibitory interneuron populations in epilepsy [98].
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPAR | α-amino-3-hydroxy-5-methyl-4-isoxazole-propionate receptor |
| NMDAR | N-methyl-D-aspartate receptor |
| KAR | kainate receptor |
| iGluR | ionotropic glutamate receptor |
| Glu | glutamic acid |
| GYKI 52466 | 4-(8-methyl-9H-[1,3]dioxolo[4,5-h][2,3]benzodiazepin-5-yl)aniline, AMPAR antagonist |
| CNQX | 7-nitro-2,3-dioxo-1,4-dihydroquinoxaline-6-carbonitrile, KAR antagonist |
| NBQX | 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzol[f]quinoxaline, AMPAR antagonist |
| 4-AP | 4-aminopyridine |
| CA1, CA2, CA3, CA4 | Cornu Ammonis regions in the hippocampal formation |
| DF | dentate fascia |
| SO | stratum oriens |
| SR | stratum radiatum |
| SLUC | stratum lucidum |
| SL | stratum lacunosum |
| SM | stratum moleculare |
| SE | status epilepticus |
| LTP | long-term potentiation |
| LTD | long-term depression |
| NeuN | neuron-specific nuclear protein |
| LEC | lateral entorhinal cortex |
| rCBF | regional cerebral blood flow |
| NMRI-strain mice | NMRI: abbreviated from Naval Medical Research Institute (inbred albino mouse line) |
| Q/R editing | RNA editing at the Q/R site: adenosine is converted into inosine by oxidative deamination. Q/R editing leads to the exchange glutamine to arginine during translation |
Appendix A
References
- Hassel, B.; Dingledine, R. Glutamate. In Basic Neurochemistry, 7th ed.; Siegel, G.J., Albers, R.W., Brady, S.T., Price, D.L., Eds.; Elsevier: London, UK, 2006; pp. 267–290. [Google Scholar]
- Pinky, N.F.; Wilkie, C.M.; Barnes, J.R.; Parsons, M.P. Region- and activity-dependent regulation of extracellular glutamate. J. Neurosci. 2018, 38. [Google Scholar] [CrossRef] [PubMed]
- Scheefhals, N.; MacGillavry, H.D. Functional organization of postsynaptic glutamate receptors. Mol. Cell. Neurosci. 2018, 91, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, M.; Gonzalez-Gonzalez, I.M.; Henley, J.M. Editorial: Ionotropic glutamate receptors trafficking in health and disease. Front. Cell. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [PubMed]
- Karpova, A.; Mikhaylova, M.; Bera, S.; Reddy, P.P.; Behnisch, T.; Rankovic, V.; Spilker, C.; Bethge, P.; Sahin, J.; Kaushik, R.; et al. Encoding and transducing the synaptic or extrasynaptic origin of NMDA receptor signals to the nucleus. Cell 2013, 152, 1119–1133. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Levitz, J. Glutamatergic signaling in the central nervous system: Ionotropic and metabotropic receptors in concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef] [PubMed]
- Volianskis, A.; France, G.; Jensen, M.S.; Bortolotto, Z.A.; Jane, D.E.; Collingridge, G.L. Long-term potentiation and the role of N-methyl-d-aspartate receptors. Brain Res. 2015, 1621, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Sloviter, R.S.; Dempster, D.W. “Epileptic” brain damage is replicated qualitatively in the rat hippocampus by central injection of glutamate or aspartate but not by GABA or acetylcholine. Brain Res. Bull. 1985, 15, 39–60. [Google Scholar] [CrossRef]
- Jakaria, Md.; Park, S.-Y.; Haque, E.; Karthivasan, G.; Kim, I.-S.; Ganesan, P.; Choi, D-K. Neurotoxic agent-induced injury in neurodegenerative disease model: Focus on involvement of glutamate receptors. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Pitkänen, A.; Lukasiuk, K.; Dudek, E.; Staley, K.J. Epileptogenesis. Cold Spring Harb. Perspect. Med. 2015, 5, a022822. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.V.P.; Collingridge, G.L.; Morris, R.G.M. Synaptic plasticity in health and disease: Introduction and overview. Philos. Trans. R. Soc. B 2013. [Google Scholar] [CrossRef] [PubMed]
- Kopniczky, Z.; Dobó, E.; Borbély, S.; Világi, I.; Détári, L.; Krisztin-Péva, B.; Bagosi, A.; Molnár, E.; Mihály, A. Lateral entorhinal cortex lesions rearrange afferents, glutamate receptors, increase seizure latency and suppress seizure-induced c-fos expression in the hippocampus of adult rat. J. Neurochem. 2005, 95, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Borbély, S.; Dobó, E.; Czégé, D.; Molnár, E.; Bakos, M.; Szűcs, B.; Vincze, A.; Világi, I.; Mihály, A. Modification of ionotropic glutamate receptor mediated processes in the rat hippocampus following repeated, brief seizures. Neuroscience 2009, 159, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Borbély, S.; Czégé, D.; Molnár, E.; Dobó, E.; Mihály, A.; Világi, I. Repeated application of 4-aminopyridine provoke an increase in entorhinal cortex excitability and rearrange AMPA and kainite receptors. Neurotox. Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Dobó, E.; Török, I.; Mihály, A.; Károly, N.; Krisztin-Péva, B. Interstrain differences of ionotropic glutamate receptor subunits in the hippocampus and induction of hippocampal sclerosis with pilocarpine. J. Chem. Neuroanat. 2015, 64–65, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mihály, A. Kainate, AMPA and NMDA receptor plasticity in animal models of epilepsy. Front. Neurosci. 2010. [Google Scholar] [CrossRef]
- Tao, W.; Diaz-Alonso, J.; Sheng, N.; Nicoll, R.A. Postsynaptic δ1 glutamate receptor assembles and maintains hippocampal synapses via Cbln2 and neurexin. Proc. Natl. Acad. Sci. USA 2018, 115, E5373–E5381. [Google Scholar] [CrossRef] [PubMed]
- Collingridge, G.L.; Olsen, R.W.; Peters, J.; Spedding, M. A nomenclature for ligand-gated ion channels. Neuropharmacology 2009, 56, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Hampson, D.R.; Huang, X.P.; Oberdorfer, M.D.; Goh, J.W.; Auyeung, A.; Wenthold, R.J. Localization of AMPA receptors in the hippocampus and cerebellum of the rat using an anti-receptor monoclonal antibody. Neuroscience 1992, 50, 11–22. [Google Scholar] [CrossRef]
- Petralia, R.S.; Wenthold, R.J. Light and electron immunocytochemical localization of AMPA-selective glutamate receptors in the rat brain. J. Comp. Neurol. 1992, 318, 329–354. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.R.; Jiang, X.; Burkhalter, A. Regional and laminar differences in synaptic localization of NMDA receptor subunit NR1 splice variants in rat visual cortex and hippocampus. J. Comp. Neurol. 1996, 368, 335–355. [Google Scholar] [CrossRef]
- Yin, H.Z.; Sensi, S.L.; Carriedo, S.G.; Weiss, J.H. Dendritic localization of Ca2+-permeable AMPA/kainate channels in hippocampal pyramidal neurons. J. Comp. Neurol. 1999, 409, 250–260. [Google Scholar] [CrossRef]
- Moga, D.; Hof, P.R.; Vissavajhala, P.; Moran, T.M.; Morrison, J.H. Parvalbumin-containing interneurons in rat hippocampus have an AMPA receptor profile suggestive of vulnerability to excitotoxicity. J. Chem. Neuroanat. 2002, 23, 249–253. [Google Scholar] [CrossRef]
- Jiao, Y.; Nadler, J.V. Stereological analysis of GluR2-immunoreactive hilar neurons in the pilocarpine model of temporal lobe epilepsy: Correlation of cell loss with mossy fiber sprouting. Exp. Neurol. 2007, 205, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Szabo, A.; Somogyi, J.; Cauli, B.; Lambolez, B.; Somogyi, P.; Lamsa, K.P. calcium-permeable AMPA receptors provide a common mechanism for LTP in glutamatergic synapses of distinct hippocampal interneuron types. J. Neurosci. 2012, 32, 6511–6516. [Google Scholar] [CrossRef] [PubMed]
- Archibald, K.; Perry, M.J.; Molnár, E.; Henley, J.M. Surface expression and metabolic half-life of AMPA receptors in cultured rat cerebellar granule cells. Neuropharmacology 1998, 37, 1345–1353. [Google Scholar] [CrossRef]
- Tönnes, J.; Stierli, B.; Cerletti, C.; Behrmann, J.T.; Molnár, E.; Streit, P. Regional distribution and developmental changes of GluR1-flop protein revelaed by monoclonal antibody in rat brain. J. Neurochem. 1999, 73, 2095–2205. [Google Scholar]
- Pickard, L.; Noel, J.; Henley, J.M.; Collingridge, G.L.; Molnar, E. Developmental changes in synaptic AMPA and NMDA receptor distribution and AMPA receptor subunit composition in living hippocampal neurons. J. Neurosci. 2000, 20, 7922–7931. [Google Scholar] [CrossRef] [PubMed]
- Scharfman, H. Advances in understanding hilar mossy cells of the dentate gyrus. Cell Tissue Res. 2018, 373, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Straub, C.; Noam, Y.; Nomura, T.; Yamasaki, M.; Yan, D.; Fernandes, H.B.; Zhang, P.; Howe, J.R.; Watanabe, M.; Contractor, A.; et al. Distinct subunit domains govern synaptic stability and specificity of the kainate receptor. Cell Rep. 2016, 16, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Wee, K.S.-L.; Zhang, Y.; Khanna, S.; Low, C.-M. Immunolocalization of NMDA receptor subunit NR3B in selected structures in the rat forebrain, cerebellum and lumbar spinal cord. J. Comp. Neurol. 2008, 509, 118–135. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.P.; Raymond, L.A. Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 2014, 82. [Google Scholar] [CrossRef] [PubMed]
- Kopniczky, Z.; Dochnal, R.; Mácsai, M.; Pál, Á.; Kiss, G.; Mihály, A.; Telegdy, G. Alterations of behavior and spatial learning after unilateral entorhinal ablation in rats. Life Sci. 2006, 78, 2683–2688. [Google Scholar] [CrossRef] [PubMed]
- Baude, A.; Nusser, Z.; Molnár, E.; McIlhinney, R.A.J.; Somogyi, P. High-resolution immunogold localization of AMPA type glutamate receptor subunits at synaptic and non-synaptic sites in rat hippocampus. Neuroscience 1995, 69, 1031–1055. [Google Scholar] [CrossRef]
- Fabian-Fine, R.; Volknandt, W.; Fine, A.; Stewart, M.G. Age-dependent pre- and postsynaptic distribution of AMPA receptors at synapses in CA3 stratum radiatum of hippocampal slice cultures compared with intact brain. Eur. J. Neurosci. 2000, 12, 3687–3700. [Google Scholar] [CrossRef] [PubMed]
- Darstein, M.; Petralia, R.S.; Swanson, G.T.; Wenthold, R.J.; Heinemann, S.F. Distribution of kainate receptor subunits at hippocampal mossy fiber synapses. J. Neurosci. 2003, 23, 8013–8019. [Google Scholar] [CrossRef] [PubMed]
- Racca, C.; Stephenson, F.A.; Streit, P.; Roberts, J.D.B.; Somogyi, P. NMDA receptor content of synapses in stratum radiatum of the hippocampal CA1 area. J. Neurosci. 2000, 20, 2512–2522. [Google Scholar] [CrossRef] [PubMed]
- Naylor, D.E.; Liu, H.; Niquet, J.; Wasterlain, C.G. Rapid surface accumulation of NMDA receptors increases glutamatergic excitation during status epilepticus. Neurobiol. Dis. 2013, 54, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Clemente, A.; Nicoll, R.A.; Roche, K.W. Diversity in NMDA receptor composition: Many regulators, many consequences. Neuroscientist 2013, 19, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.C. Pathophysiology of status epilepticus. Neurosci. Lett. 2018, 667, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Kovács, A.; Mihály, A.; Komáromi, Á.; Gyengési, E.; Szente, M.; Weiczner, R.; Krisztin-Péva, B.; Szabó, G.; Telegdy, G. Seizure, neurotransmitter release and gene expression are closely related in the striatum of 4-aminopyridine-treated rats. Epilepsy Res. 2003, 55, 117–129. [Google Scholar] [CrossRef]
- Weiczner, R.; Krisztin-Péva, B.; Mihály, A. Blockade of AMPA receptors attenuates 4-aminopyridine seizures, decreases the activation of inhibitory neurons but is ineffective against seizure-related astrocytic swelling. Epilepsy Res. 2008, 78, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Tóth, Z.; Mihály, A.; Mátyás, A.; Krisztin-Péva, B. Non-competitive antagonists of NMDA and AMPA receptors decrease seizure-induced c-fos protein expression in the cerebellum and protect against seizure symptoms in adult rats. Acta Histoch. 2018, 120, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Smolders, I.; Bortolotto, Z.A.; Clarke, V.R.; Warre, R.; Khan, G.M.; O’Neill, M.J.; Ornstein, P.L.; Bleakman, D.; Ogden, A.; Weiss, B.; et al. Antagonists of GLU(K5)-containing kainite receptors prevent pilocarpine-induced limbic seizures. Nat. Neurosci. 2002, 5, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Schidlitzki, A.; Twele, F.; Klee, R.; Waltl, I.; Römermann, K.; Bröer, S.; Meller, S.; Gerhauser, I.; Rankovic, V.; Li, D.; et al. A combination of NMDA and AMPA receptor antagonists retards granule cell dispersion and epileptogenesis in a model of acquired epilepsy. Nat. Sci. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Szakács, R.; Weiczner, R.; Mihály, A.; Krisztin-Péva, B.; Zádor, Z.; Zádor, E. Non-competitive NMDA receptor antagonists moderate seizure-induced c-fos expression in the rat cerebral cortex. Brain Res. Bull. 2003, 59, 485–493. [Google Scholar] [CrossRef]
- Fariello, R.G.; Golden, G.T.; Smith, G.G.; Reyes, P.F. Potentiation of kainic acid epileptogenicity and sparing from neuronal damage by an NMDA receptor antagonist. Epilepsy Res. 1989, 3, 206–213. [Google Scholar] [CrossRef]
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; Furokawa, H.; Wollmuth, L.P.; Gibb, A.J.; Traynelis, S.F. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 2018, 150. [Google Scholar] [CrossRef]
- Fritsch, B.; Reis, J.; Gasior, M.; Kaminski, R.M.; Rogawski, M.A. Role of GluK1 kainate receptors in seizures, epileptic discharges and epileptogenesis. J. Neurosci. 2014, 34, 5765–5775. [Google Scholar] [CrossRef] [PubMed]
- Buckmaster, P.S. Mossy fiber sprouting in the dentate gyrus. In Jasper’s Basic Mechanisms of the Epilepsies, 4th ed.; Noebels, J.L., Avoli, M., Rogawski, M.A., Eds.; National Center for Biotechnology Information: Bethesda, MD, USA, 2012. [Google Scholar]
- Vizi, S.; Bagosi, A.; Krisztin-Péva, B.; Gulya, K.; Mihály, A. Repeated 4-aminopyridine seizures reduce parvalabumin content in the medial mammillary nucleus of the rat brain. Mol. Brain Res. 2004, 131, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Fabene, P.F.; Weiczner, R.; Marzola, P.; Nicolato, E.; Calderan, L.; Andrioli, A.; Farkas, E.; Süle, Z.; Mihály, A. Structural and functional MRI following 4-aminopyridine seizures: A comparative imaging and anatomical study. Neurobiol. Dis. 2006, 21, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Károly, N.; Mihály, A.; Dobó, E. Comparative immunohistochemistry of synaptic markers in the rodent hippocampus in pilocarpine epilepsy. Acta Histochem. 2011, 113, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Capacio, B.R.; Chang, F.-C.T.; Spriggs, D.; Byers, C.E.; Matthews, R.L.; Benton, B.J. Pharmacokinetics and pharmacodynamics of 4-aminopyridine in awake guinea pigs. Drug Chem. Toxicol. 1997, 20, 151–172. [Google Scholar] [CrossRef] [PubMed]
- Van Den Herrewegen, Y.; Denewet, L.; Buckinx, A.; Albertini, G.; Van Eeckhaut, A.; Smolders, I.; De Bundel, D. The Barnes maze task reveals specific impairment of spatial learning strategy in the intrahippocampal kainic acid model for temporal lobe epilepsy. Neurochem. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Scimemi, A.; Schorge, S.; Kullmann, D.M.; Walker, M.C. Epileptogenesis is associated with enhanced glutamatergic transmission in the perforant path. J. Neurophysiol. 2006, 95, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Collingridge, G.L.; Lester, R.A.J. Excitatory amino acid receptors in the vertebrate central nervous system. Pharmacol. Rev. 1989, 40, 143–210. [Google Scholar]
- Dingledine, R. Glutamatergic mechanisms related to epilepsy. In Jasper’s Basic Mechanisms of the Epilepsies, 4th ed.; Noebels, J.L., Avoli, M., Rogawski, M.A., Eds.; National Center for Biotechnology Informatio: Bethesda, MD, USA, 2012. [Google Scholar]
- Pellegrini-Giampietro, D.E.; Gorter, J.A.; Bennett, M.V.L.; Zukin, S. The GluR2 (GluR-B) hypothesis: Ca2+-permeable AMPA receptors in neurological disorders. Trends Neurosci. 1997, 20, 464–470. [Google Scholar] [CrossRef]
- Liu, S.J.; Zukin, R.S. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007, 30. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.W.; Lopes, S.C.; Costa, A.P.; Goncalves, F.M.; Rieger, D.K.; Peres, T.V.; Eyng, H.; Prediger, R.D.; Diaz, A.P.; Nunes, J.C.; et al. Region-specific alterations of AMPA receptor phosphorylation and signaling pathways in the pilocarpine model of epilepsy. Neurochem. Int. 2015, 87, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Malkin, S.; Amakhin, D.V.; Veniaminova, E.; Kim, K.; Zubareva, O.; Magazanik, L.G.; Zaitsev, A. Changes of AMPA receptor properties in the neocortex and hippocampus following pilocarpine-induced status epilepticus in rats. Neuroscience 2016, 327, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Zubareva, O.E.; Kovalenko, A.A.; Kalemenev, S.V.; Schwarz, A.P.; Karyakin, V.B.; Zaitsev, A.V. Alterations in mRNA expression of glutamate receptor subunits and excitatory amino acid transporters following pilocarpine-induced seizures in rats. Neurosci. Lett. 2018, 686, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Pollard, H.; Héron, A.; Moreau, J.; Ben-Ari, Y.; Khrestchatisky, M. Alterations of the GluR-B AMPA receptor subunit flip/flop expression in kainate-induced epilepsy and ischemia. Neuroscience 1993, 57, 545–554. [Google Scholar] [CrossRef]
- Kitaura, H.; Sonoda, M.; Teramoto, S.; Shirozu, H.; Shimizu, H.; Kimura, T.; Masuda, H.; Ito, Y.; Takahashi, H.; Kwak, S.; et al. Ca2+-permeable AMPA receptors associated with epileptogenesis of hypothalamic hamartoma. Epilepsia 2017. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.J.; Racca, C. Differential dendritic targeting of AMPA receptor subunit mRNAs in adult rat hippocampal principal neurons and interneurons. J. Comp. Neurol. 2013, 521, 1954–2007. [Google Scholar] [CrossRef] [PubMed]
- Park, J. Phophorylation of the AMPA-TARP complex in synaptic plasticity. Proteomes 2018, 6, 40. [Google Scholar] [CrossRef] [PubMed]
- Summers, K.C.; Bogard, A.; Tavalin, S.J. Preferential generation of Ca2+-permeable AMPA receptors by AKAP79-anchored protein kinase C proceeds via GluA1 subunit phosphorylation at Ser-831. J. Biol. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Vincent, P.; Mulle, C. Kainate receptors in epilepsy and excitotoxicity. Neuroscience 2009, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Jane, D.E.; Lodge, D.; Collingridge, G.L. Kainate receptors: Pharmacology, function and therapeutic potential. Neuropharmacology 2009, 56, 90–113. [Google Scholar] [CrossRef] [PubMed]
- Falcón-Moya, R.; Sihra, T.S.; Rodriguez-Moreno, A. Kainate receptors: Role in epilepsy. Front. Mol. Neurosci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, R.A.; Schmitz, D. Synaptic plasticity at hippocampal mossy fiber synapses. Nat. Rev. Neurosci. 2005, 6, 863–876. [Google Scholar] [CrossRef] [PubMed]
- Lerma, J.; Marques, J.M. Kainate receptors in health and disease. Neuron 2013, 80, 292–311. [Google Scholar] [CrossRef] [PubMed]
- Gall, C.; Sumikawa, K.; Lynch, G. Levels of mRNA for a putative kainate receptor are affected by seizures. Proc. Natl. Acad. Sci. USA 1990, 87, 7643–7647. [Google Scholar] [CrossRef] [PubMed]
- Crépel, V.; Mulle, C. Physiopathology of kainate receptors in epilepsy. Curr. Opin. Pharmacol. 2015, 20, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Palacios-Filardo, J.; Aller, M.I.; Lerma, J. Synaptic targeting of kainate receptors. Cereb. Cortex 2016, 26, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Wyeth, M.S.; Pelkey, K.A.; Yuan, X.; Vargish, G.; Johnston, A.D.; Hunt, S.; Fang, C.; Abebe, D.; Mahadevan, V.; Fisahn, A.; et al. Neto auxiliary subunits regulate interneuron somatodendritic and presynaptic kainite receptors to control network inhibition. Cell Rep. 2017, 20, 2156–2168. [Google Scholar] [CrossRef] [PubMed]
- Tandon, P.; Yang, Y.; Stafstrom, C.E.; Holmes, G.L. Downregulation of kainate receptors in the hippocampus following repeated seizures in immature rats. Dev. Brain Res. 2002, 136, 145–150. [Google Scholar] [CrossRef]
- Peret, A.; Christie, L.A.; Ouedraogo, D.W.; Gorlewicz, A.; Epsztein, J.; Mulle, C.; Crépel, V. Contribution of aberrant GluK2-containing kainate receptors to chronic seizures in temporal lobe epilepsy. Cell Rep. 2014, 8, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Paternain, A.V.; Herrera, M.T.; Nieto, A.A.; Lerma, J. GluR5 and GluR6 kainate receptor subunits coexist in hippocampal neurons and coassemble to form functional receptors. J. Neurosci. 2000, 20, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Medina-Ceja, L.; Garcia-Barba, C. The glutamate receptor antagonists CNQX and MPEP decrease fast ripple events in rats treated with kainic acid. Neurosci. Lett. 2017, 655, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Károly, N.; Dobó, E.; Mihály, A. Comparative immunohistochemical study of the effects of pilocarpine on the mossy cells, mossy fibres and inhibitory neurons in murine dentate gyrus. Acta Neurobiol. Exp. 2015, 75, 220–237. [Google Scholar]
- Xu, X.X.; Luo, J.-H. Mutations of N-methyl-d-aspartate receptor subunits in epilepsy. Neurosci. Bull. 2018, 34, 549–565. [Google Scholar] [CrossRef] [PubMed]
- Sibarov, D.A.; Bruneau, N.; Antonov, S.A.; Szepetowski, P.; Burnashev, N.; Giniatullin, R. Functional properties of human NMDA receptors associated with epilepsy-related mutations of GluN2A subunit. Front. Cell. Neurosci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wasterlain, C.G.; Naylor, D.E.; Liu, H.; Niquet, J.; Baldwin, R. Trafficking of NMDA receptors during status epilepticus: Therapeutic implications. Epilepsia 2013, 54, 78–80. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Sun, H.; Klein, P.; Jensen, F.E. Neonatal seizures alter NMDA glutamate receptor GluN2A and 3A subunit expression and function in hippocampal CA1 neurons. Front. Cell. Neurosci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Loddenkemper, T.; Talos, D.M.; Cleary, R.; Joseph, A.; Fernández, I.S.; Alexopoulos, A.; Kotagal, P.; Najm, I.; Jensen, F.E. Subunit composition of glutamate and gamma-aminobutyric acid receptors in status epilepticus. Epilepsy Res. 2014, 108, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Punnakkal, P.; Dominic, D. NMDA receptor GluN2 subtypes control epileptiform events in the hippocampus. NeuroMol. Med. 2018, 20, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Postnikova, T.Y.; Zubareva, O.E.; Kovalenko, A.A.; Kim, K.K.; Magazanik, L.G.; Zaitsev, A.V. Status epilepticus impairs synaptic plasticity in rat hippocampus and is followed by changes in expression of NMDA receptors. Biochemistry 2017, 82, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Mikuni, N.; Babb, T.L.; Wylie, C.; Ying, Z. NMDAR1 receptor proteins and mossy fibers in the fascia dentate during rat kainate hippocampal epileptogenesis. Exp. Neurol. 2000, 163, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Peixoto-Santos, J.E.; de Carvalho, L.E.D.; Kandratavicius, L.; Diniz, P.R.B.; Scandiuzzi, R.C.; Coras, R.; Blümcke, I.; Assirati, J.A.; Carlotti, C.G.; Matias, C.C.M.S.; et al. Manual hippocampal subfield segmentation using high-field MRI: Impact of different subfields in hippocampal volume loss of temporal lobe epilepsy patients. Front. Neurol. 2018, 9, 927. [Google Scholar] [CrossRef] [PubMed]
- Stephan, H. Allocortex; Springer: Berlin/Heidelberg, Germany, 1975; pp. 494–621. [Google Scholar]
- Carta, M.; Srikumar, B.N.; Gorlewicz, A.; Rebola, N.; Mulle, C. Activity-dependent control of NMDA receptor subunit compostion at hippocampal mossy fibre synapses. J. Physiol. 2018, 596, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Ogden, K.K.; Traynelis, S.F. New advances in NMDA receptor pharmacology. Trends Pharmacol. Sci. 2011, 32, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Okuda, K.; Kobayashi, S.; Fukaya, M.; Watanabe, A.; Murakami, T.; Hagiwara, M.; Sato, T.; Ueno, H.; Ogonuki, N.; Komano-Inoue, S.; et al. CDKL5 control postsynaptic localization of GluN2B-containing NMDA receptors in the hippocampus and regulates seizure susceptibility. Neurobiol. Dis. 2017, 106, 158–170. [Google Scholar] [CrossRef] [PubMed]
- McGinnity, C.J.; Koepp, M.J.; Hammers, A.; Riano Barros, D.A.; Pressler, R.M.; Lthra, S.; Jones, P.A.; Trigg, W.; Micaleff, C.; Symms, M.R.; Brooks, D.J.; Duncan, J.S. NMDA receptor binding in focal epilepsies. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Frotscher, M.; Jonas, P.; Sloviter, R.S. Synapses formed by normal and abnormal hippocampal mossy fibers. Cell Tissue Res. 2006, 326, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Botterill, J.J.; Nogovitsyn, N.; Caruncho, H.J.; Kalynchuk, L.E. Selective plasticity of hippocampal GABAergic interneuron populations following kindling of different brain regions. J. Comp. Neurol. 2017, 525, 389–406. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mihály, A. The Reactive Plasticity of Hippocampal Ionotropic Glutamate Receptors in Animal Epilepsies. Int. J. Mol. Sci. 2019, 20, 1030. https://doi.org/10.3390/ijms20051030
Mihály A. The Reactive Plasticity of Hippocampal Ionotropic Glutamate Receptors in Animal Epilepsies. International Journal of Molecular Sciences. 2019; 20(5):1030. https://doi.org/10.3390/ijms20051030
Chicago/Turabian StyleMihály, András. 2019. "The Reactive Plasticity of Hippocampal Ionotropic Glutamate Receptors in Animal Epilepsies" International Journal of Molecular Sciences 20, no. 5: 1030. https://doi.org/10.3390/ijms20051030
APA StyleMihály, A. (2019). The Reactive Plasticity of Hippocampal Ionotropic Glutamate Receptors in Animal Epilepsies. International Journal of Molecular Sciences, 20(5), 1030. https://doi.org/10.3390/ijms20051030