Abstract
The development of leukemia is a step-wise process that is associated with molecular diversification and clonal selection of neoplastic stem cells. Depending on the number and combinations of lesions, one or more sub-clones expand/s after a variable latency period. Initial stages may develop early in life or later in adulthood and include premalignant (indolent) stages and the malignant phase, defined by an acute leukemia. We recently proposed a cancer model in which the earliest somatic lesions are often age-related early mutations detectable in apparently healthy individuals and where additional oncogenic mutations will lead to the development of an overt neoplasm that is usually a preleukemic condition such as a myelodysplastic syndrome. These neoplasms may or may not transform to overt acute leukemia over time. Thus, depending on the type and number of somatic mutations, clonal hematopoiesis (CH) can be divided into CH with indeterminate potential (CHIP) and CH with oncogenic potential (CHOP). Whereas CHIP mutations per se usually create the molecular background of a neoplastic process, CHOP mutations are disease-related or even disease-specific lesions that trigger differentiation and/or proliferation of neoplastic cells. Over time, the acquisition of additional oncogenic events converts preleukemic neoplasms into secondary acute myeloid leukemia (sAML). In the present article, recent developments in the field are discussed with a focus on CHOP mutations that lead to distinct myeloid neoplasms, their role in disease evolution, and the impact of additional lesions that can drive a preleukemic neoplasm into sAML.
1. Introduction
It is generally appreciated that cancer evolution is a step-wise process that is associated with molecular diversification and clonal selection of neoplastic stem cells [1,2,3,4,5,6]. Blood cancer evolution may begin early in life or later in adulthood and includes premalignant and malignant stages. Thus, in many instances, the development of (blood) cancer is a long-lasting process that takes several years or even decades [1,2,3,4,5,6,7,8]. In chronic (indolent) myeloid neoplasms, such as the myelodysplastic syndromes (MDS), myeloproliferative neoplasms (MPN), or chronic myeloid leukemia (CML), the disease is separable into premalignant (non-aggressive) phases and a terminal phase that resembles secondary acute myeloid leukemia (sAML) [9,10,11,12,13,14,15]. However, even before an overt myeloid neoplasm, such as a MDS, develops, a clonal pre-phase of the disease may be detected by chance or by screening apparently healthy individuals [8,16,17,18,19,20,21,22]. Such pre-phase is defined by normal or near normal blood counts and detection of one or more somatic mutations, which per se (as isolated lesion) exhibit low oncogenic potential [8,16,17,18,19,20,21,22]. Whereas, in some of these individuals, a myeloid neoplasm develops in the follow-up, others will never develop an overt myeloid disorder during their lifetime. The risk of disease evolution (risk to develop a myeloid neoplasm) depends on the type of mutations and other factors. Clonal hematopoiesis (CH) and the related mutations can thus be divided into CH with indeterminate potential (CHIP) and CH with oncogenic potential (CHOP) [8].
We and others have recently proposed a model of cancer evolution where the earliest stages of carcinogenesis are defined by expression of somatic mutations in small-sized (yet stem cell-containing) cell fractions [8,19,20,21,22,23,24,25]. Later, over time, one or more sub-clone(s) expand(s) and replace normal cells, depending on additional somatic lesions (Figure 1) [8]. As long as the neoplastic cells retain their full differentiation and maturation potential and can be controlled by the niche and the immune system, the involved clones will remain indolent and may, over time, replace or even mimic (at least to a degree) the normal organ in functional and morphological terms. However, as soon as the dominant clone(s) escape(s) most control mechanisms, the disease will further expand and progress into an aggressive malignancy. The end-stage of such malignancy (sAML) is usually resistant not only against most endogenous control systems, but also against diverse therapeutic maneuvers [9,10,11,12,13,14,15].
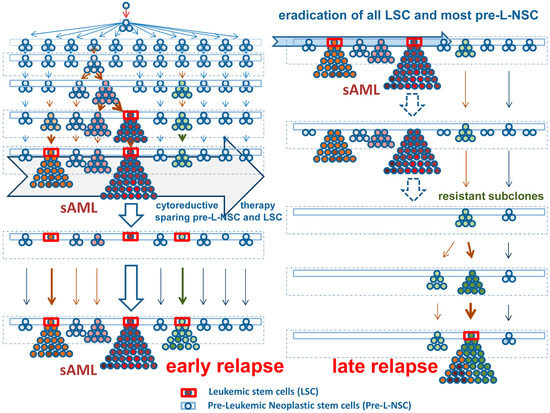
Figure 1.
Development and diversification of leukemic stem cells in secondary acute myeloid leukemia (sAML). Left panel, upper part: An initial oncogenic event transforms a normal stem cell into a premalignant (preleukemic) neoplastic stem cell (Pre-L-NSC) (blue boxes). These cells or their daughter cells acquire early somatic mutations. Usually, these have low oncogenic potential (blue-colored cells) and are thus slowly cycling or dormant cells so that the mutation is not detectable. After some time, more daughter clones develop and the somatic lesions may be detected and classified as clonal hematopoiesis with indeterminate potential (CHIP). After several years or decades, one or more daughter clones and their stem cells expand and may replace normal hematopoiesis. At that time, some of the stem cell clones may have acquired disease-specific oncogenic driver lesions (red-colored cells). Still, these cells may be indistinguishable from normal cells by morphology and in functional terms. In a next step, one or more of the sub-clones acquire additional driver mutations or lose tumor suppressor genes. As a result, the stem cells are now cycling and the neoplastic process forms a visible overt myeloid neoplasm (red-colored prominent clones—upper left panel). In most instances, these neoplasms still behave as indolent driver-positive neoplasm for some time. However, unless treated, many of these conditions will finally transform into a secondary acute myeloid leukemia (sAML). At that time, long-term disease propagating cells are called leukemic stem cells (LSC—red boxes). Note that all of the Pre-L-NSC-derived clones are also still present and can be detected (as small-sized sub-clones) in an overt sAML. Left lower panel: Nonspecific cytoreductive (palliative) therapy (example: hydroxyurea) can suppress the growth of cycling stem and progenitor cells for some time but cannot eradicate any of the Pre-L-NSC or LSC. After a variable (usually short) time period, a relapse develops. Right panel: Most interventional therapies (intensive chemotherapy, targeted drugs, or stem cell transplantation) are able to eradicate most or all of the LSC and their progeny, but not all Pre-L-NSC. When all LSC are killed, the patient enters complete remission and operational cure. In these patients, the Pre-L-NSC may or may not be detected as minimal residual disease. These Pre-L-NSC may (or may not) produce a late relapse after several months or years. Although some of the early mutations (rarely even drivers) of the original sAML clone may be detected in such relapsing disease, the molecular aberration profiles usually differ substantially from the initial molecular make-up of the sAML clone.
Depending on the disease variant and the types and numbers (combinations) of somatic mutations acquired, the premalignant phases of a myeloid neoplasm bear a low or high risk to transform to sAML [8,19,20,21,22,23,24,25]. Interestingly, certain myeloid neoplasms, such as advanced MDS, most MDS/MPN overlap disorders, and chronic phase CML bear a (relatively) high risk, whereas other clonal conditions are at a clearly lower risk, but may eventually also transform to sAML. These include, among others, chronic eosinophilic leukemia (CEL), MDS with isolated del(5q), or indolent systemic mastocytosis (ISM). So far, it remains unknown why some conditions are associated with a relatively high risk of sAML transformation. Possible explanations are the differential oncogenic potential of various driver mutations and the presence of additional germline or somatic mutations.
In the current article, we review the mutational landscape of myeloid neoplasms and try to link individual mutations and mutation-combinations to clinical phenotypes and the risk of progression. We also discuss why CHOP mutations can trigger disease evolution and why some CHOP mutants are less oncogenic than others.
2. Clonal Hematopoiesis (CH) of Indeterminate Potential (CHIP)
The term CHIP was coined to describe the presence of clonal somatic mutations (otherwise detected in myeloid neoplasms: MDS, AML, and others) in leukocytes obtained from apparently healthy individuals or subjects with minimal blood count abnormalities [8,21,22,26,27,28]. In patients with CHIP, diagnostic criteria for MDS, MPN, or other myeloid neoplasms are not fulfilled even if the size of the ’CHIP clone’ is substantial. Most patients with CHIP are older healthy individuals. Therefore, the term age-related clonal hematopoiesis (ARCH) was also proposed [19]. In patients with CHIP, the risk to develop a myeloid (hematopoietic) neoplasm is slightly elevated compared to controls without CH [18,19,20,21,26,27,28]. In addition, these patients may be at relatively high risk to develop progressive atherosclerosis and related cardiovascular disorders [19,29]. In a subset of patients with CHIP/ARCH, however, no malignancy and no severe cardiovascular disease develop.
In some individuals, the CHIP clones are small-sized and may thus escape detection by conventional screening/sequencing approaches. However, most next generation sequencing (NGS) assays have sufficient sensitivity to detect relatively small clones (mutant allele burden 1–5%) and thus represent the preferred method for the diagnostic assessment of CHIP. NGS assays can also be modified to reliably detect even smaller clones (mutant allele burden clearly <1%). These very small hematopoietic cell clones are currently not considered as CHIP per definition since their prevalence is even higher and their clinical impact remains unclear. The generally accepted definition of CHIP includes a minimal allele burden of 2%, the absence of persistent (≥4 months) cytopenia and exclusion of an underlying overt pathology associated with the somatic lesion [21,22]. The term CHIP should thus only be applied to individuals who have normal blood counts. In the case that slight cytopenia is also detected and the criteria for MDS or other myeloid neoplasms are not fulfilled, the diagnosis changes to clonal cytopenia of undetermined potential (CCUS), which is a rare condition [21,22]. When detected as an isolated defect (in the absence of other lesions or loss of tumor suppressors), CHIP mutations are indicative of a rather good prognosis regarding clonal stability, and only a small subset of these individuals will eventually develop a hematopoietic neoplasm over time [18,19,20,21]. A list of frequently reported CHIP mutations is provided in Table 1.

Table 1.
Examples of mutations that have been described in the context of clonal hematopoiesis of indeterminate potential (CHIP) or age-related clonal hematopoiesis (ARCH).
CHIP-like mutations can also be detected in patients with overt myeloid neoplasms, including MDS, AML, and mast cell neoplasms (Table 1 and Table 2) [30,31,32,33,34,35,36,37,38,39]. However, although some of these mutations are more frequently detected in certain myeloid neoplasms than others, most are not disease-specific. Another interesting aspect is that CHIP-like mutations can sometimes also be detected after successful therapy when the predominant clones have been eradicated [40,41,42,43]. This is best explained by the stem model of cancer evolution where early sub-clones are formed and acquire these mutations, but only a few of these sub-clones expand after acquiring driver lesions and additional mutations over time (Figure 1) [8,23,44,45,46,47,48]. After debulking, the early sub-clones may still be present because of their slow proliferation rate and drug resistance (Figure 1). Such early clones exhibiting CHIP-like lesions may either remain silent for the lifetime of the host or they become relevant clinically: First, they may produce a late and usually driver-negative relapse (Figure 1). Second, they may contribute to the risk to develop a severe cardiovascular (vascular occlusive) disease [19,29,40,41,42,43].
Table 2.
Some clinically relevant somatic mutations detectable in myeloid neoplasms based on oncogenic potential and risk to transform to secondary acute myeloid leukemia (sAML).
In myeloid neoplasm, isolated CHIP-like mutations may still be indicative of a rather good prognosis regarding clonal stability [30,31,32,33,34,35,36,37,38,39]. However, in the context of an overt myeloid neoplasm, CHIP-like mutations are often indicative of a poor prognosis, especially when multiple CHIP-like mutations are expressed, or the CHIP-like mutation is accompanied by clonal hematopoiesis of oncogenic potential or loss of a tumor suppressor gene (Table 2) [30,31,32,33,34,35,36,37,38,39].
It was also described that the prognostic impact of CHIP-like mutations regarding survival and AML evolution depends on (i) the type of the mutation, (ii) the dynamics of clonal evolution (rapid expansion of sub-clones carrying CHIP-like mutations is a poor prognostic sign), and (iii) the underlying primary neoplasm. For example, a stable TET2 mutation in low-risk MDS may be indicative of a good prognosis, whereas a rapidly expanding ASXL1-mutated clone in CMML-2 has to be regarded as a poor prognostic sign concerning AML evolution.
3. Clonal Hematopoiesis of Oncogenic Potential (CHOP) and Separation from CHIP
Whereas isolated CHIP-type mutations can be detected in healthy individuals who stay healthy for their lifetime, CHOP mutations are usually associated with manifestation of an overt neoplasm. In fact, most of these individuals will develop a hematopoietic malignancy, although CHOP-positive cases presenting with a long-lasting disease-free survival have been described. For example, although BCR-ABL1 can be detected in small-sized clones in a few healthy individuals [16], most patients with a persistent BCR-ABL1+ clone have or will develop a BCR-ABL1-positive leukemia. Even in those with low mutant burden, BCR-ABL1 must be regarded as a high-risk condition (CHOP) that is likely to transform to an overt leukemia after a variable latency period. As in other myeloid neoplasms, the CML clone acquires additional lesions and hits when the disease progresses into accelerated phase and blast phase [64,65,66]. In addition, early CHIP-like lesions may be detected in the CML clone [64,65,66]. These lesions may also persist during successful treatment with BCR-ABL1 tyrosine kinase inhibitors (TKI) and may confer a potential risk for the development of cardiovascular events during treatment with certain TKI, such as nilotinib [67].
In the JAK2-mutated MPN, the situation is similar compared to CML. However, in contrast to BCR-ABL1, the JAK2 V617F mutation status per se confers a high risk for the development of cardio-vascular events [17,68,69,70,71]. Therefore, JAK2 V617F+ hematopoiesis is often detected quite early, sometimes long before an overt MPN is diagnosed. However, again, the risk for the development of an MPN is high and most patients will eventually develop such a disease over time [17,68,69,70,71]. Therefore, although JAK2 V617F exhibits some features of a CHIP-like mutation, it is generally considered to belong to the group of CHOP mutations.
In patients with MPN, other driver lesions may also be detected, including mutations in the calreticulin (CALR) gene or in the MPL gene [72,73,74]. In addition, apart from these drivers and JAK2 V617F, a number of additional mutations may be detected in patients with MPN. These include, among others, mutations in TET2, ASXL1, SRSF2, DNMT3A, U2AF1, CBL, KIT, RUNX1, and EZH2 [13,75,76] (Table 1 and Table 2). Moreover, neoplastic cells in MPN patients may acquire mutations in TP53, which is usually associated with disease progression to sAML [13,75,76].
In MDS, no classical recurrent driver lesions, such as BCR-ABL1 or JAK2 V617F, have been identified. Rather, in these patients, a number of different molecular lesions and combinations of somatic mutations are found [30,31,32,33,34,35]. Based on clinical correlates regarding progression and survival, these lesions may be divided into CHIP-like mutations (Table 1 and Table 2) and more oncogenic (CHOP-like) driver mutations (Table 2 and Table 3). The latter are associated with a substantial risk to transform to AML, and include, among other, mutations in FLT3, RUNX1, WT1, NPM1, NRAS, and TP53 [30,31,32,33,34,35,77]. However, these CHOP mutations are usually not detected in a pre-diagnostic phase (in healthy subjects) but only (mostly) in the context of a full-blown MDS or AML.
Table 3.
Somatic mutations producing clonal hematopoiesis of oncogenic potential (CHOP).
In chronic myelomonocytic leukemia (CMML), a classical MPN/MDS overlap disorder, mutations can also be divided into CHIP-like and more oncogenic (CHOP-like) mutations (Table 2). Genetic lesions that are typically detectable in CMML include mutations in SRSF2 (about 50% of patients), TET2 (50–60%), and ASXL1 (35–49%) (Table 1). Mutations associated with disease progression to sAML include mutations in CBL, NRAS, KRAS, RUNX1, and SETBP1 [55,78,79,80,81,82]. These mutations are detectable in at least 10% of all patients with CMML and are associated with poor prognoses. Especially RAS-pathway mutations and complex mutation patterns are highly predictive for progression to sAML. Mutations in ASXL1 and TET2 are also detected frequently in CMML. As an isolated lesion in CMML, a TET2 mutation may be regarded as a CHIP-like mutation. However, in the CMML-context, ASXL1 mutations are indicative of a poor prognosis, especially when other mutations are also expressed in CMML cells [83,84,85]. The same holds true for other rare driver mutations, such as BRAF mutations or FLT3 mutations [86,87]. An overview of CHOP-type mutations is provided in Table 2.
In de novo AML, a number of disease-related or even AML-specific driver mutations, such as RUNX1-RUNX1T1 (CBFB-MYH11), PML-RARa, FLT3 mutations, KIT D816V, or NPM1 mutations, have been identified [37,88,89,90,91,92]. Some of these mutations are even diagnostic and serve as diagnostic AML criteria in the World Health Organization (WHO) classification [37,89,90]. Other mutations contribute to clonal expansion of leukemic cells and are indicative of a poor prognosis (examples: TP53 mutations, RAS pathway mutations, multiple somatic mutations) similar to the situation in CMML [37,89,90]. In addition, CHIP-like mutations can be detected in both de novo AML patients and (even more frequently) in patients with sAML [37,89,90,91].
Based on preclinical data and clinical studies, the low oncogenic potential of individual CHIP-like mutations may change to a higher oncogenic potential when the lesion is expressed together with other CHIP-like mutations or with a CHOP-type driver lesion. Indeed, in most disease models analyzed, the presence of multimutated sub-clones is usually associated with disease progression and an unfavorable outcome [36,37,38,39,82,83,84,85,89,90]. An exception may be the presence of two CHIP-like mutations in two coexisting and clearly separable (independent) sub-clones.
Another important aspect is that oncogenic (CHOP-like) mutations may also be expressed in the germline, thereby leading to a familial predisposition to develop a myeloid neoplasm, including AML. Such germline mutations were described in RUNX1, CBL, KIT, and other target genes [92,93,94,95]. In addition, it was described that certain gene constellations (genetic patterns or gene variations) predispose for the acquisition of CHOP mutations, such as JAK2 V617F (MPN) or KIT D816V (mastocytosis) [96,97,98,99]. In these conditions, familial clustering of MPN or mastocytosis is found, but the oncogenic driver lesion is a somatic defect [96,97,98,99].
Finally, inherited gene defects may be associated with loss of tumor suppressor genes or tumor suppressor function and may thereby contribute to the development of myeloid neoplasms, including MDS, CMML, and AML.
4. Classification of CHOP: Drivers of Differentiation, Proliferation, Maturation, and/or Oncogenesis
The term ‘oncogenic’ is often used to describe a somatic process that converts a non-neoplastic or a premalignant neoplastic condition into an overt malignancy. This definition implies that these mutations (or other events) provide a growth and survival advantage over normal cells. However, in many instances, the driver mutation per se is promoting differentiation and maturation rather than proliferation, unless additional mutations are also expressed by neoplastic cells.
In general, CHOP mutations can be divided into (a) disease specific drivers that trigger differentiation and maturation without promoting substantial proliferation (weak oncogenes) like KIT D816V, (b) disease-specific drivers of differentiation and proliferation of hematopoietic (stem) cells, like BCR-ABL1, and (c) drivers that preferentially trigger the proliferation of hematopoietic stem and progenitor cells but do not or only marginally promote differentiation, such as mutant forms of RAS. In most instances, tumor suppressor loss (functionally by mutations or loss of genetic material) is also associated with enhanced proliferation of neoplastic (stem) cells.
The Ba/F3 model with doxycycline-inducible expression of oncogenes is a useful tool to define the differentiation and proliferation capacity of disease-related drivers. For example, expression of KIT D816V induces histamine production and (mast cell) differentiation, but not proliferation in Ba/F3 cells, whereas expression of BCR-ABL1 induces both histamine production and proliferation in Ba/F3 cells [100]. Correspondingly, patients with indolent systemic mastocytosis, where KIT D816V is usually the only driver lesion, accumulate their mast cell aggregates over years or even decades, and have a stable disease course with normal life expectancy. In these patients, most neoplastic mast cells are mature cells that do not proliferate and even the immature mast cell progenitor cells show no enhanced proliferative capacity over normal cells (the total burden of mast cells remains stable) unless additional mutations are acquired. By contrast, in BCR-ABL1+ chronic phase CML, uncontrolled proliferation of myelopoietic cells is a key feature, and when untreated, the disease rapidly transforms to accelerated phase and blast phase with short survival times.
Another important aspect is that some of the oncogenic driver mutants, such as BCR-ABL1 or JAK2 V617F, are per se capable of promoting clonal instability. For example, it was described that BCR-ABL1 promotes the acquisition of secondary mutations by a pathway involving STAT5 and increased production of reactive oxygen species (ROS), with consecutive DNA damage and clonal instability [101]. Similarly, JAK2 V617F was described to induce clonal instability and loss of heterozygosity via mutant-induced generation of ROS [102].
These additional lesions and hits are required for leukemogenesis and are indeed found in high-risk patients and those who actually progress to sAML. Sometimes, the primary driver may even suppress evolution to sAML because of its differentiation-inducing (and thus proliferation-hindering) effects. Therefore, it is not unexpected that in patients with ISM or MPN who progress to sAML, the AML clone often becomes or is negative for the molecular driver lesion. Especially when these patients are treated with drugs directed against these drivers, progression to sAML is often accompanied by a ‘loss’ of the driver, which can be explained by the selection of more malignant, ‘initial driver-negative’ sub-clones. A summary of CHOP mutations and secondary driver lesions and their functional impact in oncogenesis in myeloid neoplasms is provided in Table 2.
5. CHIP and CHOP in the Context of Leukemic Stem Cells
The concept of leukemic stem cells (LSC) has been coined to explain cellular hierarchies, sub-clone formation, and the directed diversification of clonal cell populations in diverse malignancies [44,45,46,47,48,103,104]. In addition, the LSC concept was propagated with the idea to focus research work and translational approaches on disease-initiating and -propagating cells. In fact, it is clear that antineoplastic therapies can only be curative in nature when eliminating most or all neoplastic stem cells fractions in a given malignancy [103,104,105].
In the past few years, neoplastic stem cells have been classified into premalignant (preleukemic) neoplastic stem cells (pre-L-NSC) and leukemic (malignant) neoplastic stem cells (LSC), also termed cancer stem cells in the context of a solid cancer [45,46,47,48]. Pre-L-NSC give rise to small-sized clones and usually behave very similar compared to normal hematopoietic stem cells. By contrast, LSC give rise to an overt leukemia or another advanced blood cell cancer. It is generally accepted that at least one or more CHIP mutations are required to convert a normal (stem) cell into a Pre-L-NSC. CHOP mutations may also be expressed in Pre-L-NSC, especially in chronic myeloid neoplasms. However, in these patients, Pre-L-NSC may convert into LSC within short time. Based on the model of step-wise evolution of cancer/leukemic stem cells (Figure 1), LSC and their progeny (= leukemic cells) must be expected to contain a mixture of CHIP and CHOP mutations. However, in rare cases, CHOP-negative sub-clones expand, especially under targeted therapy (Figure 1 and Figure 2). In these cases, other oncogenic mutant forms may be expressed or the loss of certain tumor suppressors may introduce an additional oncogenic player.
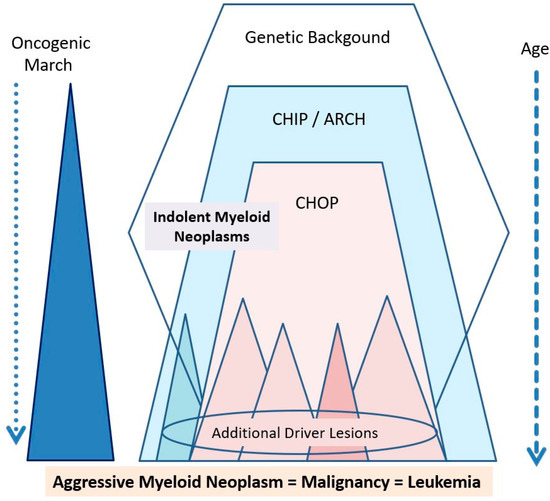
Figure 2.
Major players contributing to the ‘oncogenic march’ in myeloid neoplasms. The genetic background may form the basis of a familiar predisposition to the development of hematopoietic (and thus also myeloid) neoplasms. In some of these families, more or less specific of even disease-related mutations (with low or even high oncogenic potential) are found. CHIP develops later during lifetime—the related somatic mutations per se (as isolated lesions) have a low oncogenic potential and are more frequently detectable at higher age. Therefore, these lesions are also called age-related clonal hematopoiesis (ARCH). Later, somatic mutations with CHOP may be acquired and usually lead to an overt myeloid neoplasm (at least after some time). This neoplasm may manifest as an indolent (chronic) myeloid neoplasm unless additional drivers (driver lesions) and other oncogenic hits (loss of tumor suppressors) are acquired. In a few cases, such additional driver lesions may be acquired in a CHIP status (blue triangle) or even a pre-CHIP status and may then lead to the immediate formation of primary (de novo) AML.
All in all, the CHIP vs. CHOP concept is in line with and nicely explains the classification of NSC into Pre-L-NSC and fully malignant/leukemic NSC = LSC. Whereas Pre-L-NSC are more likely to contain one CHIP mutation, LSC are more likely to express two or more CHIP mutations or (more frequently) at least one CHOP mutation.
An important aspect, when considering potentially curative concepts, is LSC resistance. In fact, LSC are known to exhibit multiple forms of drug resistance, including intrinsic stem cell resistance (natural defense against toxins), acquired drug resistance (often mediated by secondary mutations in driver target genes or mutations in additional genes), niche-mediated resistance, and immune-mediated resistance (often triggered by checkpoint molecules). In addition, pharmacologic resistance may occur. Finally, LSC sub-populations may confer resistance because of the lack of major drug targets [45,46].
6. CHOP-Mutant Forms as Major Targets of Therapy
Because of their specificity and obvious function as major drivers of oncogenesis in various myeloid neoplasms, CHOP mutant forms were recognized as major targets of therapy. It started with the notion that BCR-ABL1-targeting TKI, such as imatinib, could effectively suppress growth and survival of leukemic cells in patients with Ph+ CML [106]. Later, imatinib was also found to block the kinase activity of FIP1L1-PDGFRA, the major driver of oncogenesis in chronic eosinophilic leukemia (CEL) and in a subset of related myeloid malignancies [107,108]. Subsequently, a number of different driver mutants and drugs directed against these drivers have been examined. In the classical MPN, inhibitors of JAK2 V617F were applied with considerable success [109]. In advanced systemic mastocytosis, drugs targeting the oncogenic KIT D816V mutant have been developed [110,111]. Finally, in AML, drugs directed against FLT3 ITD, IDH2 mutants and other oncogenic mutants were developed and are applied together with poly-chemotherapy in these patients.
All in all, targeting of CHOP-like mutant forms seems to be an effective approach in many patients. However, not all patients respond or show long lasting remissions. Rather, neoplastic cells are often resistant or develop resistance against these drugs. A number of different mechanisms of resistance against driver mutants have been deciphered in recent years. A full description and review of these mechanisms is beyond the scope of this article. The reader is referred to the available literature. In general, fusion genes encoding for CHOP-like drivers can acquire secondary mutations through which drug resistance develops. Second, driver-negative sub-clones can emerge. Third, niche-related and immunological forms of resistance may contribute to overall drug resistance. Finally, intrinsic stem cell resistance and pharmacological resistance may occur [45,46]. All these forms of resistance can be found in myeloid neoplasms and are often critically involved in MDS, CMML, and AML.
A logical way to overcome the multiple forms of resistance against CHOP driver-directed drugs is the application of drug combinations. Such combinations are currently being tested in preclinical and clinical studies.
7. Concluding Remarks and Future Perspectives
The term CHOP was proposed for somatic mutations that drive oncogenesis in various hematopoietic neoplasms as a single hit or cooperative hit that acts pro-oncogenic in somatic aberration networks. Whereas some of these drivers may directly induce the proliferation of neoplastic stem and progenitor cells, others induce differentiation in distinct hematopoietic lineages and are therefore disease-specific and lineage-related and often detected in premalignant chronic neoplasms. Some of these drivers may per se promote oncogenesis through the induction of clonal instability. Finally, in the context of multimutated sub-clones, oncogenic drivers contribute to the transformation to sAML. CHOP-related mutants have also been recognized as promising targets of therapy in myeloid neoplasms. However, complete suppression of oncogenesis and eradication to cure require the elimination of all premalignant and malignant neoplastic stem cells.
Author Contributions
All authors of this article contributed equally by participating in project discussion by writing parts of the draft and by reading and reviewing the final manuscript. All authors approved the final version of the document.
Funding
This study was supported by the Austrian Science Fund (FWF), grants F4701-B20 and Herzfelder´sche Familienstiftung grant P30627-B25.
Acknowledgments
All sources of funding of the study should be disclosed. Please clearly indicate grants that you have received in support of your research work. Clearly state if you received funds for covering the costs to publish in open access.
Conflicts of Interest
The authors declare that they have no conflict to disclose in this study.
References
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Cahill, D.P.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Genetic instability and darwinian selection in tumours. Trends Cell Biol. 1999, 9, 57–60. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. The multistep nature of cancer. Trends Genet. 1993, 9, 138–141. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; McGranahan, N.; Dewhurst, S.M.; Burrell, R.A.; Tomlinson, I.; Swanton, C. Cancer: Evolution within a lifetime. Annu. Rev. Genet. 2014, 48, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Wright, N.A. Boveri at 100: Cancer evolution, from preneoplasia to malignancy. J. Pathol. 2014, 234, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Curtius, K.; Wright, N.A.; Graham, T.A. Evolution of Premalignant Disease. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Arock, M.; Bock, C.; George, T.I.; Galli, S.J.; Gotlib, J.; Haferlach, T.; Hoermann, G.; Hermine, O.; et al. Proposed Terminology and Classification of Pre-Malignant Neoplastic Conditions: A Consensus Proposal. EBioMedicine 2017, 26, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Dutcher, J.P.; Wiernik, P.H. Accelerated and blastic phase of chronic myeloid leukemia. Curr. Treat. Options Oncol. 2000, 1, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Preisler, H.D. Evolution of secondary hematologic disorders: preMDS-->MDS-->sAML. Cancer Treat. Res. 2001, 108, 185–230. [Google Scholar] [PubMed]
- Melo, J.V.; Barnes, D.J. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer. 2007, 7, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, O.; Manshouri, T.; Patel, J.; Harris, K.; Yao, J.; Hedvat, C.; Heguy, A.; Bueso-Ramos, C.; Kantarjian, H.; Levine, R.L.; et al. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res. 2010, 70, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 2014, 123, 2220–2228. [Google Scholar] [CrossRef] [PubMed]
- Cargo, C.A.; Rowbotham, N.; Evans, P.A.; Barrans, S.L.; Bowen, D.T.; Crouch, S.; Jack, A.S. Targeted sequencing identifies patients with pre-clinical MDS at high risk of disease progression. Blood 2015, 126, 2362–2365. [Google Scholar] [CrossRef] [PubMed]
- Pfeilstöcker, M.; Tuechler, H.; Sanz, G.; Schanz, J.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Time-dependent changes in mortality and transformation risk in MDS. Blood 2016, 128, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Biernaux, C.; Loos, M.; Sels, A.; Huez, G.; Stryckmans, P. Detection of major bcr-abl gene expression at a very low level in blood cells of some healthy individuals. Blood 1995, 86, 3118–3122. [Google Scholar] [PubMed]
- Passamonti, F.; Rumi, E.; Pietra, D.; Lazzarino, M.; Cazzola, M. JAK2 (V617F) mutation in healthy individuals. Br. J. Haematol. 2007, 136, 678–679. [Google Scholar] [CrossRef] [PubMed]
- Busque, L.; Patel, J.P.; Figueroa, M.E.; Vasanthakumar, A.; Provost, S.; Hamilou, Z.; Mollica, L.; Li, J.; Viale, A.; Heguy, A.; et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet. 2012, 44, 1179–1181. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Orazi, A.; Steensma, D.P.; Ebert, B.L.; Haase, D.; Malcovati, L.; van de Loosdrecht, A.A.; Haferlach, T.; Westers, T.M.; Wells, D.A.; et al. Proposed minimal diagnostic criteria for myelodysplastic syndromes (MDS) and potential pre-MDS conditions. Oncotarget 2017, 8, 73483–73500. [Google Scholar] [CrossRef]
- Corces-Zimmerman, M.R.; Majeti, R. Pre-leukemic evolution of hematopoietic stem cells: The importance of early mutations in leukemogenesis. Leukemia 2014, 28, 2276–2282. [Google Scholar] [CrossRef] [PubMed]
- Shlush, L.I.; Zandi, S.; Itzkovitz, S.; Schuh, A.C. Aging, clonal hematopoiesis and preleukemia: Not just bad luck? Int. J. Hematol. 2015, 102, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Sykes, S.M.; Kokkaliaris, K.D.; Milsom, M.D.; Levine, R.L.; Majeti, R. Clonal evolution of preleukemic hematopoietic stem cells in acute myeloid leukemia. Exp. Hematol. 2015, 43, 989–992. [Google Scholar] [CrossRef] [PubMed]
- Malcovati, L.; Cazzola, M. The shadowlands of MDS: Idiopathic cytopenias of undetermined significance (ICUS) and clonal hematopoiesis of indeterminate potential (CHIP). Hematology Am. Soc. Hematol. Educ. Program 2015, 2015, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Sperling, A.S.; Gibson, C.J.; Ebert, B.L. The genetics of myelodysplastic syndrome: From clonal haematopoiesis to secondary leukaemia. Nat. Rev. Cancer 2017, 17, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Valent, P. ICUS, IDUS, CHIP and CCUS: Diagnostic Criteria, Separation from MDS and Clinical Implications. Pathobiology 2018, 1, 1–9. [Google Scholar] [CrossRef]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Itzykson, R.; Kosmider, O.; Fenaux, P. Somatic mutations and epigenetic abnormalities in myelodysplastic syndromes. Best Pract. Res. Clin. Haematol. 2013, 26, 355–364. [Google Scholar] [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Bacher, U.; Haferlach, T.; Schnittger, S.; Zenger, M.; Meggendorfer, M.; Jeromin, S.; Roller, A.; Grossmann, V.; Krauth, M.T.; Alpermann, T.; et al. Investigation of 305 patients with myelodysplastic syndromes and 20q deletion for associated cytogenetic and molecular genetic lesions and their prognostic impact. Br. J. Haematol. 2014, 164, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Jeromin, S.; Haferlach, T.; Weissmann, S.; Meggendorfer, M.; Eder, C.; Nadarajah, N.; Alpermann, T.; Kohlmann, A.; Kern, W.; Haferlach, C.; et al. Refractory anemia with ring sideroblasts and marked thrombocytosis cases harbor mutations in SF3B1 or other spliceosome genes accompanied by JAK2V617F and ASXL1 mutations. Haematologica 2015, 100, e125–e127. [Google Scholar] [CrossRef] [PubMed]
- Makishima, H.; Yoshizato, T.; Yoshida, K.; Sekeres, M.A.; Radivoyevitch, T.; Suzuki, H.; Przychodzen, B.; Nagata, Y.; Meggendorfer, M.; Sanada, M.; et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat. Genet. 2017, 49, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, C.M.; Nazha, A.; Kneen, K.; Abazeed, M.E.; Meggendorfer, M.; Przychodzen, B.P.; Nadarajah, N.; Adema, V.; Nagata, Y.; Goyal, A.; et al. Consequences of mutant TET2 on clonality and subclonal hierarchy. Leukemia 2018, 32, 1751–1761. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, S.; Alpermann, T.; Grossmann, V.; Kowarsch, A.; Nadarajah, N.; Eder, C.; Dicker, F.; Fasan, A.; Haferlach, C.; Haferlach, T.; et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia 2012, 26, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Rose, D.; Haferlach, T.; Schnittger, S.; Perglerova, K.; Kern, W.; Haferlach, C. Subtype-specific patterns of molecular mutations in acute myeloid leukemia. Leukemia 2017, 31, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Schwaab, J.; Schnittger, S.; Sotlar, K.; Walz, C.; Fabarius, A.; Pfirrmann, M.; Kohlmann, A.; Grossmann, V.; Meggendorfer, M.; Horny, H.P.; et al. Comprehensive mutational profiling in advanced systemic mastocytosis. Blood 2013, 122, 2460–2466. [Google Scholar] [CrossRef] [PubMed]
- Jawhar, M.; Schwaab, J.; Schnittger, S.; Sotlar, K.; Horny, H.P.; Metzgeroth, G.; Müller, N.; Schneider, S.; Naumann, N.; Walz, C.; et al. Molecular profiling of myeloid progenitor cells in multi-mutated advanced systemic mastocytosis identifies KIT D816V as a distinct and late event. Leukemia 2015, 29, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.N.; Miller, C.A.; Klco, J.M.; Petti, A.; Demeter, R.; Helton, N.M.; Li, T.; Fulton, R.S.; Heath, S.E.; Mardis, E.R.; et al. Rapid expansion of preexisting nonleukemic hematopoietic clones frequently follows induction therapy for de novo AML. Blood 2016, 127, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.J.; Lindsley, R.C.; Tchekmedyian, V.; Mar, B.G.; Shi, J.; Jaiswal, S.; Bosworth, A.; Francisco, L.; He, J.; Bansal, A.; et al. Clonal Hematopoiesis Associated With Adverse Outcomes After Autologous Stem-Cell Transplantation for Lymphoma. J. Clin. Oncol. 2017, 35, 1598–1605. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Wang, F.; Kantarjian, H.; Doss, D.; Khanna, K.; Thompson, E.; Zhao, L.; Patel, K.; Neelapu, S.; Gumbs, C.; et al. Preleukaemic clonal haemopoiesis and risk of therapy-related myeloid neoplasms: A case-control study. Lancet Oncol. 2017, 18, 100–111. [Google Scholar] [CrossRef]
- Jongen-Lavrencic, M.; Grob, T.; Hanekamp, D.; Kavelaars, F.G.; Al Hinai, A.; Zeilemaker, A.; Erpelinck-Verschueren, C.A.J.; Gradowska, P.L.; Meijer, R.; Cloos, J.; et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. N. Engl. J. Med. 2018, 378, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Hope, K.J.; Jin, L.; Dick, J.E. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat. Immunol. 2004, 5, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Valent, P. Targeting of leukemia-initiating cells to develop curative drug therapies: Straightforward but nontrivial concept. Curr. Cancer Drug Targets 2011, 11, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Bonnet, D.; De Maria, R.; Lapidot, T.; Copland, M.; Melo, J.V.; Chomienne, C.; Ishikawa, F.; Schuringa, J.J.; Stassi, G.; et al. Cancer stem cell definitions and terminology: The devil is in the details. Nat. Rev. Cancer 2012, 12, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, A.; Barreyro, L.; Steidl, U. Concise review: Preleukemic stem cells: Molecular biology and clinical implications of the precursors to leukemia stem cells. Stem Cells Transl. Med. 2013, 2, 143–150. [Google Scholar] [CrossRef]
- Valent, P.; Bonnet, D.; Wöhrer, S.; Andreeff, M.; Copland, M.; Chomienne, C.; Eaves, C. Heterogeneity of neoplastic stem cells: Theoretical, functional, and clinical implications. Cancer Res. 2013, 73, 1037–1045. [Google Scholar] [CrossRef]
- Walter, M.J.; Shen, D.; Ding, L.; Shao, J.; Koboldt, D.C.; Chen, K.; Larson, D.E.; McLellan, M.D.; Dooling, D.; Abbott, R.; et al. Clonal architecture of secondary acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1090–1098. [Google Scholar] [CrossRef]
- Cazzola, M.; Della Porta, M.G.; Malcovati, L. The genetic basis of myelodysplasia and its clinical relevance. Blood 2013, 122, 4021–4034. [Google Scholar] [CrossRef]
- Milosevic, J.D.; Puda, A.; Malcovati, L.; Berg, T.; Hofbauer, M.; Stukalov, A.; Klampfl, T.; Harutyunyan, A.S.; Gisslinger, H.; Gisslinger, B.; et al. Clinical significance of genetic aberrations in secondary acute myeloid leukemia. Am. J. Hematol. 2012, 87, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Nagata, Y.; Makishima, H.; Sanada, M.; Shiozawa, Y.; Kon, A.; Yoshizato, T.; Sato-Otsubo, A.; Kataoka, K.; Shiraishi, Y.; et al. Somatic PHF6 mutations in 1760 cases with various myeloid neoplasms. Leukemia 2016, 30, 2270–2273. [Google Scholar] [CrossRef] [PubMed]
- Nangalia, J.; Green, T.R. The evolving genomic landscape of myeloproliferative neoplasms. Hematology Am. Soc. Hematol. Educ. Program 2014, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Rampal, R.; Ahn, J.; Abdel-Wahab, O.; Nahas, M.; Wang, K.; Lipson, D.; Otto, G.A.; Yelensky, R.; Hricik, T.; McKenney, A.S.; et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA 2014, 111, E5401–E5410. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.C.; Liang, D.C.; Huang, C.F.; Shih, Y.S.; Wu, J.H.; Lin, T.L.; Shih, L.Y. RUNX1 mutations are frequent in chronic myelomonocytic leukemia and mutations at the C-terminal region might predict acute myeloid leukemia transformation. Leukemia 2009, 23, 1426–1431. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.J.; Przychodzen, B.; Thota, S.; Radivoyevitch, T.; Visconte, V.; Kuzmanovic, T.; Clemente, M.; Hirsch, C.; Morawski, A.; Souaid, R.; et al. Genomic determinants of chronic myelomonocytic leukemia. Leukemia 2017, 31, 2815–2823. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, M.M.; Wassie, E.A.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.; Tefferi, A. Blast transformation in chronic myelomonocytic leukemia: Risk factors, genetic features, survival, and treatment outcome. Am. J. Hematol. 2015, 90, 411–416. [Google Scholar] [CrossRef]
- Elena, C.; Galli, A.; Such, E.; Meggendorfer, M.; Germing, U.; Rizzo, E.; Cervera, J.; Molteni, E.; Fasan, A.; Schuler, E.; et al. Integrating clinical features and genetic lesions in the risk assessment of patients with chronic myelomonocytic leukemia. Blood 2016, 128, 1408–1417. [Google Scholar] [CrossRef]
- Jan, M.; Snyder, T.M.; Corces-Zimmerman, M.R.; Vyas, P.; Weissman, I.L.; Quake, S.R.; Majeti, R. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci. Transl. Med. 2012, 4, 149ra118. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Malcovati, L.; Galli, A.; Travaglino, E.; Ambaglio, I.; Rizzo, E.; Molteni, E.; Elena, C.; Ferretti, V.V.; Catricala, S.; Bono, E.; et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood 2017, 129, 3371–3378. [Google Scholar] [CrossRef] [PubMed]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of mutation order on myeloproliferative neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Boultwood, J.; Perry, J.; Zaman, R.; Fernandez-Santamaria, C.; Littlewood, T.; Kusec, R.; Pellagatti, A.; Wang, L.; Clark, R.E.; Wainscoat, J.S. High-density single nucleotide polymorphism array analysis and ASXL1 gene mutation screening in chronic myeloid leukemia during disease progression. Leukemia 2010, 24, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, V.; Kohlmann, A.; Zenger, M.; Schindela, S.; Eder, C.; Weissmann, S.; Schnittger, S.; Kern, W.; Müller, M.C.; Hochhaus, A.; et al. A deep-sequencing study of chronic myeloid leukemia patients in blast crisis (BC-CML) detects mutations in 76.9% of cases. Leukemia 2011, 25, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Makishima, H.; Jankowska, A.M.; McDevitt, M.A.; O’Keefe, C.; Dujardin, S.; Cazzolli, H.; Przychodzen, B.; Prince, C.; Nicoll, J.; Siddaiah, H.; et al. CBL, CBLB, TET2, ASXL1, and IDH1/2 mutations and additional chromosomal aberrations constitute molecular events in chronic myelogenous leukemia. Blood 2011, 117, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Hadzijusufovic, E.; Albrecht-Schgoer, K.; Huber, K.; Hoermann, G.; Grebien, F.; Eisenwort, G.; Schgoer, W.; Herndlhofer, S.; Kaun, C.; Theurl, M.; et al. Nilotinib-induced vasculopathy: Identification of vascular endothelial cells as a primary target site. Leukemia 2017, 31, 2388–2397. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.K.; Lea, N.C.; Heneghan, M.A.; Westwood, N.B.; Milojkovic, D.; Thanigaikumar, M.; Yallop, D.; Arya, R.; Pagliuca, A.; Gäken, J.; et al. Prevalence of the activating JAK2 tyrosine kinase mutation V617F in the Budd-Chiari syndrome. Gastroenterology 2006, 130, 2031–2038. [Google Scholar] [CrossRef]
- Colaizzo, D.; Amitrano, L.; Tiscia, G.L.; Scenna, G.; Grandone, E.; Guardascione, M.A.; Brancaccio, V.; Margaglione, M. The JAK2 V617F mutation frequently occurs in patients with portal and mesenteric venous thrombosis. J. Thromb. Haemost. 2007, 5, 55–61. [Google Scholar] [CrossRef]
- De Stefano, V.; Fiorini, A.; Rossi, E.; Za, T.; Farina, G.; Chiusolo, P.; Sica, S.; Leone, G. Incidence of the JAK2 V617F mutation among patients with splanchnic or cerebral venous thrombosis and without overt chronic myeloproliferative disorders. J. Thromb. Haemost. 2007, 5, 708–714. [Google Scholar] [CrossRef]
- Colaizzo, D.; Amitrano, L.; Guardascione, M.A.; Tiscia, G.L.; D’Andrea, G.; Longo, V.A.; Grandone, E.; Margaglione, M. Outcome of patients with splanchnic venous thrombosis presenting without overt MPN: A role for the JAK2 V617F mutation re-evaluation. Thromb. Res. 2013, 132, e99–e104. [Google Scholar] [CrossRef] [PubMed]
- Kilpivaara, O.; Levine, R.L. JAK2 and MPL mutations in myeloproliferative neoplasms: Discovery and science. Leukemia 2008, 22, 1813–1817. [Google Scholar] [CrossRef] [PubMed]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [PubMed]
- Ferreira Cristina, S.; Polo, B.; Lacerda, J.F. Somatic Mutations in Philadelphia Chromosome-Negative Myeloproliferative Neoplasms. Semin Hematol 2018, 55, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Delic, S.; Rose, D.; Kern, W.; Nadarajah, N.; Haferlach, C.; Haferlach, T.; Meggendorfer, M. Application of an NGS-based 28-gene panel in myeloproliferative neoplasms reveals distinct mutation patterns in essential thrombocythaemia, primary myelofibrosis and polycythaemia vera. Br. J. Haematol. 2016, 175, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Senín, A.; Fernández-Rodríguez, C.; Bellosillo, B.; Camacho, L.; Longarón, R.; Angona, A.; Besses, C.; Álvarez-Larrán, A. Non-driver mutations in patients with JAK2V617F-mutated polycythemia vera or essential thrombocythemia with long-term molecular follow-up. Ann. Hematol. 2018, 97, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Meggendorfer, M.; de Albuquerque, A.; Nadarajah, N.; Alpermann, T.; Kern, W.; Steuer, K.; Perglerová, K.; Haferlach, C.; Schnittger, S.; Haferlach, T. Karyotype evolution and acquisition of FLT3 or RAS pathway alterations drive progression of myelodysplastic syndrome to acute myeloid leukemia. Haematologica 2015, 100, e487–e490. [Google Scholar] [CrossRef] [PubMed]
- Padua, R.A.; Guinn, B.A.; Al-Sabah, A.I.; Smith, M.; Taylor, C.; Pettersson, T.; Ridge, S.; Carter, G.; White, D.; Oscier, D.; et al. RAS, FMS and p53 mutations and poor clinical outcome in myelodysplasias: A 10-year follow-up. Leukemia 1998, 12, 887–892. [Google Scholar] [CrossRef]
- Gelsi-Boyer, V.; Trouplin, V.; Adélaïde, J.; Aceto, N.; Remy, V.; Pinson, S.; Houdayer, C.; Arnoulet, C.; Sainty, D.; Bentires-Alj, M.; et al. Genome profiling of chronic myelomonocytic leukemia: Frequent alterations of RAS and RUNX1 genes. B.M.C. Cancer 2008, 8, 299. [Google Scholar] [CrossRef]
- Ricci, C.; Fermo, E.; Corti, S.; Molteni, M.; Faricciotti, A.; Cortelezzi, A.; Deliliers, G.L.; Beran, M.; Onida, F. RAS mutations contribute to evolution of chronic myelomonocytic leukemia to the proliferative variant. Clin. Cancer Res. 2010, 16, 2246–2256. [Google Scholar] [CrossRef]
- Peng, J.; Zuo, Z.; Fu, B.; Oki, Y.; Tang, G.; Goswami, M.; Priyanka, P.; Muzzafar, T.; Medeiros, L.J.; Luthra, R.; et al. Chronic myelomonocytic leukemia with nucleophosmin (NPM1) mutation. Eur. J. Haematol. 2016, 96, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Palomo, L.; Garcia, O.; Arnan, M.; Xicoy, B.; Fuster, F.; Cabezón, M.; Coll, R.; Ademà, V.; Grau, J.; Jiménez, M.J.; et al. Targeted deep sequencing improves outcome stratification in chronic myelomonocytic leukemia with low risk cytogenetic features. Oncotarget 2016, 7, 57021–57035. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zheng, Y.; Wang, Z.C.; Wang, S.Y. Prognostic significance of ASXL1 mutations in myelodysplastic syndromes and chronic myelomonocytic leukemia: A meta-analysis. Hematology 2016, 21, 454–461. [Google Scholar] [CrossRef]
- Sallman, D.A.; Komrokji, R.; Cluzeau, T.; Vaupel, C.; Al Ali, N.H.; Lancet, J.; Hall, J.; List, A.; Padron, E.; Song, J. ASXL1 frameshift mutations drive inferior outcomes in CMML without negative impact in MDS. Blood Cancer J. 2017, 7, 633. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Lee, K.O.; Park, S.; Jang, J.H.; Jung, C.W.; Kim, S.H.; Kim, H.J. Poor Prognostic Implication of ASXL1 Mutations in Korean Patients With Chronic Myelomonocytic Leukemia. Ann. Lab. Med. 2018, 38, 495–502. [Google Scholar] [CrossRef]
- Daver, N.; Strati, P.; Jabbour, E.; Kadia, T.; Luthra, R.; Wang, S.; Patel, K.; Ravandi, F.; Cortes, J.; Qin Dong, X.; et al. FLT3 mutations in myelodysplastic syndrome and chronic myelomonocytic leukemia. Am. J. Hematol. 2013, 88, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Singh, R.R.; Patel, K.P.; Stingo, F.; Routbort, M.; You, M.J.; Miranda, R.N.; Garcia-Manero, G.; Kantarjian, H.M.; Medeiros, L.J.; et al. BRAF kinase domain mutations are present in a subset of chronic myelomonocytic leukemia with wild-type RAS. Am. J. Hematol. 2014, 89, 499–504. [Google Scholar] [CrossRef]
- Mrózek, K.; Marcucci, G.; Paschka, P.; Bloomfield, C.D. Advances in molecular genetics and treatment of core-binding factor acute myeloid leukemia. Curr. Opin. Oncol. 2008, 20, 711–718. [Google Scholar] [CrossRef]
- Marcucci, G.; Haferlach, T.; Döhner, H. Molecular genetics of adult acute myeloid leukemia: Prognostic and therapeutic implications. J. Clin. Oncol. 2011, 29, 475–486. [Google Scholar] [CrossRef]
- Bullinger, L.; Döhner, K.; Döhner, H. Genomics of Acute Myeloid Leukemia Diagnosis and Pathways. J. Clin. Oncol. 2017, 35, 934–946. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Roloff, G.W.; Griffiths, E.A. When to obtain genomic data in acute myeloid leukemia (AML) and which mutations matter. Blood Adv. 2018, 2, 3070–3080. [Google Scholar] [CrossRef] [PubMed]
- Schlegelberger, B.; Heller, P.G. RUNX1 deficiency (familial platelet disorder with predisposition to myeloid leukemia, FPDMM). Semin. Hematol. 2017, 54, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Bellissimo, D.C.; Speck, N.A. RUNX1 Mutations in Inherited and Sporadic Leukemia. Front. Cell. Dev. Biol. 2017, 5, 111. [Google Scholar] [CrossRef] [PubMed]
- Tawana, K.; Rio-Machin, A.; Preudhomme, C.; Fitzgibbon, J. Familial CEBPA-mutated acute myeloid leukemia. Semin. Hematol. 2017, 54, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.; Kazi, J.U.; Zhao, H.; Sun, J. Germline mutations of KIT in gastrointestinal stromal tumor (GIST) and mastocytosis. Cell. Biosci. 2016, 6, 55. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, R.; Simioni, L.; Garcia-Montero, A.C.; Perbellini, O.; Bonadonna, P.; Caruso, B.; Jara-Acevedo, M.; Bonifacio, M.; De Matteis, G. Somatic D816V KIT mutation in a case of adult-onset familial mastocytosis. J. Allergy Clin. Immunol. 2013, 131, 605–607. [Google Scholar] [CrossRef]
- Hinds, D.A.; Barnholt, K.E.; Mesa, R.A.; Kiefer, A.K.; Do, C.B.; Eriksson, N.; Mountain, J.L.; Francke, U.; Tung, J.Y.; Nguyen, H.M.; et al. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood 2016, 128, 1121–1128. [Google Scholar] [CrossRef]
- Tashi, T.; Swierczek, S.; Prchal, J.T. Familial MPN Predisposition. Curr. Hematol. Malig. Rep. 2017, 12, 442–447. [Google Scholar] [CrossRef]
- Mayerhofer, M.; Gleixner, K.V.; Hoelbl, A.; Florian, S.; Hoermann, G.; Aichberger, K.J.; Bilban, M.; Esterbauer, H.; Krauth, M.T.; Sperr, W.R.; et al. Unique effects of KIT D816V in BaF3 cells: Induction of cluster formation, histamine synthesis, and early mast cell differentiation antigens. J. Immunol. 2008, 180, 5466–5476. [Google Scholar] [CrossRef]
- Warsch, W.; Grundschober, E.; Berger, A.; Gille, L.; Cerny-Reiterer, S.; Tigan, A.S.; Hoelbl-Kovacic, A.; Valent, P.; Moriggl, R.; Sexl, V. STAT5 triggers BCR-ABL1 mutation by mediating ROS production in chronic myeloid leukaemia. Oncotarget 2012, 3, 1669–1687. [Google Scholar] [CrossRef] [PubMed]
- Marty, C.; Lacout, C.; Droin, N.; Le Couédic, J.P.; Ribrag, V.; Solary, E.; Vainchenker, W.; Villeval, J.L.; Plo, I. A role for reactive oxygen species in JAK2 V617F myeloproliferative neoplasm progression. Leukemia 2013, 27, 2187–2195. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; van Etten, R.A. Right on target: Eradicating leukemic stem cells. Trends Mol. Med. 2007, 13, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, E.; Castelli, G.; Testa, U. Targeting LSCs through membrane antigens selectively or preferentially expressed on these cells. Blood Cells Mol. Dis. 2015, 55, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Schulenburg, A.; Blatt, K.; Cerny-Reiterer, S.; Sadovnik, I.; Herrmann, H.; Marian, B.; Grunt, T.W.; Zielinski, C.C.; Valent, P. Cancer stem cells in basic science and in translational oncology: Can we translate into clinical application? J. Hematol. Oncol. 2015, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J. Translation of the Philadelphia chromosome into therapy for CML. Blood 2008, 112, 4808–4817. [Google Scholar] [CrossRef] [PubMed]
- Gotlib, J.; Cools, J. Five years since the discovery of FIP1L1-PDGFRA: What we have learned about the fusion and other molecularly defined eosinophilias. Leukemia 2008, 22, 1999–2010. [Google Scholar] [CrossRef] [PubMed]
- Metzgeroth, G.; Schwaab, J.; Gosenca, D.; Fabarius, A.; Haferlach, C.; Hochhaus, A.; Cross, N.C.; Hofmann, W.K.; Reiter, A. Long-term follow-up of treatment with imatinib in eosinophilia-associated myeloid/lymphoid neoplasms with PDGFR rearrangements in blast phase. Leukemia 2013, 27, 2254–2256. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Harrison, C.N. Emerging treatments for classical myeloproliferative neoplasms. Blood 2017, 129, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Gotlib, J.; Kluin-Nelemans, H.C.; George, T.I.; Akin, C.; Sotlar, K.; Hermine, O.; Awan, F.T.; Hexner, E.; Mauro, M.J.; Sternberg, D.W.; et al. Efficacy and Safety of Midostaurin in Advanced Systemic Mastocytosis. N. Engl. J. Med. 2016, 374, 2530–2541. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Hartmann, K.; George, T.I.; Sotlar, K.; Peter, B.; Gleixner, K.V.; Blatt, K.; Sperr, W.R.; Manley, P.W.; et al. Midostaurin: A magic bullet that blocks mast cell expansion and activation. Ann. Oncol. 2017, 28, 2367–2376. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).