Aberrant Calcium Signals in Reactive Astrocytes: A Key Process in Neurological Disorders



{kind=link}
Abstract
1. Introduction
2. Measurement of Ca2+ Signals in Astrocytes
3. Reactive Astrocytes in Disease
4. Ca2+ Signals in Reactive Astrocytes
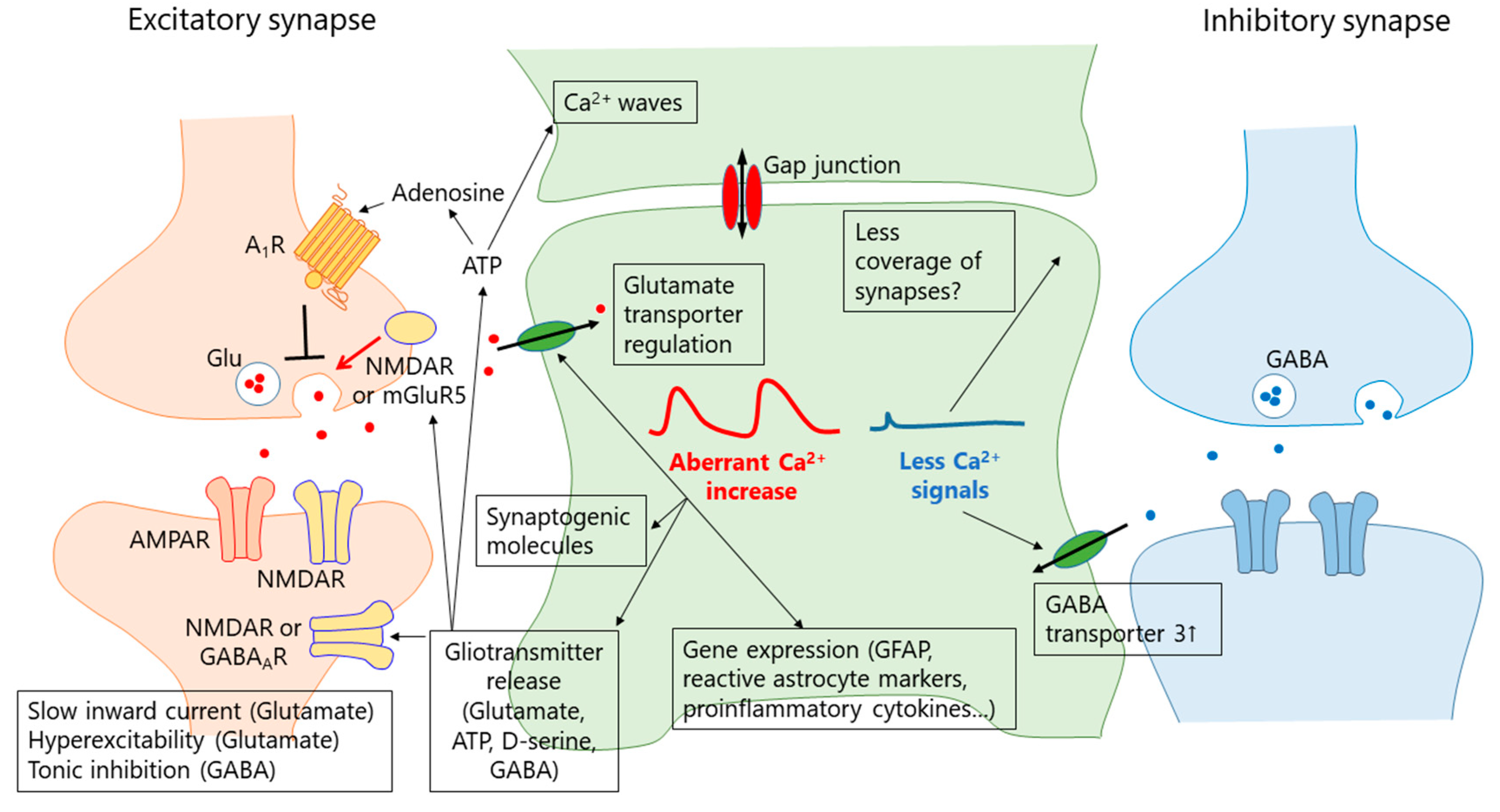
5. Mechanisms of Aberrant Ca2+ Signals
5.1. Receptor-Mediated Ca2+ Signals
5.2. Transmembrane Ca2+ Pathways
5.3. Ca2+ Release from Mitochondria
5.4. Other Mechanisms
6. What Is the Function of the Aberrant Ca2+ Signal in Reactive Astrocytes?
6.1. Gliotransmission
6.2. Synapse Remodeling
6.3. GFAP Upregulation
6.4. Neuronal Damage
7. Role of Reduced Astrocyte Ca2+ Signals in Disease
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| AM | Acetoxymethyl |
| AxCa | Aberrant extra-large Ca2+ signal |
| AxD | Alexander disease |
| CSD | Cortical spreading depression |
| DS | Down syndrome |
| EAE | Experimental autoimmune encephalitis |
| ER | Endoplasmic reticulum |
| GAT-3 | GABA transporter 3 |
| GECI | Genetically encoded Ca2+ indicator |
| GFAP | Glial fibrillary acidic protein |
| GqPCR | Gq-protein coupled receptor |
| hPMCA2w/b | Human PMCA2w/b |
| IP3R2 | Inositol-1,4,5 trisphosphate receptor type2 |
| IP3R2KO | IP3R type2 knockout |
| MCAO | Middle cerebral artery occlusion |
| mGluR5 | Metabotropic glutamate receptor 5 |
| MSN | Medium spiny neuron |
| mPTP | Mitochondrial permeability transition pore |
| MS | Multiple sclerosis |
| OGD | Oxygen glucose deprivation |
| PID | Peri-infarct depolarization |
| RTT | Rett syndrome |
| SCI | Spinal cord injury |
| SE | Status epilepticus |
| SIC | Slow inward current |
| TBI | Traumatic brain injury |
| TSP1 | Thrombospondin 1 |
References
- Khakh, B.S.; Sofroniew, M.V. Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 2015, 18, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Nedergaard, M. Physiology of astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- Dallerac, G.; Zapata, J.; Rouach, N. Versatile control of synaptic circuits by astrocytes: Where, when and how? Nat. Rev. Neurosci. 2018, 19, 729–743. [Google Scholar] [CrossRef] [PubMed]
- Ben Haim, L.; Rowitch, D.H. Functional diversity of astrocytes in neural circuit regulation. Nat. Rev. Neurosci. 2017, 18, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Araque, A.; Carmignoto, G.; Haydon, P.G.; Oliet, S.H.; Robitaille, R.; Volterra, A. Gliotransmitters travel in time and space. Neuron 2014, 81, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Shigetomi, E.; Patel, S.; Khakh, B.S. Probing the complexities of astrocyte calcium signaling. Trends Cell Biol. 2016, 26, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Pekna, M.; Messing, A.; Steinhauser, C.; Lee, J.M.; Parpura, V.; Hol, E.M.; Sofroniew, M.V.; Verkhratsky, A. Astrocytes: A central element in neurological diseases. Acta Neuropathol. 2016, 131, 323–345. [Google Scholar] [CrossRef] [PubMed]
- Bazargani, N.; Attwell, D. Astrocyte calcium signaling: The third wave. Nat. Neurosci. 2016, 19, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Burda, J.E.; Sofroniew, M.V. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 2014, 81, 229–248. [Google Scholar] [CrossRef] [PubMed]
- Reeves, A.M.; Shigetomi, E.; Khakh, B.S. Bulk loading of calcium indicator dyes to study astrocyte physiology: Key limitations and improvements using morphological maps. J. Neurosci. 2011, 31, 9353–9358. [Google Scholar] [CrossRef] [PubMed]
- Shigetomi, E.; Bushong, E.A.; Haustein, M.D.; Tong, X.; Jackson-Weaver, O.; Kracun, S.; Xu, J.; Sofroniew, M.V.; Ellisman, M.H.; Khakh, B.S. Imaging calcium microdomains within entire astrocyte territories and endfeet with GCaMPs expressed using adeno-associated viruses. J. Gen. Physiol. 2013, 141, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Kanemaru, K.; Sekiya, H.; Xu, M.; Satoh, K.; Kitajima, N.; Yoshida, K.; Okubo, Y.; Sasaki, T.; Moritoh, S.; Hasuwa, H.; et al. In vivo visualization of subtle, transient, and local activity of astrocytes using an ultrasensitive Ca(2+) indicator. Cell Rep. 2014, 8, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Gee, J.M.; Gibbons, M.B.; Taheri, M.; Palumbos, S.; Morris, S.C.; Smeal, R.M.; Flynn, K.F.; Economo, M.N.; Cizek, C.G.; Capecchi, M.R.; et al. Imaging activity in astrocytes and neurons with genetically encoded calcium indicators following in utero electroporation. Front. Mol. Neurosci. 2015, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, R.; Huang, B.S.; Venugopal, S.; Johnston, A.D.; Chai, H.; Zeng, H.; Golshani, P.; Khakh, B.S. Ca(2+) signaling in astrocytes from Ip3r2(-/-) mice in brain slices and during startle responses in vivo. Nat. Neurosci. 2015, 18, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Wu, P.H.; Hughes, E.G.; Fukaya, M.; Tischfield, M.A.; Langseth, A.J.; Wirtz, D.; Bergles, D.E. Transient opening of the mitochondrial permeability transition pore induces microdomain calcium transients in astrocyte processes. Neuron 2017, 93, 587.e7–605.e7. [Google Scholar] [CrossRef] [PubMed]
- Bindocci, E.; Savtchouk, I.; Liaudet, N.; Becker, D.; Carriero, G.; Volterra, A. Three-dimensional Ca2+ imaging advances understanding of astrocyte biology. Science 2017, 356, eaai8185. [Google Scholar] [CrossRef] [PubMed]
- Stobart, J.L.; Ferrari, K.D.; Barrett, M.J.P.; Gluck, C.; Stobart, M.J.; Zuend, M.; Weber, B. Cortical circuit activity evokes rapid astrocyte calcium signals on a similar timescale to neurons. Neuron 2018, 98, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.A.; Kress, B.T.; Lu, Y.; Chandler-Militello, D.; Benraiss, A.; Nedergaard, M. Fluorescent Ca(2+) indicators directly inhibit the Na,K-ATPase and disrupt cellular functions. Sci. Signal. 2018, 11, eaal2039. [Google Scholar] [CrossRef] [PubMed]
- Zariwala, H.A.; Borghuis, B.G.; Hoogland, T.M.; Madisen, L.; Tian, L.; De Zeeuw, C.I.; Zeng, H.; Looger, L.L.; Svoboda, K.; Chen, T.W. A Cre-dependent GCaMP3 reporter mouse for neuronal imaging in vivo. J. Neurosci. 2012, 32, 3131–3141. [Google Scholar] [CrossRef] [PubMed]
- Monai, H.; Ohkura, M.; Tanaka, M.; Oe, Y.; Konno, A.; Hirai, H.; Mikoshiba, K.; Itohara, S.; Nakai, J.; Iwai, Y.; et al. Calcium imaging reveals glial involvement in transcranial direct current stimulation-induced plasticity in mouse brain. Nat. Commun. 2016, 7, 11100. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; O’Donnell, J.; Thrane, A.S.; Zeppenfeld, D.; Kang, H.; Xie, L.; Wang, F.; Nedergaard, M. Alpha1-adrenergic receptors mediate coordinated Ca2+ signaling of cortical astrocytes in awake, behaving mice. Cell Calcium 2013, 54, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Paukert, M.; Agarwal, A.; Cha, J.; Doze, V.A.; Kang, J.U.; Bergles, D.E. Norepinephrine controls astroglial responsiveness to local circuit activity. Neuron 2014, 82, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Pekna, M. Astrocyte reactivity and reactive astrogliosis: Costs and benefits. Physiol. Rev. 2014, 94, 1077–1098. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.; Kobayakawa, K.; Ohkawa, Y.; Kumamaru, H.; Yokota, K.; Saito, T.; Kijima, K.; Yoshizaki, S.; Harimaya, K.; Nakashima, Y.; et al. Interaction of reactive astrocytes with type I collagen induces astrocytic scar formation through the integrin-N-cadherin pathway after spinal cord injury. Nat. Med. 2017, 23, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.A.; Burda, J.E.; Ren, Y.; Ao, Y.; O’Shea, T.M.; Kawaguchi, R.; Coppola, G.; Khakh, B.S.; Deming, T.J.; Sofroniew, M.V. Astrocyte scar formation aids central nervous system axon regeneration. Nature 2016, 532, 195. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.L.; Gallo, V. The diversity and disparity of the glial scar. Nat. Neurosci. 2018, 21, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Chai, H.; Diaz-Castro, B.; Shigetomi, E.; Monte, E.; Octeau, J.C.; Yu, X.; Cohn, W.; Rajendran, P.S.; Vondriska, T.M.; Whitelegge, J.P.; et al. Neural circuit-specialized astrocytes: Transcriptomic, proteomic, morphological, and functional evidence. Neuron 2017, 95, 531.e9–549.e9. [Google Scholar] [CrossRef] [PubMed]
- John Lin, C.C.; Yu, K.; Hatcher, A.; Huang, T.W.; Lee, H.K.; Carlson, J.; Weston, M.C.; Chen, F.; Zhang, Y.; Zhu, W.; et al. Identification of diverse astrocyte populations and their malignant analogs. Nat. Neurosci. 2017, 20, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Morel, L.; Chiang, M.S.R.; Higashimori, H.; Shoneye, T.; Iyer, L.K.; Yelick, J.; Tai, A.; Yang, Y. Molecular and functional properties of regional astrocytes in the adult brain. J. Neurosci. 2017, 37, 8706–8717. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Volterra, A. Astrocytic dysfunction: Insights on the role in neurodegeneration. Brain Res. Bull. 2009, 80, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; MacVicar, B.A. In vitro ischemia promotes calcium influx and intracellular calcium release in hippocampal astrocytes. J. Neurosci. 1996, 16, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.P.; He, J.Q.; Chai, Z. Astrocytic Ca(2+) waves mediate activation of extrasynaptic nmda receptors in hippocampal neurons to aggravate brain damage during ischemia. Neurobiol. Dis. 2013, 58, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Choo, A.M.; Miller, W.J.; Chen, Y.C.; Nibley, P.; Patel, T.P.; Goletiani, C.; Morrison, B., 3rd; Kutzing, M.K.; Firestein, B.L.; Sul, J.Y.; et al. Antagonism of purinergic signalling improves recovery from traumatic brain injury. Brain 2013, 136, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.F.; Azmi, H.; Takano, T.; Xu, Q.; Peng, W.; Lin, J.; Oberheim, N.; Lou, N.; Wang, X.; Zielke, H.R.; et al. An astrocytic basis of epilepsy. Nat. Med. 2005, 11, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Kanemaru, K.; Kubota, J.; Sekiya, H.; Hirose, K.; Okubo, Y.; Iino, M. Calcium-dependent n-cadherin up-regulation mediates reactive astrogliosis and neuroprotection after brain injury. Proc. Natl. Acad. Sci. USA 2013, 110, 11612–11617. [Google Scholar] [CrossRef] [PubMed]
- Heuser, K.; Nome, C.G.; Pettersen, K.H.; Abjorsbraten, K.S.; Jensen, V.; Tang, W.; Sprengel, R.; Tauboll, E.; Nagelhus, E.A.; Enger, R. Ca2+ signals in astrocytes facilitate spread of epileptiform activity. Cereb. Cortex 2018, 28, 4036–4048. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Fellin, T.; Zhu, Y.; Lee, S.Y.; Auberson, Y.P.; Meaney, D.F.; Coulter, D.A.; Carmignoto, G.; Haydon, P.G. Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J. Neurosci. 2007, 27, 10674–10684. [Google Scholar] [CrossRef] [PubMed]
- Plata, A.; Lebedeva, A.; Denisov, P.; Nosova, O.; Postnikova, T.Y.; Pimashkin, A.; Brazhe, A.; Zaitsev, A.V.; Rusakov, D.A.; Semyanov, A. Astrocytic atrophy following status epilepticus parallels reduced Ca(2+) activity and impaired synaptic plasticity in the rat hippocampus. Front. Mol. Neurosci. 2018, 11, 215. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Shih, P.Y.; Gomi, H.; Yoshida, T.; Nakai, J.; Ando, R.; Furuichi, T.; Mikoshiba, K.; Semyanov, A.; Itohara, S. Astrocytic Ca2+ signals are required for the functional integrity of tripartite synapses. Mol. Brain 2013, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Delekate, A.; Fuchtemeier, M.; Schumacher, T.; Ulbrich, C.; Foddis, M.; Petzold, G.C. Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer’s disease mouse model. Nat. Commun. 2014, 5, 5422. [Google Scholar] [CrossRef] [PubMed]
- Kuchibhotla, K.V.; Lattarulo, C.R.; Hyman, B.T.; Bacskai, B.J. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 2009, 323, 1211–1215. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Wang, T.; Cui, W.; Haydon, P.G. Photothrombosis ischemia stimulates a sustained astrocytic Ca2+ signaling in vivo. Glia 2009, 57, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Fordsmann, J.C.; Murmu, R.P.; Cai, C.; Brazhe, A.; Thomsen, K.J.; Zambach, S.A.; Lonstrup, M.; Lind, B.L.; Lauritzen, M. Spontaneous astrocytic Ca(2+) activity abounds in electrically suppressed ischemic penumbra of aged mice. Glia 2019, 67, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Shigetomi, E.; Yasuda, R.; Sato, R.; Nakano, M.; Tashiro, K.; Tanaka, K.F.; Ikenaka, K.; Mikoshiba, K.; Mizuta, I.; et al. Aberrant astrocyte Ca(2+) signals “axca signals” exacerbate pathological alterations in an Alexander disease model. Glia 2018, 66, 1053–1067. [Google Scholar] [CrossRef] [PubMed]
- Shigetomi, E.; Bowser, D.N.; Sofroniew, M.V.; Khakh, B.S. Two forms of astrocyte calcium excitability have distinct effects on NMDA receptor-mediated slow inward currents in pyramidal neurons. J. Neurosci. 2008, 28, 6659–6663. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S.; McCarthy, K.D. Astrocyte calcium signaling: From observations to functions and the challenges therein. Cold Spring Harb. Perspect. Biol. 2015, 7, a020404. [Google Scholar] [CrossRef] [PubMed]
- Ujita, S.; Sasaki, T.; Asada, A.; Funayama, K.; Gao, M.; Mikoshiba, K.; Matsuki, N.; Ikegaya, Y. cAMP-dependent calcium oscillations of astrocytes: An implication for pathology. Cereb. Cortex 2017, 27, 1602–1614. [Google Scholar] [CrossRef] [PubMed]
- Petravicz, J.; Fiacco, T.A.; McCarthy, K.D. Loss of IP3 receptor-dependent Ca2+ increases in hippocampal astrocytes does not affect baseline Ca1 pyramidal neuron synaptic activity. J. Neurosci. 2008, 28, 4967–4973. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, M.W.; Arizono, M.; Hisatsune, C.; Bannai, H.; Ebisui, E.; Sherwood, J.L.; Panatier, A.; Oliet, S.H.; Mikoshiba, K. Astrocytic IP3 Rs: Contribution to Ca(2+) signalling and hippocampal LTP. Glia 2017, 65, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Rungta, R.L.; Bernier, L.P.; Dissing-Olesen, L.; Groten, C.J.; LeDue, J.M.; Ko, R.; Drissler, S.; MacVicar, B.A. Ca2+ transients in astrocyte fine processes occur via Ca2+ influx in the adult mouse hippocampus. Glia 2016, 64, 2093–2103. [Google Scholar] [CrossRef] [PubMed]
- Okubo, Y.; Kanemaru, K.; Suzuki, J.; Kobayashi, K.; Hirose, K.; Iino, M. Inositol 1,4,5-trisphosphate receptor type 2-independent Ca(2+) release from the endoplasmic reticulum in astrocytes. Glia 2018, 67, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Staats, K.A.; Humblet-Baron, S.; Bento-Abreu, A.; Scheveneels, W.; Nikolaou, A.; Deckers, K.; Lemmens, R.; Goris, A.; Van Ginderachter, J.A.; Van Damme, P.; et al. Genetic ablation of IP3 receptor 2 increases cytokines and decreases survival of SOD1G93A mice. Hum. Mol. Genet. 2016, 25, 3491–3499. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; McConnell, E.; Pare, J.F.; Xu, Q.; Chen, M.; Peng, W.; Lovatt, D.; Han, X.; Smith, Y.; Nedergaard, M. Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science 2013, 339, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Haustein, M.D.; Kracun, S.; Lu, X.H.; Shih, T.; Jackson-Weaver, O.; Tong, X.; Xu, J.; Yang, X.W.; O’Dell, T.J.; Marvin, J.S.; et al. Conditions and constraints for astrocyte calcium signaling in the hippocampal mossy fiber pathway. Neuron 2014, 82, 413–429. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, A.N.; Kowalewski, J.M.; Renner, M.; Bousset, L.; Koulakoff, A.; Melki, R.; Giaume, C.; Triller, A. Beta-amyloid and ATP-induced diffusional trapping of astrocyte and neuronal metabotropic glutamate type-5 receptors. Glia 2013, 61, 1673–1686. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Iyer, A.; Ronco, V.; Grolla, A.A.; Canonico, P.L.; Aronica, E.; Genazzani, A.A. Amyloid beta deregulates astroglial mGluR5-mediated calcium signaling via calcineurin and nf-kb. Glia 2013, 61, 1134–1145. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; van Vliet, E.A.; Mayboroda, O.A.; Troost, D.; da Silva, F.H.; Gorter, J.A. Upregulation of metabotropic glutamate receptor subtype mGluR3 and mGluR5 in reactive astrocytes in a rat model of mesial temporal lobe epilepsy. Eur. J. Neurosci. 2000, 12, 2333–2344. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Hayashi, H.; Ishikawa, T.; Shibata, K.; Shigetomi, E.; Shinozaki, Y.; Inada, H.; Roh, S.E.; Kim, S.J.; Lee, G.; et al. Cortical astrocytes rewire somatosensory cortical circuits for peripheral neuropathic pain. J. Clin. Investig. 2016, 126, 1983–1997. [Google Scholar] [CrossRef] [PubMed]
- Umpierre, A.D.; West, P.J.; White, J.A.; Wilcox, K.S. Conditional knockout of mGluR5 from astrocytes during epilepsy development impairs high-frequency glutamate uptake. J. Neurosci. 2019, 39, 727–742. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Romano, C.; Cotman, C.W. Growth factor upregulation of a phosphoinositide-coupled metabotropic glutamate receptor in cortical astrocytes. J. Neurosci. 1995, 15, 6103–6109. [Google Scholar] [CrossRef] [PubMed]
- Buscemi, L.; Ginet, V.; Lopatar, J.; Montana, V.; Pucci, L.; Spagnuolo, P.; Zehnder, T.; Grubisic, V.; Truttman, A.; Sala, C.; et al. Homer1 scaffold proteins govern Ca2+ dynamics in normal and reactive astrocytes. Cereb. Cortex 2017, 27, 2365–2384. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, S. Synchronization of Ca2+ oscillations: Involvement of atp release in astrocytes. FEBS J. 2010, 277, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Shigetomi, E.; Hirayama, Y.J.; Ikenaka, K.; Tanaka, K.F.; Koizumi, S. Role of purinergic receptor P2Y1 in spatiotemporal Ca(2+) dynamics in astrocytes. J. Neurosci. 2018, 38, 1383–1395. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Ferradas, C.; Morales, J.C.; Wellmann, M.; Nualart, F.; Roncagliolo, M.; Fuenzalida, M.; Bonansco, C. Enhanced astroglial Ca2+ signaling increases excitatory synaptic strength in the epileptic brain. Glia 2015, 63, 1507–1521. [Google Scholar] [CrossRef] [PubMed]
- Kuboyama, K.; Harada, H.; Tozaki-Saitoh, H.; Tsuda, M.; Ushijima, K.; Inoue, K. Astrocytic P2y(1) receptor is involved in the regulation of cytokine/chemokine transcription and cerebral damage in a rat model of cerebral ischemia. J. Cereb. Blood Flow Metab. 2011, 31, 1930–1941. [Google Scholar] [CrossRef] [PubMed]
- Reichenbach, N.; Delekate, A.; Breithausen, B.; Keppler, K.; Poll, S.; Schulte, T.; Peter, J.; Plescher, M.; Hansen, J.N.; Blank, N.; et al. P2Y1 receptor blockade normalizes network dysfunction and cognition in an Alzheimer’s disease model. J. Exp. Med. 2018, 215, 1649–1663. [Google Scholar] [CrossRef] [PubMed]
- Wellmann, M.; Alvarez-Ferradas, C.; Maturana, C.J.; Saez, J.C.; Bonansco, C. Astroglial Ca(2+)-dependent hyperexcitability requires P2Y1 purinergic receptors and pannexin-1 channel activation in a chronic model of epilepsy. Front. Cell. Neurosci. 2018, 12, 446. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, L.; Shen, W.; Nobili, P.; Virenque, A.; Ulmann, L.; Audinat, E. Blocking TNFα-driven astrocyte purinergic signaling restores normal synaptic activity during epileptogenesis. Glia 2018, 66, 2673–2683. [Google Scholar] [CrossRef] [PubMed]
- Perea, G.; Gomez, R.; Mederos, S.; Covelo, A.; Ballesteros, J.J.; Schlosser, L.; Hernandez-Vivanco, A.; Martin-Fernandez, M.; Quintana, R.; Rayan, A.; et al. Activity-dependent switch of gabaergic inhibition into glutamatergic excitation in astrocyte-neuron networks. Elife 2016, 5, e20362. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, L.; Losi, G.; Lia, A.; Melone, M.; Chiavegato, A.; Gomez-Gonzalo, M.; Sessolo, M.; Bovetti, S.; Forli, A.; Zonta, M.; et al. Interneuron-specific signaling evokes distinctive somatostatin-mediated responses in adult cortical astrocytes. Nat. Commun. 2018, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.C.; Chen, R.Y.; Cheng, T.C.; Chiang, Y.C.; Shen, M.L.; Hsu, L.L.; Zhou, N. Spreading depression promotes astrocytic calcium oscillations and enhances gliotransmission to hippocampal neurons. Cereb. Cortex 2018, 28, 3204–3216. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.G.; Hsiao, E.C.; Wang, M.M.; Ho, K.; Kim, D.H.; Wang, X.; Guo, W.; Kang, J.; Yu, G.Q.; Adame, A.; et al. Astrocytic adenosine receptor A2A and GS-coupled signaling regulate memory. Nat. Neurosci. 2015, 18, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Shigetomi, E.; Jackson-Weaver, O.; Huckstepp, R.T.; O’Dell, T.J.; Khakh, B.S. TRPA1 channels are regulators of astrocyte basal calcium levels and long-term potentiation via constitutive D-serine release. J. Neurosci. 2013, 33, 10143–10153. [Google Scholar] [CrossRef] [PubMed]
- Bosson, A.; Paumier, A.; Boisseau, S.; Jacquier-Sarlin, M.; Buisson, A.; Albrieux, M. TRPA1 channels promote astrocytic Ca(2+) hyperactivity and synaptic dysfunction mediated by oligomeric forms of amyloid-beta peptide. Mol. Neurodegener. 2017, 12, 53. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.I.; Lee, H.T.; Lin, H.C.; Tsay, H.J.; Tsai, F.C.; Shyue, S.K.; Lee, T.S. Role of transient receptor potential ankyrin 1 channels in Alzheimer’s disease. J. Neuroinflamm. 2016, 13, 92. [Google Scholar] [CrossRef] [PubMed]
- Pirttimaki, T.M.; Codadu, N.K.; Awni, A.; Pratik, P.; Nagel, D.A.; Hill, E.J.; Dineley, K.T.; Parri, H.R. Alpha7 nicotinic receptor-mediated astrocytic gliotransmitter release: Abeta effects in a preclinical alzheimer’s mouse model. PLoS ONE 2013, 8, e81828. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Liu, Q.; Li, R.; Wang, A.; Bu, Q.; Wang, K.H.; Chang, Q. Mechanism and consequence of abnormal calcium homeostasis in rett syndrome astrocytes. Elife 2018, 7, e33417. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, Y.; Ikeda-Matsuo, Y.; Notomi, S.; Enaida, H.; Kinouchi, H.; Koizumi, S. Astrocyte-mediated ischemic tolerance. J. Neurosci. 2015, 35, 3794–3805. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, Y.; Koizumi, S. Hypoxia-independent mechanisms of HIF-1alpha expression in astrocytes after ischemic preconditioning. Glia 2017, 65, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.R.; Kong, L.; Hanna, M.G.T.; Hoffman, B.; Krencik, R.; Bradley, R.; Hagemann, T.; Choi, J.; Doers, M.; Dubovis, M.; et al. Mutations in GFAP disrupt the distribution and function of organelles in human astrocytes. Cell Rep. 2018, 25, 947.e4–958.e4. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, G.O.; Wang, Y.; Shi, G.; Wang, Y.; Sun, J.; Papadopoulos, S.; Broussard, G.J.; Unger, E.K.; Deng, W.; Weick, J.; et al. Aberrant calcium signaling in astrocytes inhibits neuronal excitability in a human down syndrome stem cell model. Cell Rep. 2018, 24, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Fiacco, T.A.; McCarthy, K.D. Multiple lines of evidence indicate that gliotransmission does not occur under physiological conditions. J. Neurosci. 2018, 38, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Savtchouk, I.; Volterra, A. Gliotransmission: Beyond black-and-white. J. Neurosci. 2018, 38, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Carmignoto, G.; Haydon, P.G. Astrocyte calcium signaling and epilepsy. Glia 2012, 60, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Rakers, C.; Petzold, G.C. Astrocytic calcium release mediates peri-infarct depolarizations in a rodent stroke model. J. Clin. Investig. 2017, 127, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Habbas, S.; Santello, M.; Becker, D.; Stubbe, H.; Zappia, G.; Liaudet, N.; Klaus, F.R.; Kollias, G.; Fontana, A.; Pryce, C.R.; et al. Neuroinflammatory TNFalpha impairs memory via astrocyte signaling. Cell 2015, 163, 1730–1741. [Google Scholar] [CrossRef] [PubMed]
- Pascual, O.; Ben Achour, S.; Rostaing, P.; Triller, A.; Bessis, A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. USA 2012, 109, E197–E205. [Google Scholar] [CrossRef] [PubMed]
- Santello, M.; Bezzi, P.; Volterra, A. TNFalpha controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron 2011, 69, 988–1001. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, N.B.; Attwell, D. Do astrocytes really exocytose neurotransmitters? Nat. Rev. Neurosci. 2010, 11, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Agulhon, C.; Sun, M.Y.; Murphy, T.; Myers, T.; Lauderdale, K.; Fiacco, T.A. Calcium signaling and gliotransmission in normal vs. Reactive astrocytes. Front. Pharmacol. 2012, 3, 139. [Google Scholar] [CrossRef] [PubMed]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Gonzalo, M.; Zehnder, T.; Requie, L.M.; Bezzi, P.; Carmignoto, G. Insights into the release mechanism of astrocytic glutamate evoking in neurons NMDA receptor-mediated slow depolarizing inward currents. Glia 2018, 66, 2188–2199. [Google Scholar] [CrossRef] [PubMed]
- Beppu, K.; Sasaki, T.; Tanaka, K.F.; Yamanaka, A.; Fukazawa, Y.; Shigemoto, R.; Matsui, K. Optogenetic countering of glial acidosis suppresses glial glutamate release and ischemic brain damage. Neuron 2014, 81, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. Gaba from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Guo, Z.; Gearing, M.; Chen, G. Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an alzheimer’s [corrected] disease model. Nat. Commun. 2014, 5, 4159. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.; An, H.; Lim, J.; Woo, J.; Lee, J.; Ryu, H.; Lee, C.J. Astrocytic proBDNF and tonic GABA distinguish active versus reactive astrocytes in hippocampus. Exp. Neurobiol. 2018, 27, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Schipke, C.G.; Boucsein, C.; Ohlemeyer, C.; Kirchhoff, F.; Kettenmann, H. Astrocyte Ca2+ waves trigger responses in microglial cells in brain slices. FASEB J. 2002, 16, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.J.; Eroglu, C. Cell biology of astrocyte-synapse interactions. Neuron 2017, 96, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Nabekura, J.; Koizumi, S. Astrocyte-mediated synapse remodeling in the pathological brain. Glia 2017, 65, 1719–1727. [Google Scholar] [CrossRef] [PubMed]
- Middeldorp, J.; Hol, E.M. Gfap in health and disease. Prog. Neurobiol. 2011, 93, 421–443. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xie, Y.; Zhang, N.; Yu, Y.; Zhang, Q.; Ding, S. Disruption of IP(3)R2-mediated Ca(2)(+) signaling pathway in astrocytes ameliorates neuronal death and brain damage while reducing behavioral deficits after focal ischemic stroke. Cell Calcium 2015, 58, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Rakers, C.; Schmid, M.; Petzold, G.C. TRPV4 channels contribute to calcium transients in astrocytes and neurons during peri-infarct depolarizations in a stroke model. Glia 2017, 65, 1550–1561. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, Y.; Okabe, K.; Shibasaki, K.; Funatsu, T.; Matsuki, N.; Ikegaya, Y.; Koyama, R. Ischemic brain injury leads to brain edema via hyperthermia-induced TRPV4 activation. J. Neurosci. 2018, 38, 5700–5709. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Diaz-Castro, B.; Looger, L.L.; Khakh, B.S. Dysfunctional calcium and glutamate signaling in striatal astrocytes from huntington’s disease model mice. J. Neurosci. 2016, 36, 3453–3470. [Google Scholar] [CrossRef] [PubMed]
- Kanemaru, K.; Okubo, Y.; Hirose, K.; Iino, M. Regulation of neurite growth by spontaneous Ca2+ oscillations in astrocytes. J. Neurosci. 2007, 27, 8957–8966. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Taylor, A.M.W.; Nagai, J.; Golshani, P.; Evans, C.J.; Coppola, G.; Khakh, B.S. Reducing astrocyte calcium signaling in vivo alters striatal microcircuits and causes repetitive behavior. Neuron 2018, 99, 1170–1187. [Google Scholar] [CrossRef] [PubMed]
- Takano, T.; He, W.; Han, X.; Wang, F.; Xu, Q.; Wang, X.; Oberheim Bush, N.A.; Cruz, N.; Dienel, G.A.; Nedergaard, M. Rapid manifestation of reactive astrogliosis in acute hippocampal brain slices. Glia 2014, 62, 78–95. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shigetomi, E.; Saito, K.; Sano, F.; Koizumi, S. Aberrant Calcium Signals in Reactive Astrocytes: A Key Process in Neurological Disorders. Int. J. Mol. Sci. 2019, 20, 996. https://doi.org/10.3390/ijms20040996
Shigetomi E, Saito K, Sano F, Koizumi S. Aberrant Calcium Signals in Reactive Astrocytes: A Key Process in Neurological Disorders. International Journal of Molecular Sciences. 2019; 20(4):996. https://doi.org/10.3390/ijms20040996
Chicago/Turabian StyleShigetomi, Eiji, Kozo Saito, Fumikazu Sano, and Schuichi Koizumi. 2019. "Aberrant Calcium Signals in Reactive Astrocytes: A Key Process in Neurological Disorders" International Journal of Molecular Sciences 20, no. 4: 996. https://doi.org/10.3390/ijms20040996
APA StyleShigetomi, E., Saito, K., Sano, F., & Koizumi, S. (2019). Aberrant Calcium Signals in Reactive Astrocytes: A Key Process in Neurological Disorders. International Journal of Molecular Sciences, 20(4), 996. https://doi.org/10.3390/ijms20040996