Targeting Autophagy to Overcome Human Diseases





Abstract
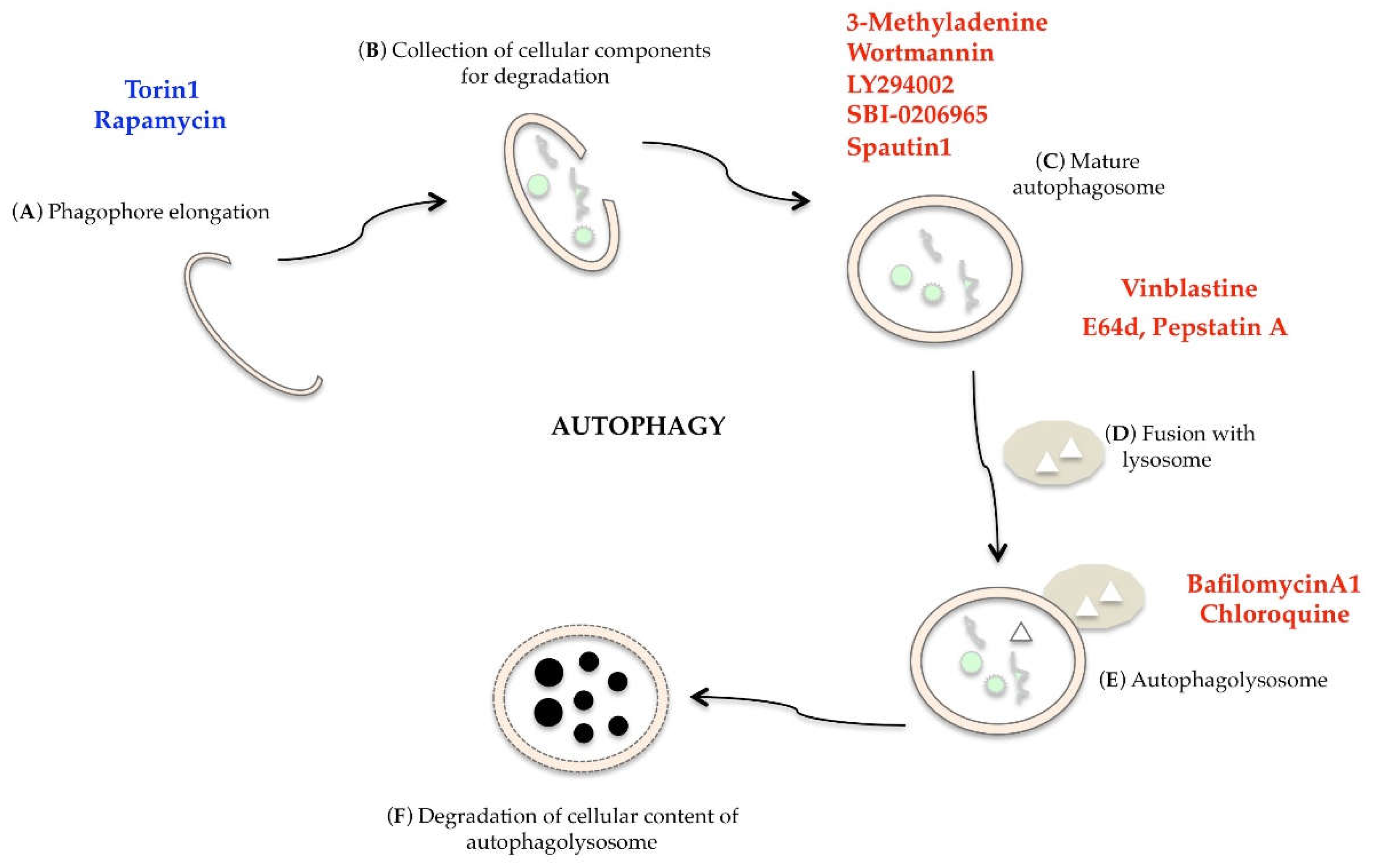
:1. Introduction
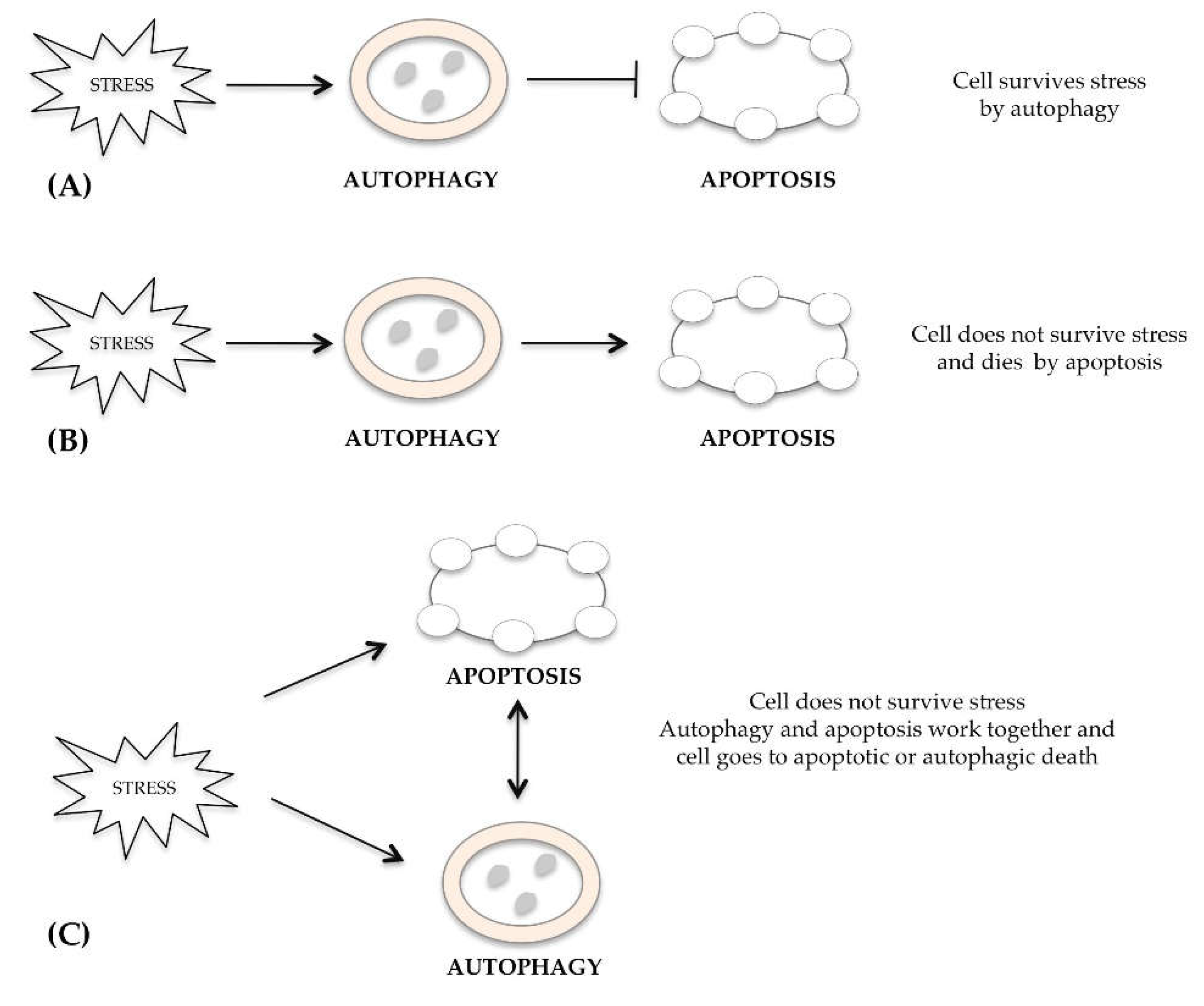
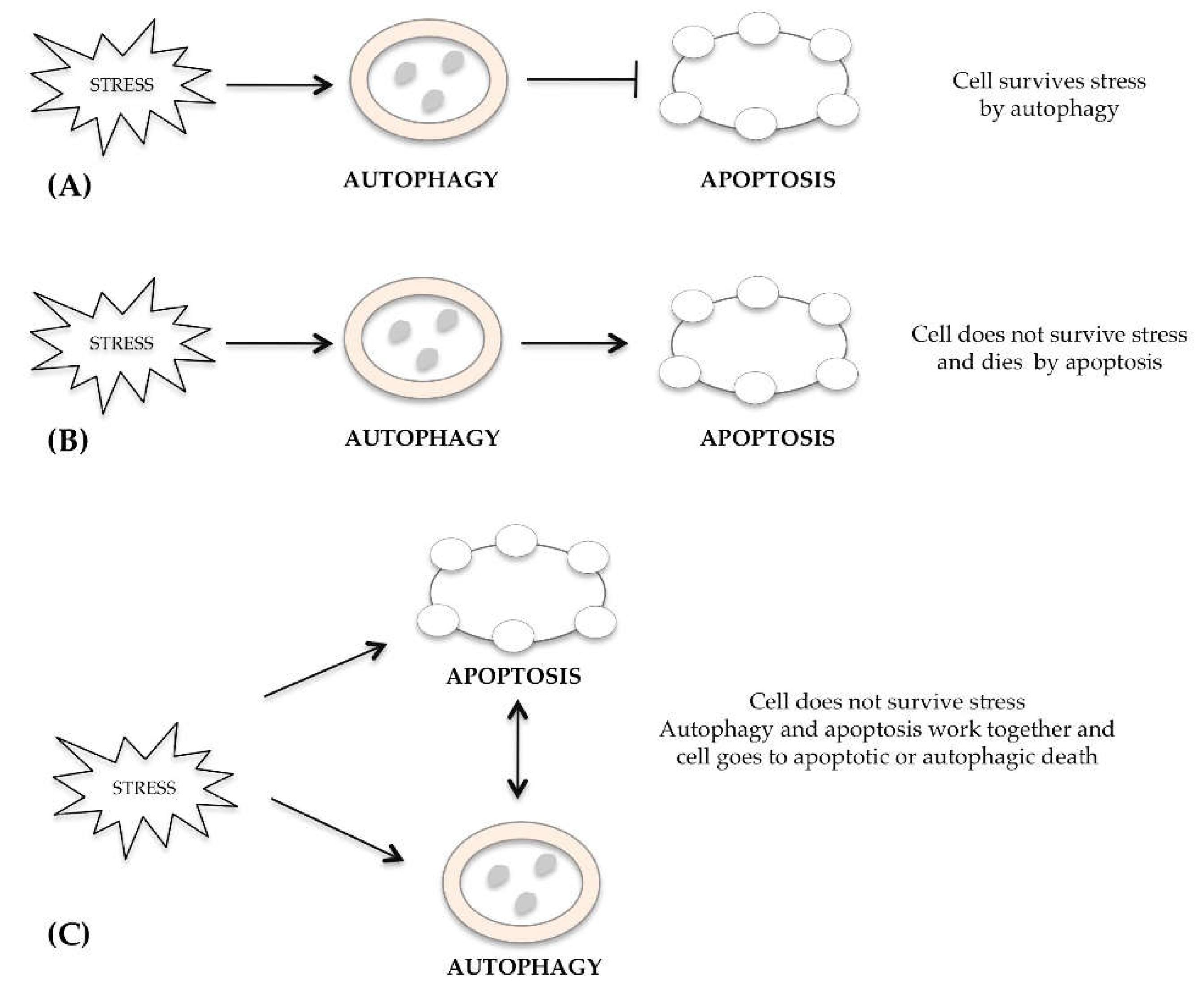
2. Cross-Talk between Autophagic, Apoptotic and Necrotic Pathways
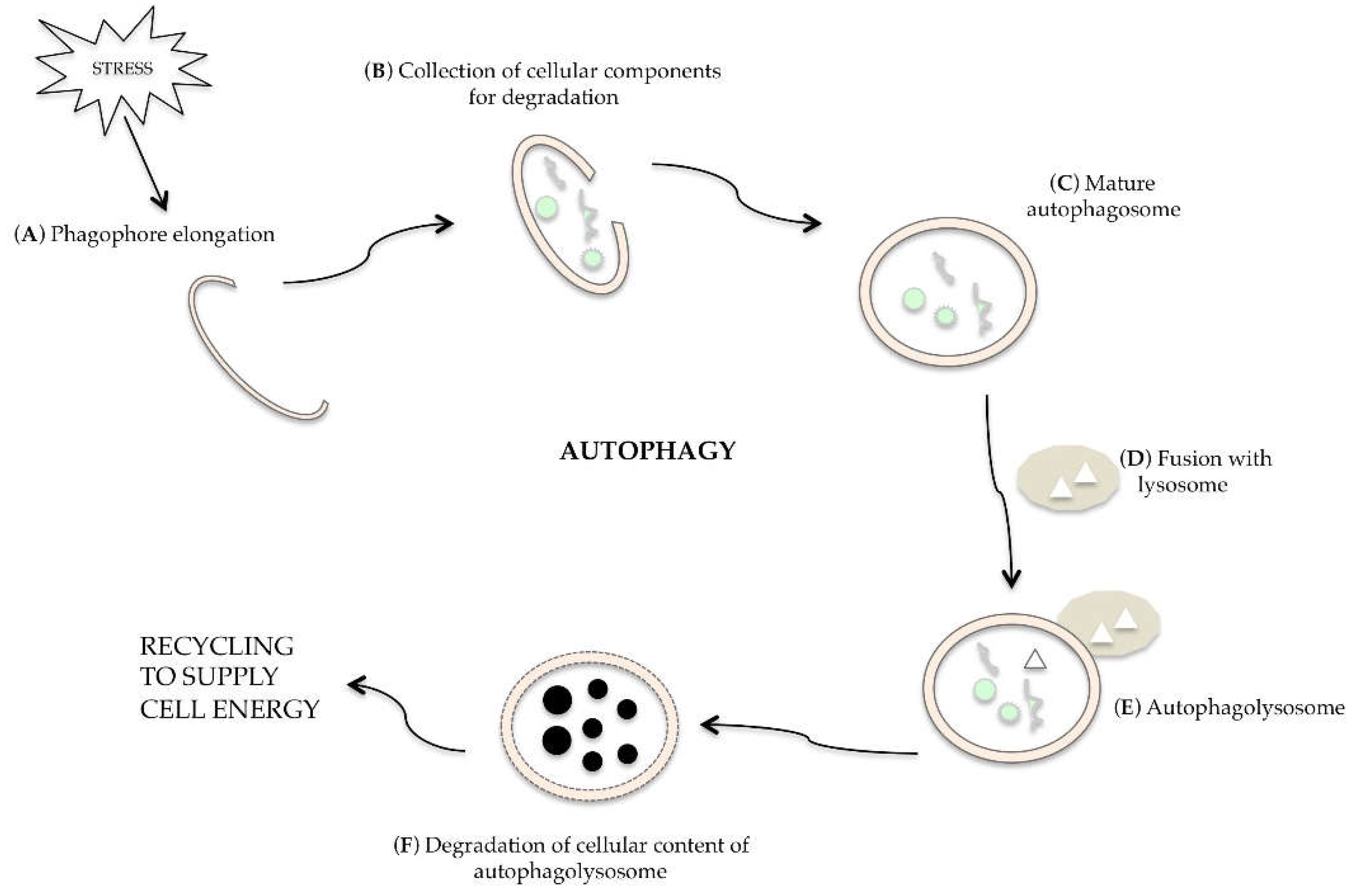
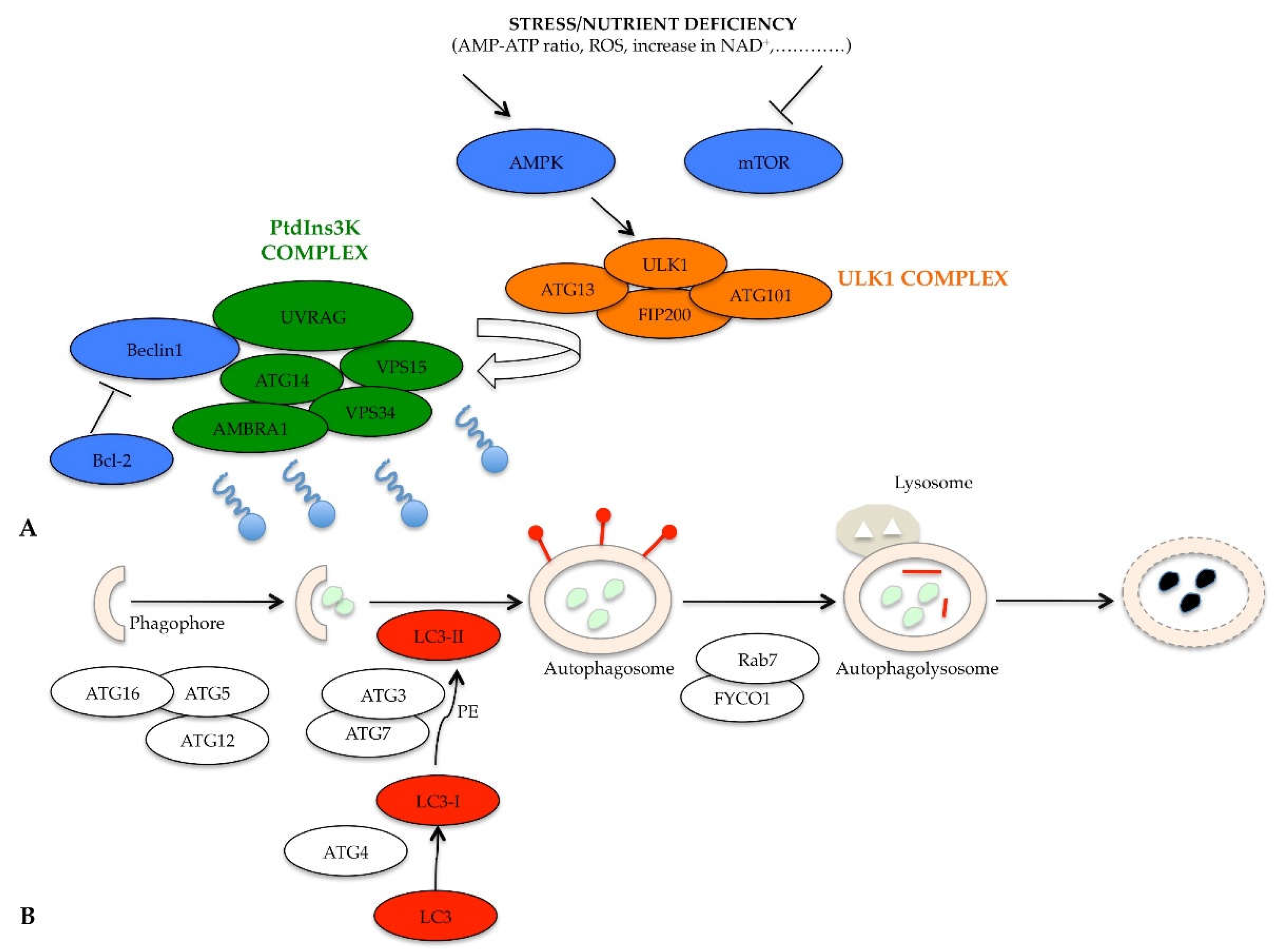
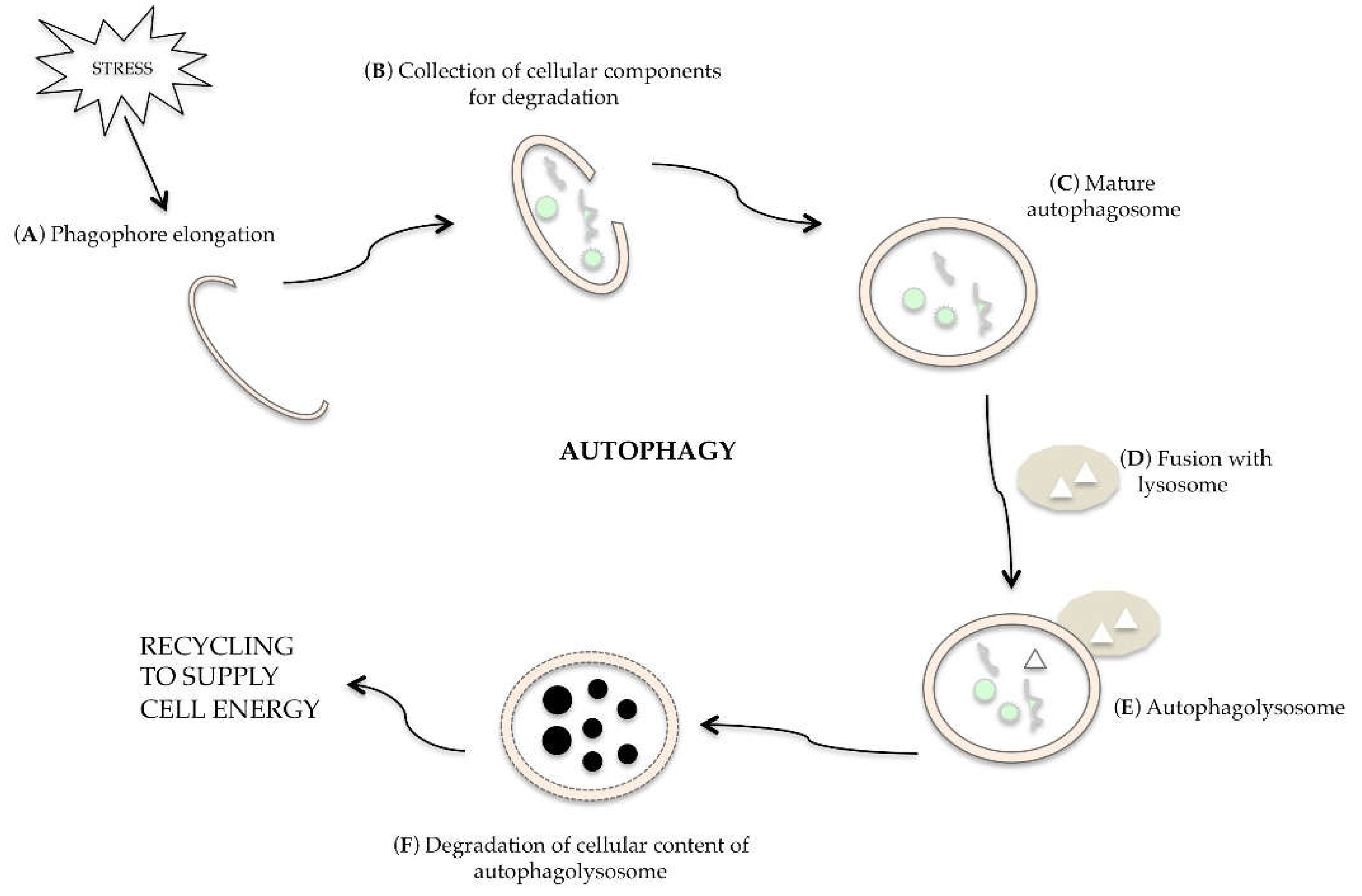
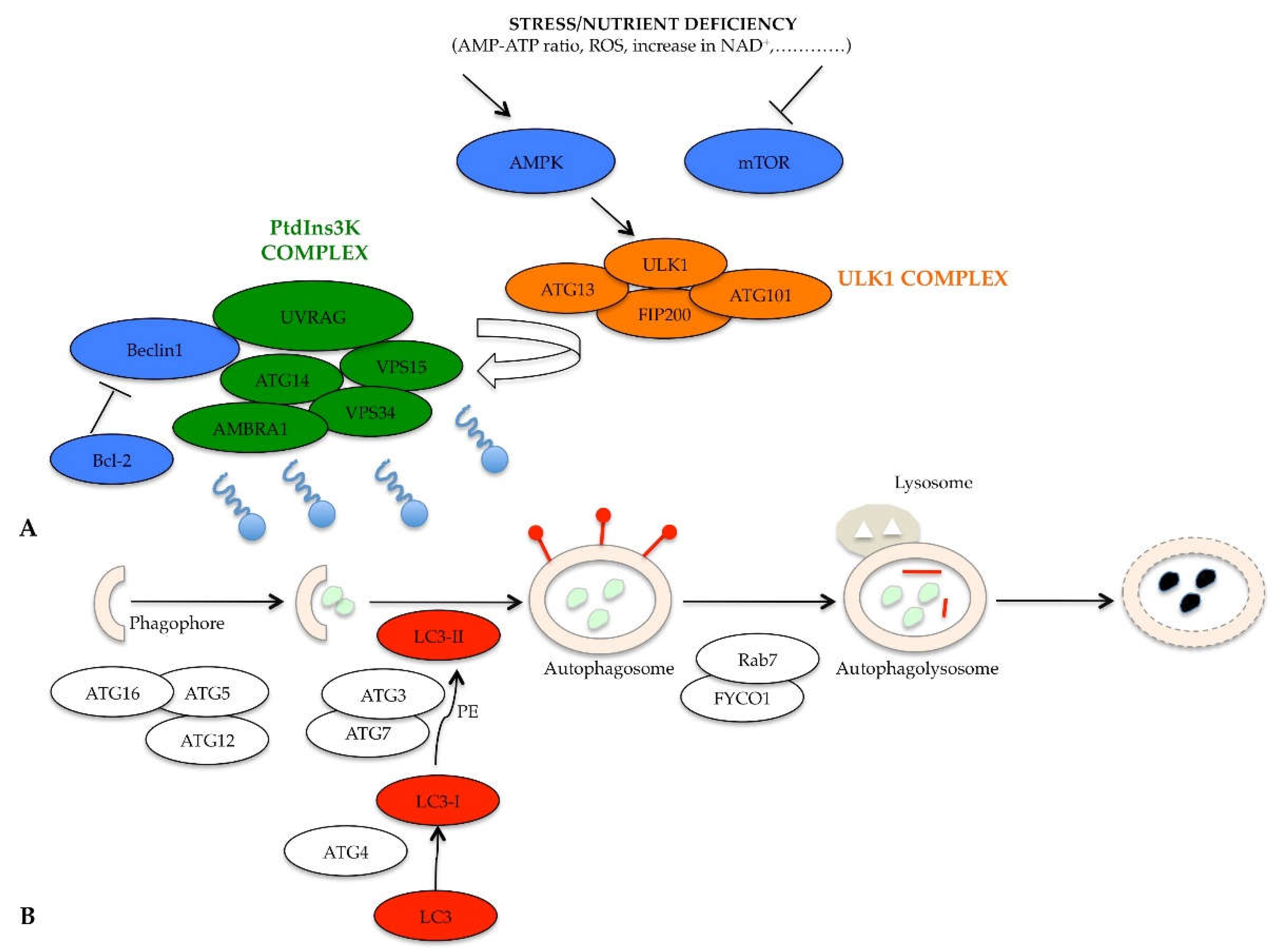
3. Molecular Regulation of Autophagy
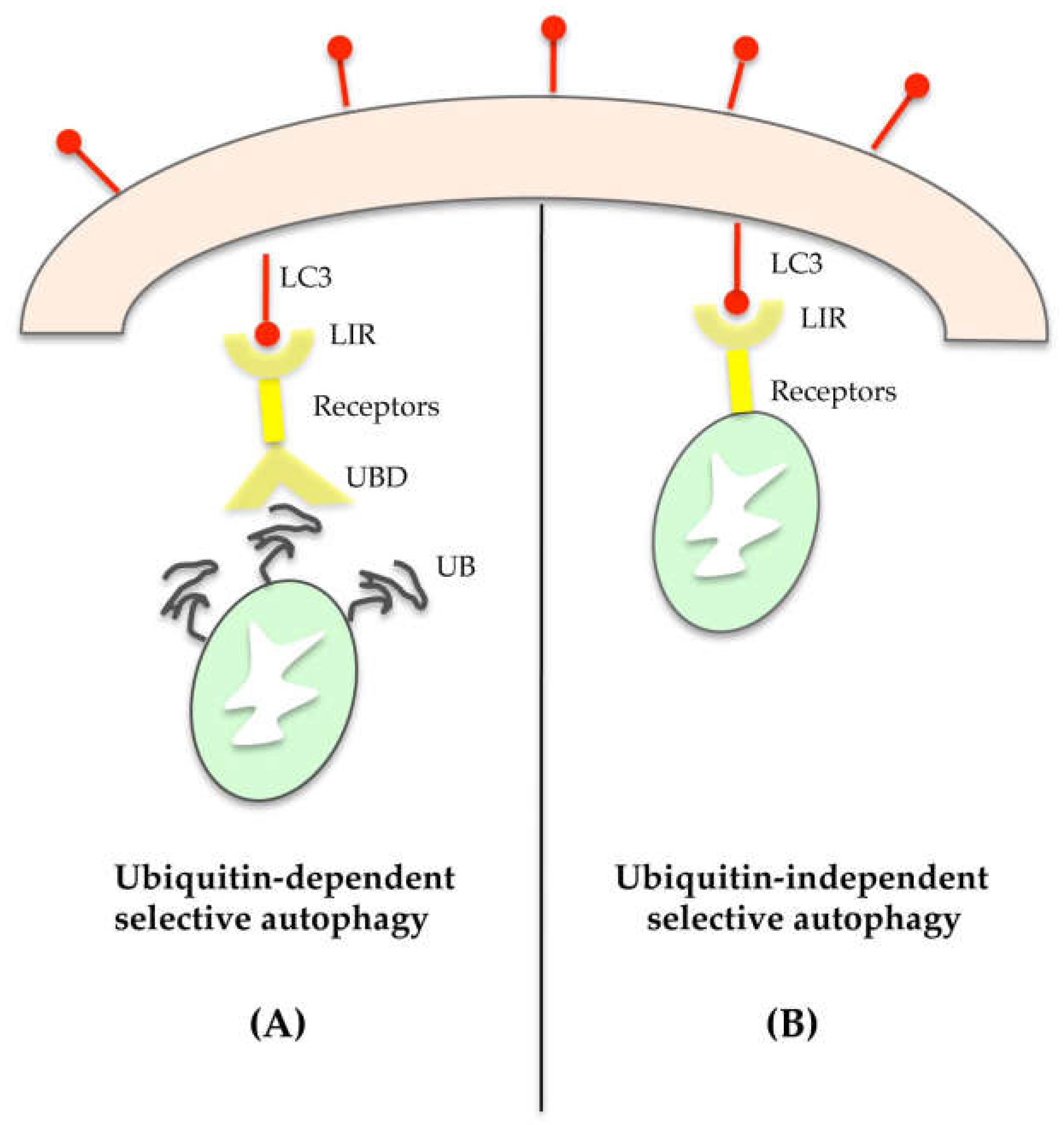
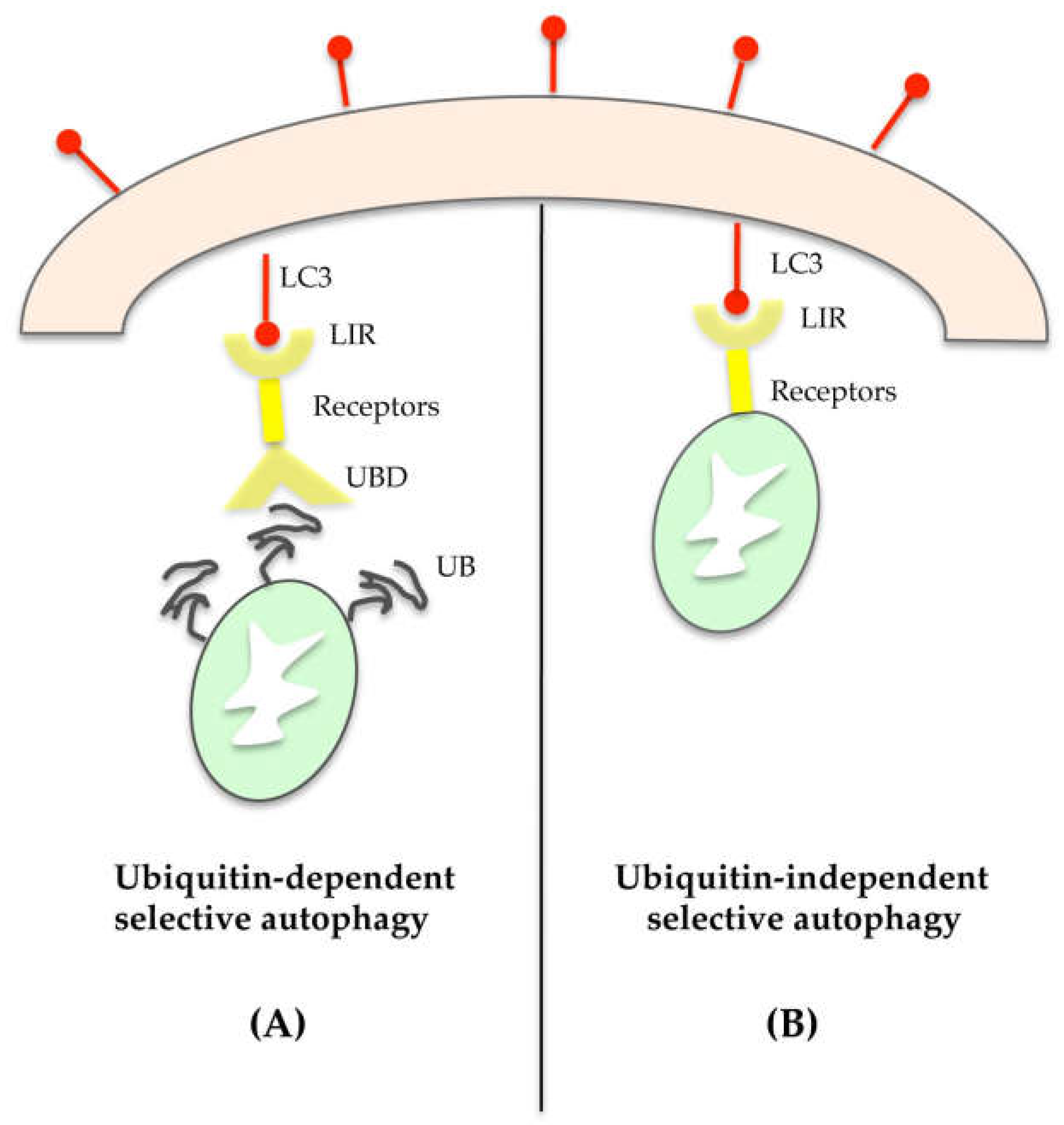
4. Selective Autophagy
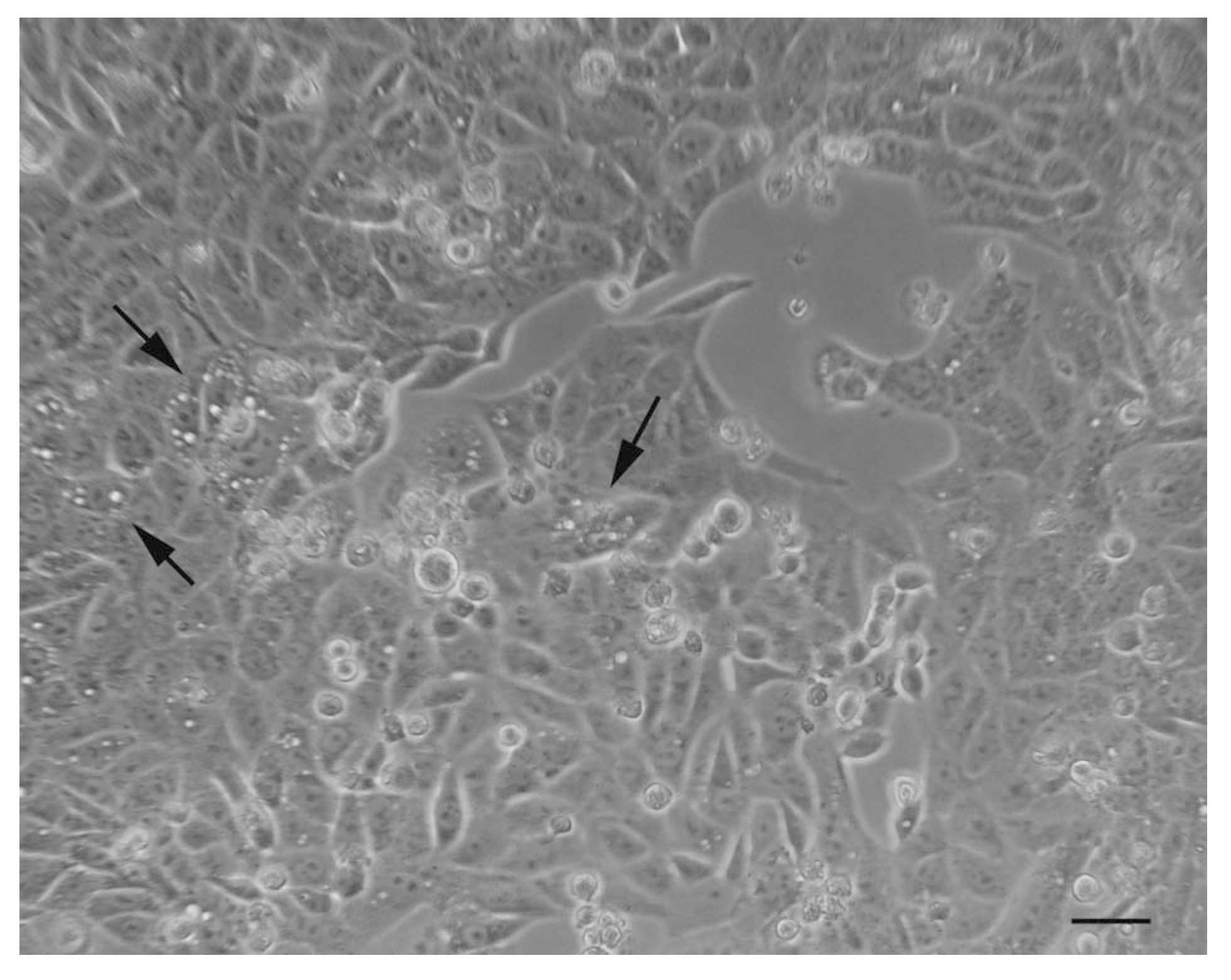
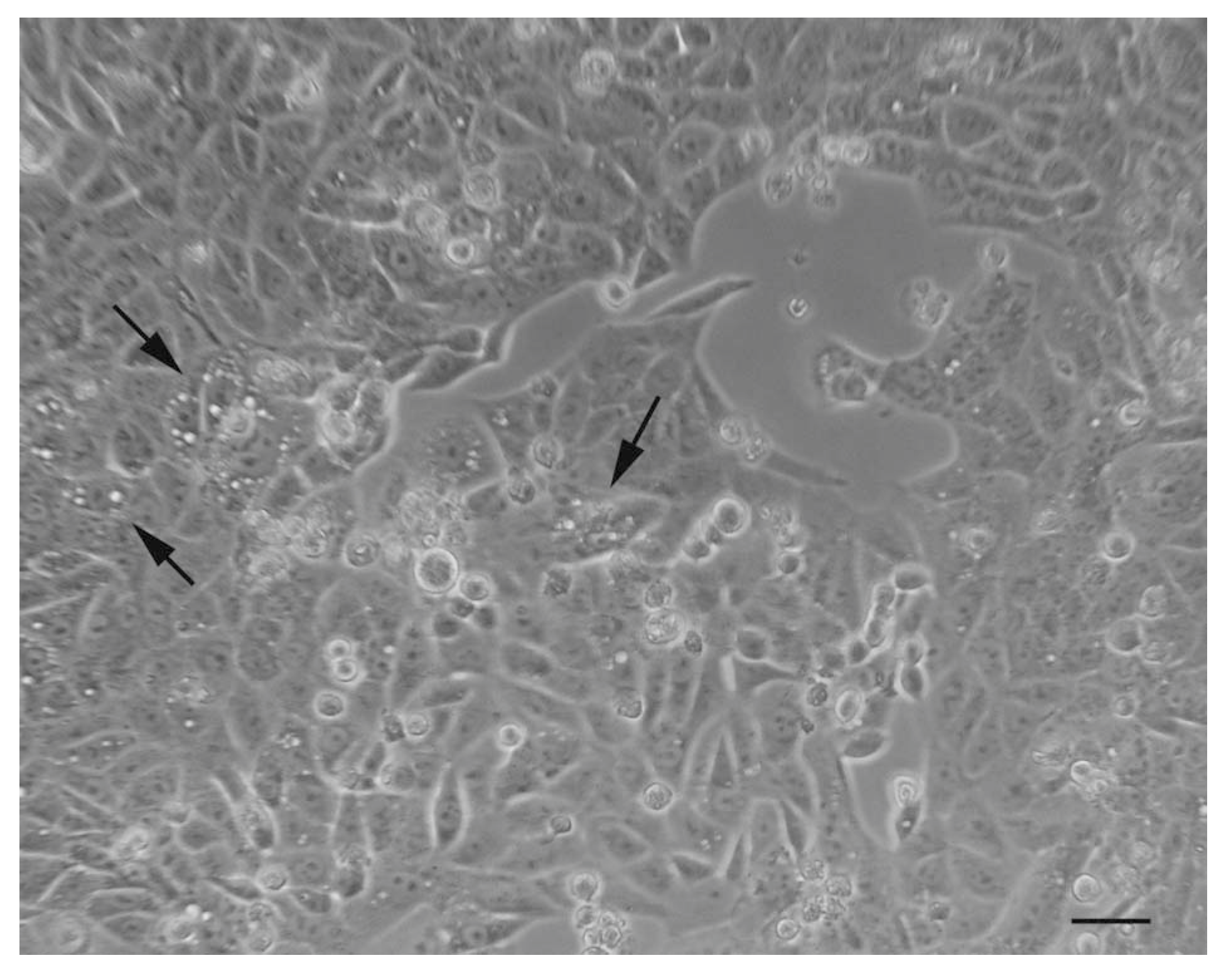
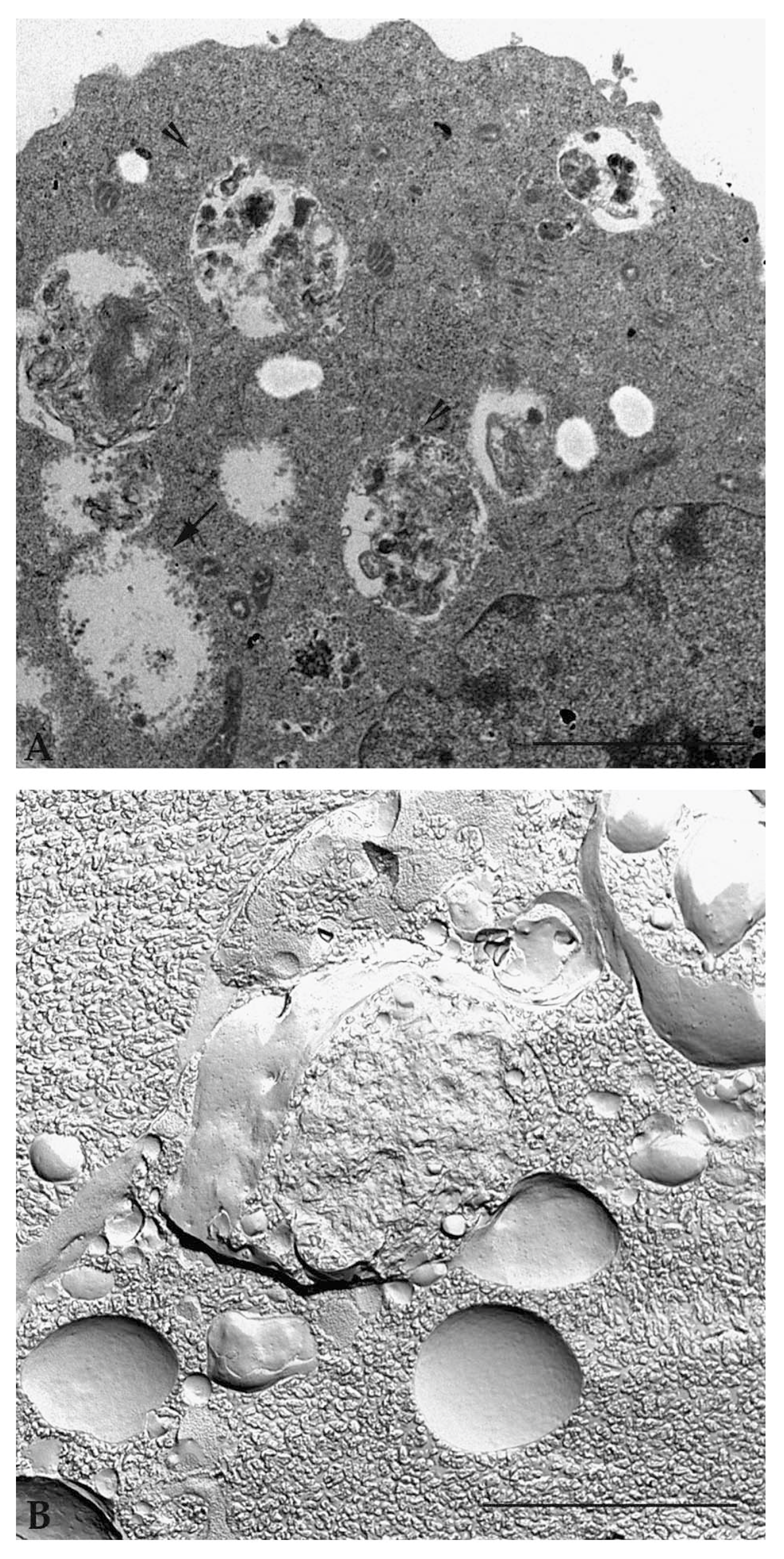
5. Autophagy Detection Methods in Mammalian Cells
6. Autophagy, Inflammation and Aging
- by removing damaged organelles such as mitochondria, leading to reduced release of ROS and subsequent suppression of inflammasome activation;
- by p62-dependent degradation of inflammasome complexes and mitochondria;
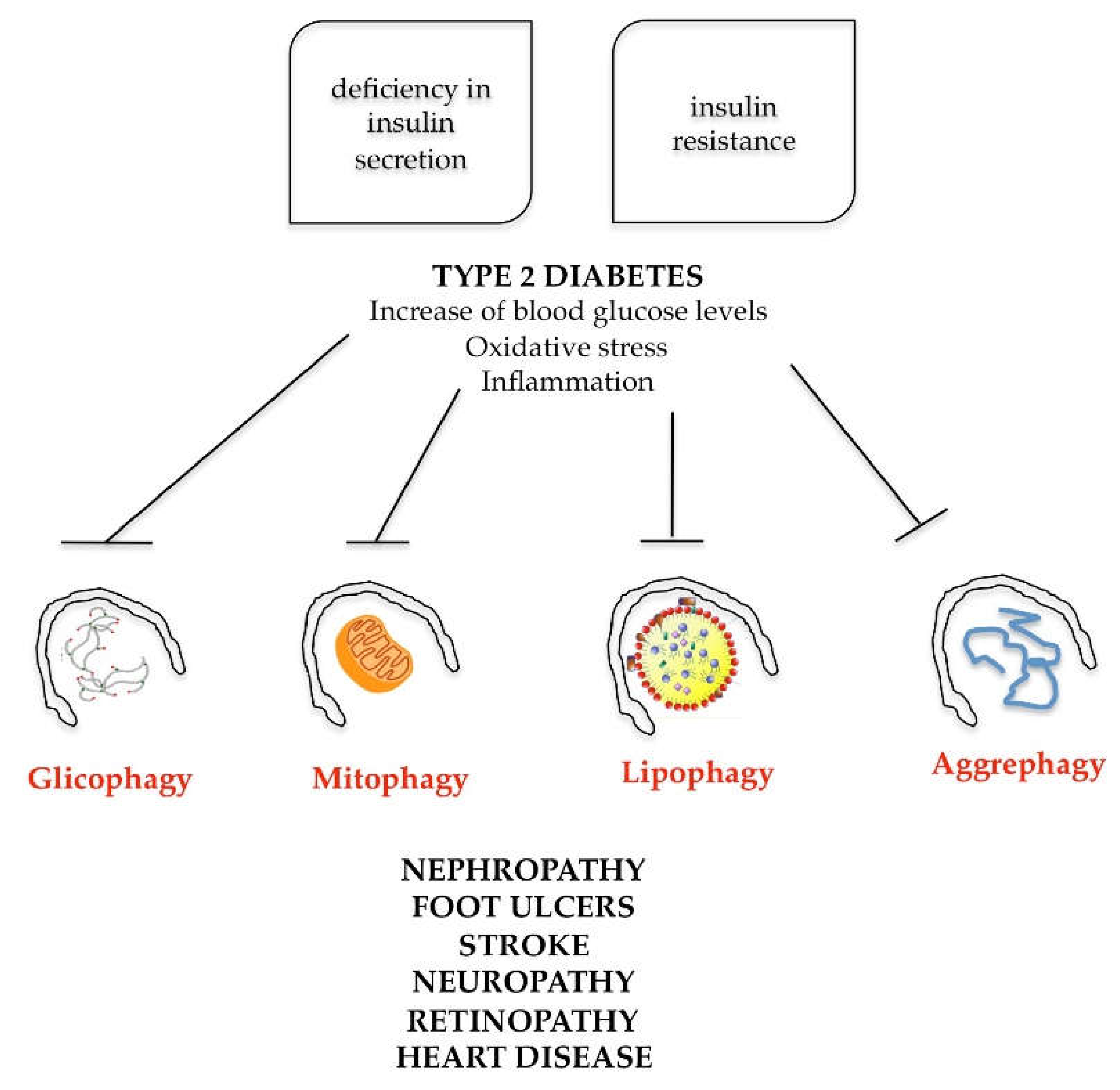
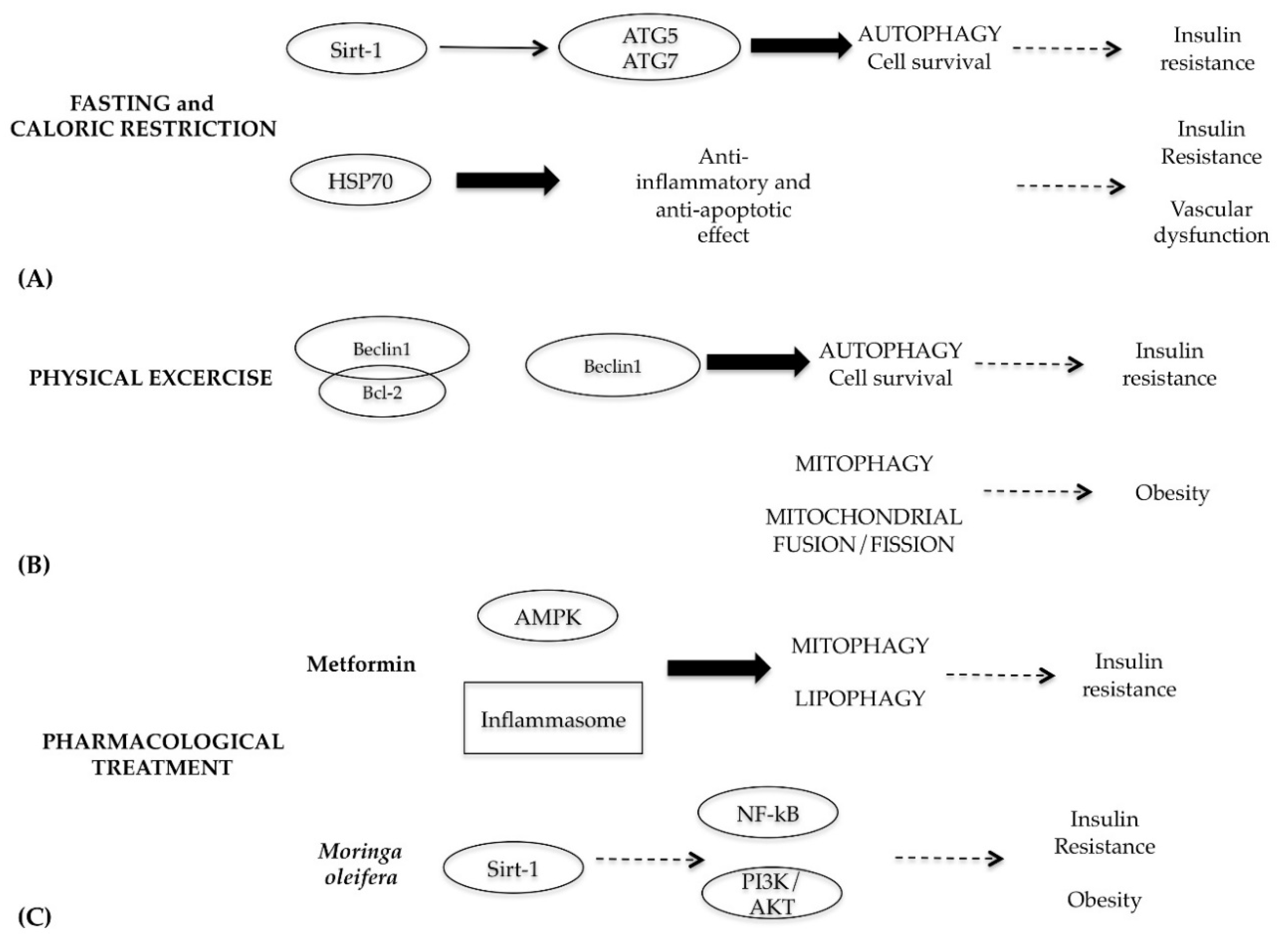
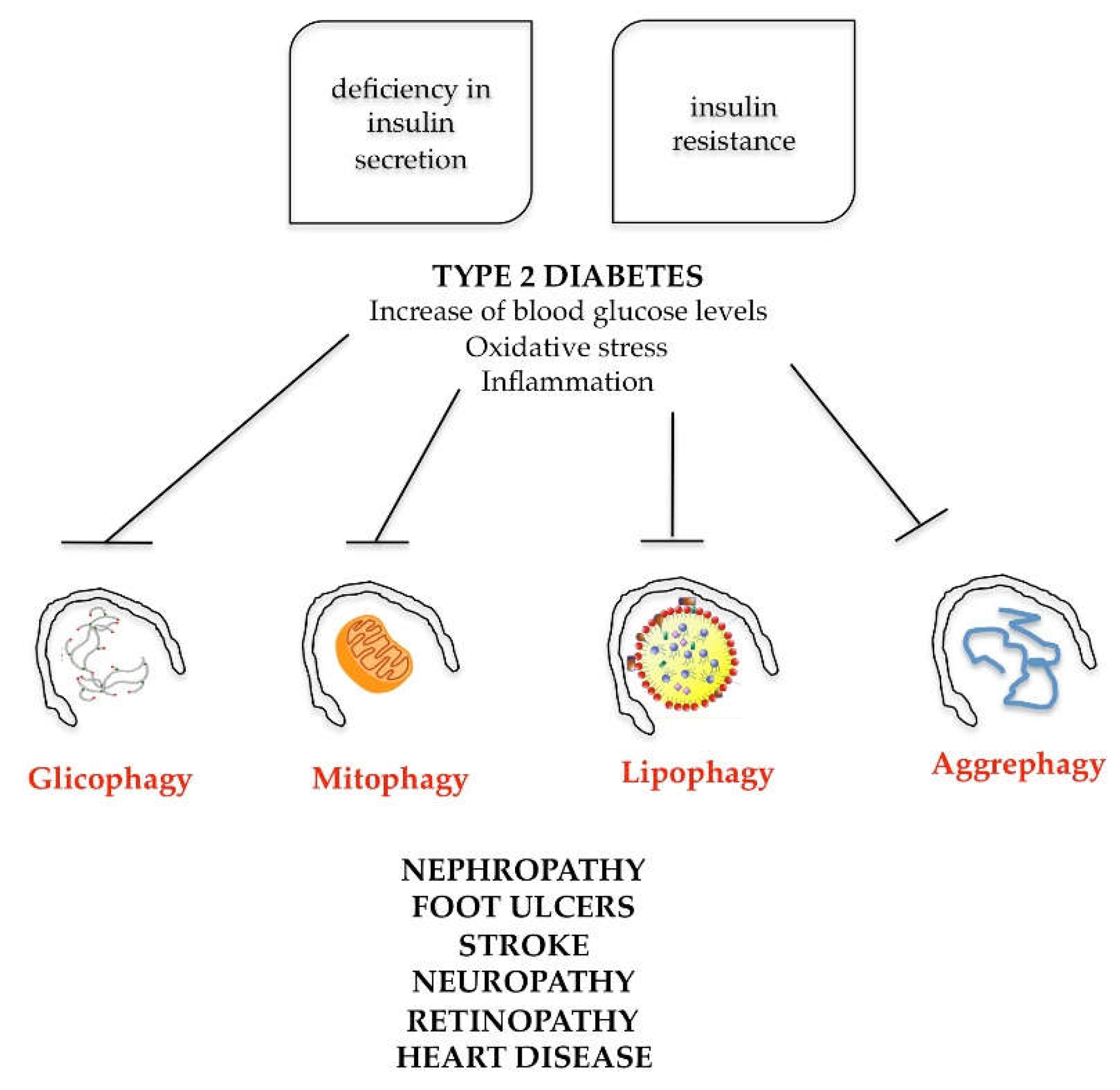
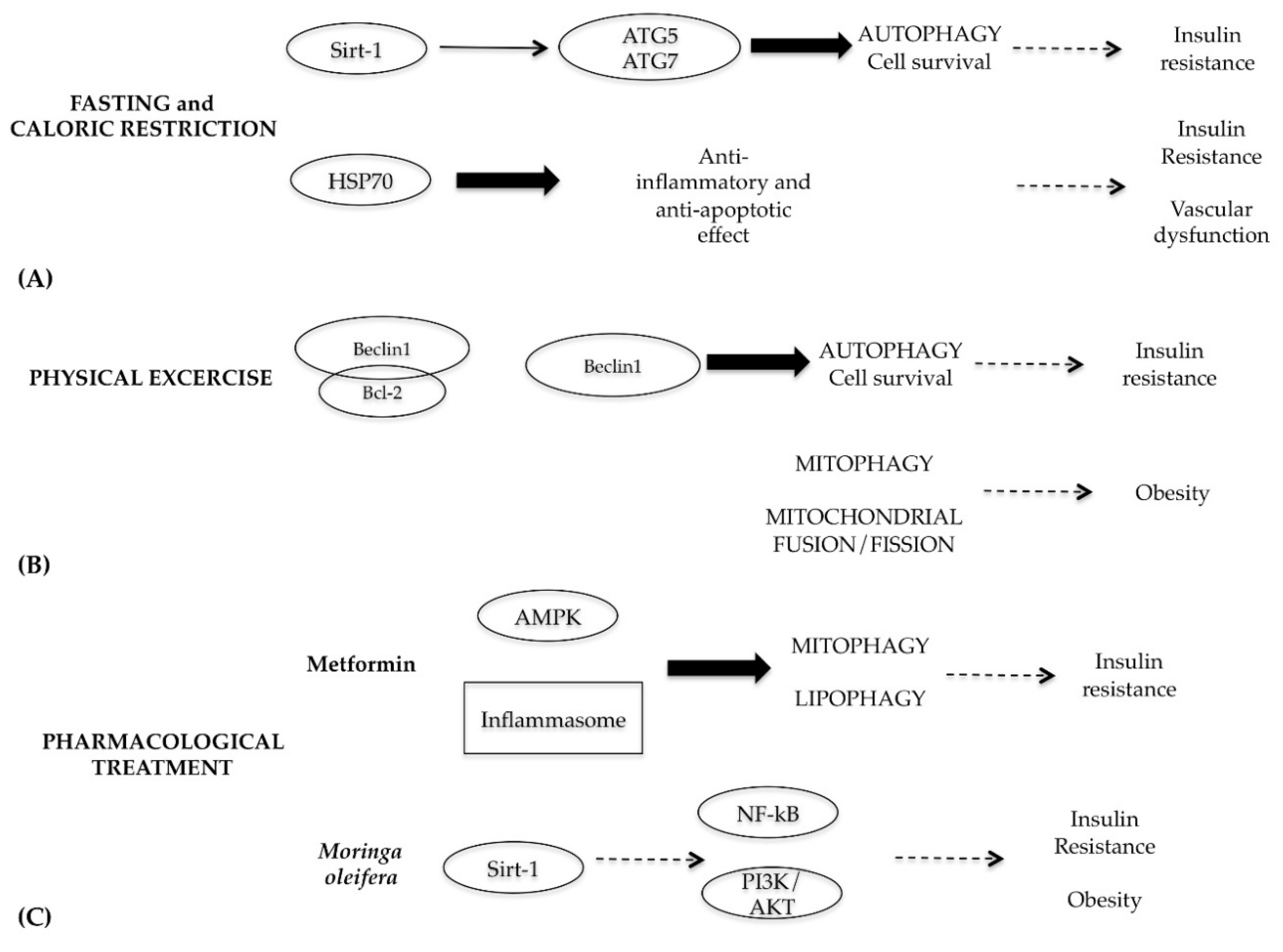
7. Role of Autophagy in Metabolic Syndrome Diseases
- high blood pressure (above 85/130 mmHg);
- high triglycerides (above 150 mg/dL);
- low levels of high-density lipoprotein (HDL) cholesterol (lower than 40/50 mg/dL);
- high blood sugar (above 100 mg/dL);
- visceral distribution of body fat [72].
8. Autophagy and Neurodegenerative Disorders
9. Pro-Survival Autophagy in Cancer
10. Autophagy and Autoimmune Diseases
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATG | Autophagy |
| mTOR | Mammalian target of rapamycin |
| PKA | Protein kinase A |
| ULK1 | Unc-51-like kinase 1 |
| NAD+ | Nicotinamide adenine dinucleotide |
| AMPK | AMP-activated protein kinase |
| FIP200 | Focal adhesion kinase family interacting protein of 200 kDa |
| PtdIns3K | Phosphatidylinositol 3-kinase |
| VPS | Vacuolar protein sorting |
| AMBRA1 | Activating molecule in BECN1 regulated autophagy protein 1 |
| UVRAG | Ultraviolet irradiation resistance-associated gene |
| PI3P | Phosphatidylinositol-3-phosphate |
| WIPI | WD-repeat protein interacting with phosphoinositides |
| MAP1-LC3 | Microtubule-associated protein 1 light chain 3 |
| PE | Phosphatidylethanolamine |
| NBR1 | Neighbor of BRCA1 gene 1 |
| NDP52 | Nuclear domain 10 protein 52 |
| UB | Polyubiquitinated chain |
| UBD | UB-binding domain |
| LIR | LC3-interacting regions |
| SQSTM1 | Sequestosome 1 |
| BNIP3 | Bcl-2/adenovirus E1B 19-kDa interacting protein 3 |
| NLR | (NOD)-like receptor |
| IL | Interleukin |
| IGF-1 | Insulin-like growth factor 1 |
| Sirt-1 | Sirtuin-1 |
| HDL | High density lipoprotein |
| EGFR | Epidermal growth factor receptor |
| HSP70 | Heat shock protein 70 |
| TPD-43 | TAR DNA-binding protein 43 |
| PS1 | Presenilin 1 |
| Nrf2 | Nuclear factor erythroid derived 2 like 2 |
| TFEB | EB transcription factor |
| LRRK2 | Leucine rich repeat Kinase 2 |
| PARK2 | Parkin RBR E3 ubiquitin protein ligase |
| PINK1 | PTEN induced putative kinase 1 |
| HTT | Huntingtin |
| CSC | Cancer stem cells |
| SLE | Systemic lupus erythematosus |
| Treg | Regulatory T |
| Drp1 | Dynamin-related protein 1 |
| Foxp3 | Forkhead box protein 3 |
References
- Schneider, J.L.; Cuervo, A.M. Autophagy and human disease: Emerging themes. Curr. Opin. Genet. Dev. 2014, 26, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S. A key role for autophagy and the autophagy gene Atg16l in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Gupta, J.; Jyothula, S.S.; Butsch Kovacic, M.; Biagini Myers, J.M.; Pattersonm, T.L.; Ericksen, M.B.; He, H.; Gibsonm, A.M.; Baye, T.M. Functional variant in the autophagy-related 5 gene promotor is associated with childhood asmha. PLoS ONE 2012, 7, e33454. [Google Scholar]
- Zhou, X.J.; Lu, X.L.; Lv, J.C.; Yang, H.Z.; Qin, L.X.; Zhao, M.H.; Su, Y.; Li, Z.G.; Zhang, H. Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann. Rheum. Dis. 2011, 70, 1330–1337. [Google Scholar] [CrossRef] [PubMed]
- Magistrelli, C.; Samoilova, E.; Agarwal, R.K. Polymorphic genotypes of the HRES-1 human endogenous retrovirus locus correlate with systemic lupus erythematosus and autoreactivity. Immunogenetics 1999, 49, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Rea, S.L.; Majcherm, V.; Searle, M.S.; Layfieldm, R. SQSTM1 mutations-bridging Paget disease of bone and ALS/FTLD. Exp. Cell Res. 2014, 325, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fieselm, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Xu, W.; Tian, M.; Zhou, Y. The relationship between insulin resistance, adiponectin and C-reactive protein and vascular endothelial injurj in diabetic patients with coronary hearth disease. Exp. Ther. Med. 2018, 3, 2022–2026. [Google Scholar]
- Singh, S.S.; Vats, S.; Chia, A.Y.; Tanm, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh Sethi, G.; Huang, R.Y.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pedro, J.M.B.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Shravage, B.; Simin, R.; Mills, K.; Berrym, D.L.; Baehrecke, E.H.; Kumar, S. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr. Biol. 2009, 19, 1741–1746. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Nicolson, S.; Kumar, S. Cell death by autophagy: Facts and apparent artefacts. Cell Death Differ. 2012, 19, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.; Abdelmonhsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Arozena, A.A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–122. [Google Scholar] [PubMed]
- Chen, Q.; Kang, J.; Fu, C. The independence and associations among apoptosis, autophagy and necrosis. Signal Transduct. Target Ther. 2018, 3, 18. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, H.; Nishitoh, H. Endoplasmic reticulum quality control by garbage disposal. FEBS J. 2018, 286, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, J.; Janikiewicz, J.; Michalska, B.; Patas-Krawczyk, P.; Perrone, M.; Ziolkowsky, W.; Duszynski, J.; Pinton, P.; Dobrzyn, A.; Wieckowski, M.R. Interaction of mithocondria with endoplasmic reticulum in plasma membrane in calcium homeostasis, lipid trafficking and mitochondrial structure. Int. J. Mol. Sci. 2017, 18, e1576. [Google Scholar] [CrossRef]
- Domagala, A.; Fidyt, K.; Bobrowicz, M.; Stachura, J.; Szczygiel, K.; Firczuk, M. Typical and atypical inducers of lysosomal cell death: A promising anticancer strategy. Int. J. Mol. Sci. 2018, 19, e2256. [Google Scholar] [CrossRef]
- Mizushima, N.; Ohsumi, Y.; Yoshimori, T. Autophagosome formation in mammalian cells. Cell Struct. Funct. 2002, 27, 421–429. [Google Scholar] [CrossRef]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef]
- Klionsky, D.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef]
- Wang, C.W.; Klionsky, D.J. The molecular mechanism of autophagy. Mol. Med. 2003, 9, 65–76. [Google Scholar] [CrossRef]
- Stephan, J.S.; Yeh, Y.Y.; Ramachandran, V.; Deminoff, S.J.; Herman, P.K. The Tor and PKA signaling pathways independently target the Atg1/Atg13 protein Kinase complex to control autophagy. Proc. Natl. Acad. Sci. USA 2009, 106, 17049–17054. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.M.; Puente, C.; Ganley, I.C.; Jiang, X. The ULK1 complex: Sensing nutrient signals for autophagy activation. Autophagy 2013, 9, 124–137. [Google Scholar] [CrossRef]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energysensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Burman, C.; Kitstakis, N.T. Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett. 2010, 584, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Erlich, S.; Mizrachy, L.; Segev, O.; Lindenboim, L.; Zmira, O.; Adi-Harel, S.; Hirsch, J.A.; Stein, R.; Pinkas-Kramarski, R. Differential interactions between Beclin 1 and Bcl2 family members. Autophagy 2007, 3, 561–568. [Google Scholar] [CrossRef]
- Mizushima, N.; Kuma, A.; Kobayashi, Y.; Yamamoto, A.; Matsubae, M.; Takao, T.; Natsume, T.; Ohsumi, Y.; Yoshimori, T. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J. Cell Sci. 2003, 116, 1679–1688. [Google Scholar] [CrossRef]
- Hurley, J.H.; Young, L.N.J. Mechanisms of autophagy initiation. Ann. Rev. Biochem. 2017, 86, 225–244. [Google Scholar] [CrossRef]
- Kirisako, T.; Ichimura, Y.; Okada, H.; Kabeya, Y.; Mizushima, N.; Yoshimori, T.; Ohsumi, M.; Takao, T.; Noda, T.; Ohsumi, Y. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J. Cell Biol. 2000, 151, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Satoo, K.; Noda, N.N.; Kumeta, H.; Fujioka, Y.; Mizushima, N.; Oshumi, Y.; Inagaki, F. The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J. 2009, 28, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Kochl, R.; Hu, X.W.; Chan, E.Y.W.; Tooze, S.A. Microtubules facilitate autophagosomes formation and fusion of autophagosomes with endosomes. Traffic 2006, 7, 12–145. [Google Scholar] [CrossRef]
- Pankiv, S.; Alemu, E.A.; Brech, A.; Bruun, J.A.; Lamark, T.; Overvatn, A.; Bjorkov, G.; Johansen, T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell. Biol. 2010, 188, 253–269. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar]
- Zaffagnini, G.; Martens, S. Mechanisms of selective autophagy. J. Mol. Biol. 2016, 428, 1714–1724. [Google Scholar] [CrossRef]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef]
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-dependent and independent signals in selective autophagy. Trends Cell. Biol. 2016, 26, 6–16. [Google Scholar] [CrossRef]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef]
- Kirkin, V.; Lamark, T.; Sou, Y.S.; Bjørkøy, G.; Nunn, J.L.; Bruun, J.A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell. 2009, 33, 505–516. [Google Scholar] [CrossRef]
- Thurston, T.L.; Ryzhakov, G.; Bloor, S.; von Muhlinen, N.; Randow, F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 2009, 10, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef]
- Shin, J. p62 and the sequestosome, a novel mechanism for protein metabolism. Arch. Pharm. Res. 1998, 21, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiquitin-proteasome system upon ubiquinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Mizushima, N. p62 targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J. Cell. Biol. 2011, 192, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Waters, S.; Marchblanck, K.; Solomon, E.; Whitehouse, C.; Gautel, M. Interactions with LC3 and polyubiquitin chains link nbr1 to autophagic protein turnover. FEBS Lett. 2009, 583, 1846–1852. [Google Scholar] [CrossRef]
- Kirkin, V.; Lamark, T.; Johansen, T.; Dikic, I. NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy 2009, 5, 732–733. [Google Scholar] [CrossRef]
- Deosaran, E.; Larsen, K.B.; Hua, R.; Sargent, G.; Wang, Y.; Kim, S.; Lamark, T.; Jauregui, M.; Law, K.; Lippincott-Schwartz, J.; et al. NBR1 acts as an autophagy receptor for peroxisomes. J. Cell Sci. 2013, 126, 939–952. [Google Scholar] [CrossRef]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, A.B. Microtubule-associated protein 1 light chain 3 (LC3) interscts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 2012, 287, 19094–19104. [Google Scholar] [CrossRef]
- Chu, C.T. Mechanisms of selective autophagy and mitophagy: Implications for neurodegenerative diseases. Neurobiol. Dis. 2018, 18, 30275–30284. [Google Scholar] [CrossRef]
- Meschini, S.; Condello, M.; Lista, P.; Arancia, G. Autophagy: Molecular mechanisms and their implications for anticancer therapies. Curr. Cancer Drug. Targets 2011, 11, 357–379. [Google Scholar] [CrossRef]
- Meschini, S.; Condello, M.; Calcabrini, A.; Marra, M.; Formisano, G.; Lista, P.; De Milito, A.; Federici, E.; Arancia, G. The plant alkaloid voacamine induces apoptosis-independent autopahgic cell death on both sensitive and multidrug resistant human osteosarcoma cells. Autophagy 2008, 4, 1020–1033. [Google Scholar] [CrossRef]
- Saitoh, T.; Akira, S. Regulation of inflammasomes by autophagy. J. Allergy Clin. Immunol. 2016, 138, 28–36. [Google Scholar] [CrossRef]
- Vanaja, S.K.; Rathinam, V.A.; Fitzgerald, K.A. Mechanisms of inflammasome activation: Recent advances and novel insights. Trends Cell Biol. 2015, 25, 308–315. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; De La Vega, F.M.; Briggs, J.; et al. A genome-wide association scan of nonsynonoymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2007, 3, 207–211. [Google Scholar] [CrossRef]
- Lassen, K.G.; Kuballa, P.; Conway, K.L.; Patel, K.K.; Becker, C.E.; Peloquin, J.M.; Villablanca, E.J.; Norman, J.M.; Liu, T.C.; Heath, R.J.; et al. ATG16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc. Natl. Acad. Sci. USA 2014, 111, 7741–7746. [Google Scholar] [CrossRef]
- Jounai, N.; Kobiyama, K.; Shiina, M.; Ogata, K.; Ishii, K.J.; Takeshita, F. NRLP4 negatively regulates autophagic processes through an association with beclin1. J. Immunol. 2011, 186, 1646–1655. [Google Scholar] [CrossRef]
- Collij, V.; Imhann, F.; Vich, V.A.; Fu, J.; Dijkstra, G.; Festen, E.A.M.; Voskuil, M.D.; Daly, M.J.; Xavier, R.J.; Wijmenga, C.; et al. SLC39A8 missense variant is associated with Crohn’s disease but does not have a major impact on gut microbiome composition in healthy subjects. PLoS ONE 2019, 14, e0211328. [Google Scholar] [CrossRef]
- Babayeva, G.H.; Babayev, Z.M. Frequency of detection of some markers of endothelial dysfunction in patients with inflammatory bowel diseases. Ter. Arkh. 2018, 90, 12–16. [Google Scholar] [CrossRef]
- Yang, S.K.; Hong, M.; Zhao, W.; Jung, Y.; Baek, J.; Tayebi, N.; Kim, K.M.; Ye, B.D.; Kim, K.J.; Park, S.H. Genome-wide association study of Crohn’s disease in koreans revealed three new susceptibility loci and common attributes of genetic susceptibility across ethnic populations. Gut 2014, 63, 80–87. [Google Scholar] [CrossRef]
- Yang, S.K.; Ye, B.D.; Song, K. Atg16l1 contributes to Crohn’s disease susceptibility in koreans: Overmuch concern for ethnic difference? Gut 2015, 64, 687–688. [Google Scholar] [CrossRef]
- Sun, Q.; Fan, J.; Billiar, T.R.; Scott, M.J. Inflammasome and autophagy regulation: A two-way street. Mol. Med. 2017, 23, 188–195. [Google Scholar] [CrossRef]
- Davis, B.K.; Wen, H.; Ting, J.P. The inflammasome NLRs in immunity, inflammation, and associated diseases. Ann. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef]
- Catana, C.S.; Atanasov, A.G.; Berindan-Neagoe, I. Natural products with anti-aging potential: Affected targets and molecular mechanisms. Biotechnol. Adv. 2018, 36, 1649–1656. [Google Scholar] [CrossRef]
- Hou, X.; Rooklin, D.; Fang, H.; Zhang, Y. Resveratrol serves as protein-substrate interaction stabilizer in human SIRT1 activation. Sci. Rep. 2016, 6, 38186. [Google Scholar] [CrossRef]
- Si, H.; Liu, D. Dietary antiaging phytochemicals and mechanisms associated with prolonged survival. J. Nutr. Biochem. 2014, 25, 581–591. [Google Scholar] [CrossRef]
- NHS: Home. Available online: https://www.nhs.uk/conditions/metabolic-syndrome/ (accessed on 18 October 2018).
- Yang, J.S.; Lu, C.C.; Kuo, S.C.; Hsu, Y.M.; Tsai, S.C.; Chen, S.Y.; Chen, Y.T.; Lin, Y.J.; Huang, Y.C.; Chen, C.J.; et al. Autophagy and its link to type II diabetes mellitus. Biomedicine 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Rocchi, A.; He, C. Emerging roles of autophagy in metabolism and metabolic disorders. Front. Biol. 2015, 10, 154–164. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Oshumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef]
- Jung, H.S.; Chung, K.W.; Won Kim, J.; Kim, J.; Komatsu, M.; Tanaka, K.; Nguyen, Y.H.; Kang, T.M.; Yoon, K.H.; Kim, J.W.; et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008, 8, 318–324. [Google Scholar] [CrossRef]
- Zhang, Y.; Goldman, S.; Baerga, R.; Zhao, Y.; Komatsu, M.; Jin, S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 19860–19865. [Google Scholar] [CrossRef]
- Kim, K.M.; Jeong, Y.T.; Oh, H.; Kim, S.H.; Cho, J.M.; Kim, Y.N.; Kim, S.S.; Kim, D.H.; Hur, K.Y.; Kim, H.K.; et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing FGF21 as mitokine. Nat. Med. 2013, 19, 83–92. [Google Scholar] [CrossRef]
- Long, M.; Li, X.; Li, L.; Dodson, M.; Zhang, D.D.; Zheng, H. MuLtifunctional p62 effcts underlie diverse metabolic diseases. Trends Endocrinol. Metab. 2017, 28, 818–830. [Google Scholar] [CrossRef]
- Zhang, Y.; Sowers, J.R.; Ren, J. Targeting autophagy in obesity. From pathophysiology to management. Nat. Rev. Endocrinol. 2018, 14, 356–376. [Google Scholar] [CrossRef]
- Golbidi, S.; Daiber, A.; Korac, B.; Li, H.; Essop, M.F.; Laher, I. Health benefits of fasting and caloric restriction. Curr. Diab. Rep. 2017, 17, 123. [Google Scholar] [CrossRef]
- Cohen, H.Y.; Miller, C.; Bitterman, K.J.; Wall, N.R.; Hekking, B.; Kessler, B.; Howitz, K.T.; Gorospe, M.; de Cabo, R.; Sinclair, D.A. Caloric restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 2004, 305, 390–392. [Google Scholar] [CrossRef]
- Cantò, C.; Auwerx, J. Calorie restrcition: Is AMPK a kaey sensor and effector? Physiology 2011, 26, 214–224. [Google Scholar] [CrossRef]
- Zabolotny, J.M.; Kim, Y.B. Silencing insulin resistance through SIRT1. Cell Metab. 2007, 6, 247–249. [Google Scholar] [CrossRef]
- Morselli, E.; Maiuri, M.C.; Markaki, M.; Megalou, E.; Pasparaki, A.; Palikaras, K.; Criollo, A.; Galluzzi, L.; Malik, S.A.; Vitale, I.; et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1 dependent induction of autophagy. Cell Death Dis. 2010, 1, e10. [Google Scholar] [CrossRef]
- Weiss, E.P.; Fontana, L. Caloric restriction: Oowerful protection for the aging heart and vasculature. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1205–1219. [Google Scholar] [CrossRef]
- Atalay, M.; Oksala, N.; Lappalainen, J.; Laaksonen, D.E.; Sen, C.K.; Roy, S. Heat shock proteins in diabetes and wound healing. Curr. Protein Pept. Sci. 2009, 10, 85–95. [Google Scholar] [CrossRef]
- Vainshetein, A.; Hood, D.A. The regulation of autophagy during exercise in skeletal muscle. J. Appl. Physiol. 2016, 120, 664–673. [Google Scholar] [CrossRef]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise–induced BCL2–regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar] [CrossRef]
- Heo, J.W.; No, M.H.; Park, D.H.; Kang, J.H.; Seo, D.Y.; Han, J.; Neufer, P.D.; Kwak, H.B. Effects of exercise on obesity-induced mitochondrial dysfunction in skeletal muscle. Korean J. Physiol. Pharmacol. 2017, 21, 567–577. [Google Scholar] [CrossRef]
- Hur, K.Y.; Lee, M.S. New mechanisms of metformin action: Focusing on mitochondria and the gut. J. Diabetes Investig. 2015, 6, 600–609. [Google Scholar] [CrossRef]
- Tabatabaei-Malazy, O.; Larijani, B.; Abdollahi, M. Targeting metabolic disorders by natural products. J. Diabetes Metab. Disord. 2015, 14, 57–78. [Google Scholar] [CrossRef]
- Kou, X.; Li, B.; Olayanju, J.B.; Drake, J.M.; Chen, N. Nutraceutical or pharmacological potential of Moringa Oleifera Lam. Nutrients 2018, 10, 343–355. [Google Scholar] [CrossRef]
- Sosa-Gutiérrez, J.A.; Valdéz-Solana, M.A.; Forbes-Hernàndez, T.Y.; Avitia-Domìnguez, C.I.; Garcia-Vargas, G.G.; Salas-Pacheco, J.M.; Flores-Herrera, O.; Téllez-Valencia, A.; Battino, M.; Sierra-Campos, E. Effects of Moringa oleifera Leaves Extract on High Glucose-Induced Metabolic Changes in HepG2 Cells. Biology 2018, 7, 37–56. [Google Scholar] [CrossRef]
- Crews, L.; Spencer, B.; Desplats, P.; Patrick, C.; Paulino, A.; Rockenstein, E.; Hansen, L.; Adame, A.; Galasko, D.; Masliaet, E. Selective molecular alteration in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS ONE 2010, 5, e9313. [Google Scholar] [CrossRef]
- Blennow, K.; Zetterberg, H. Biomarker for Alzehimer’s disease: Current status and prospects for the future. J. Intern. Med. 2018, 284, 643–663. [Google Scholar] [CrossRef]
- Hsu, S.; Gordon, B.A.; Hornbeck, R.; Norton, J.B.; Levitch, D.; Louden, A.; Ziegemeier, E.; Laforce, R., Jr.; Chhatwal, J.; Day, G.S.; et al. Discovery and validation of autosomal dominant Alzheimer’s disease mutations. Alzehimers Res. Ther. 2018, 10, 67. [Google Scholar] [CrossRef]
- Tong, B.C.; Wu, A.J.; Li, M.; Cheung, K.H. Calcium signalling in Alzehimer’s disease and therapies. Biochim. Biophy. Acta Mol. Cell Res. 2018, 1865, 1745–1760. [Google Scholar] [CrossRef]
- Prasad, K.N. Simultaneous activation of Nrf2 and elevation of antioxidant compounds for reducing oxidative stress and chronic inflammationin human Alzheimer’s disease. Mech. Ageing Dev. 2016, 153, 41–47. [Google Scholar] [CrossRef]
- Kim, B.W.; Hong, S.B.; Kim, J.H.; Kwon, D.H.; Song, H.K. Structural basic for recognition of autophagic receptor NDP52 by the sugar recepto galectin-B. Nat. Commun. 2013, 4, 1613. [Google Scholar] [CrossRef]
- Spagnuolo, M.S.; Bergamo, P.; Crescenzo, R.; Iannotta, L.; Treppiccione, L.; Iossa, S.; Cigliano, L. Brain Nrf2 pathway, autophagy and synaptic function proteins are modulated by a short-term fructose feeding in young and adult rats. Nutr. Neurosci. 2018, 1–12. [Google Scholar] [CrossRef]
- Guo, F.; Liu, X.; Cai, H.; Le, W. Autophagy in neurodegenerative diseases: Pathogenesis and therapy. Brain Pathol. 2018, 28, 3–13. [Google Scholar] [CrossRef]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Martini-Stoica, H.; Xu, Y.; Ballabio, A.; Zheng, H. The autophagy-lysosomal pathway in neurodegeneration: A TFEB perspective. Trends Neurosci. 2016, 39, 221–234. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef]
- Kazlauskaite, A.; Muqit, M.M. PINK1 and Parkin-mitochondrial interplay between phosphorylation and ubiquitylation in Parkinson’s disease. FEBS J. 2015, 282, 215–223. [Google Scholar] [CrossRef]
- Matsumoto, G.; Wada, K.; Okuno, M.; Kurosawa, M.; Nukina, N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol. Cell 2011, 44, 279–289. [Google Scholar] [CrossRef]
- Pasinelli, P.; Brown, R.H. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat. Rev. Neurosci. 2006, 7, 710–723. [Google Scholar] [CrossRef]
- Poillet-Perez, L.; Despouy, G.; Delage-Mourroux, R.; Boyer-Guittaut, M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015, 4, 184–192. [Google Scholar] [CrossRef]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin-1, an autophagy gene essential for early embryonic development, is a haploinsuffient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Autophagy in cancer metastasis. Oncogene 2017, 36, 1619–1630. [Google Scholar] [CrossRef]
- Ojha, R.; Jha, V.; Singh, S.K.; Bhattacharyya, S. Autophagy inhibition suppresses the tumorigenic potential of cancer stem cell enriched side population in bladder cancer. Biochim. Biophys. Acta 2014, 1842, 2073–2086. [Google Scholar] [CrossRef]
- Gong, C.; Bauvy, C.; Tonelli, G.; Yue, W.; Deloménie, C.; Nicolas, V.; Zhu, Y.; Domergue, V.; Marin-Esteban, V.; Tharinger, H.; Delbos, L.; et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene 2013, 32, 2261–2272. [Google Scholar] [CrossRef]
- Tan, Q.; Wang, M.; Yu, M.; Zhang, J.; Bristow, R.G.; Hill, R.P.; Tannock, I.F. Role of autophagy as a survival mechanism for hypoxic cells in tumors. Neoplasia 2016, 18, 347–355. [Google Scholar] [CrossRef]
- Li, Y.J.; Lei, Y.H.; Yao, N.; Wang, C.R.; Hu, N.; Ye, W.C.; Zhang, D.M.; Chen, Z.S. Autophagy and multidrug resistance in cancer. Chin. J. Cancer 2017, 36, 52–62. [Google Scholar] [CrossRef]
- Qiu, S.; Sun, L.; Jin, Y.; An, Q.; Weng, C.; Zheng, J. Silencing of BAG3 promotes the sensitivity of ovarian cancer cells to cisplatin via inhibition of autophagy. Oncol. Rep. 2017, 38, 309–316. [Google Scholar] [CrossRef]
- Shimizu, S. Autophagic cell death and cancer chemotherapeutics. In Innovative Medicine: Basic Research and Development (Internet); Nakao, K., Minato, N., Uemoto, S., Eds.; Springer: Tokyo, Japan, 2015. [Google Scholar]
- Diederich, M.; Cerella, C. Non-canonical programmed cell desth mechanisms triggered by natural compounds. Semin. Cancer Biol. 2016, 40, 4–34. [Google Scholar] [CrossRef]
- Zhao, G.X.; Pan, H.; Ouyang, D.Y.; He, X.H. The critical molecular interconnections in regulating apoptosis and autophagy. Ann. Med. 2015, 47, 305–315. [Google Scholar] [CrossRef]
- Islam, M.A.; Sooro, M.A.; Zhang, P. Autophagic regulation of p62 is critical for cancer therapy. Int. J. Mol. Sci. 2018, 19, 1405. [Google Scholar] [CrossRef]
- De Roock, W.; De Vriendt, V.; Normanno, N.; Ciardiello, F.; Tejpar, S. KRAS, BRAF, PIK3CA, and PTEN mutations: Implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011, 12, 594–603. [Google Scholar] [CrossRef]
- Wiersma, V.R.; de Bruyn, M.; Wei, Y.; van Ginkel, R.J.; Hirashima, M.; Niki, T.; Nishi, N.; Zhou, J.; Pouwels, S.D.; Samplonius, D.F.; et al. The epithelial polarity regulator LGALS9/galectin-9 induces fatal frustrated autophagy in KRAS mutant colon carcinoma that depends on elevated basal autophagic flux. Autophagy 2015, 11, 1373–1388. [Google Scholar] [CrossRef]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef]
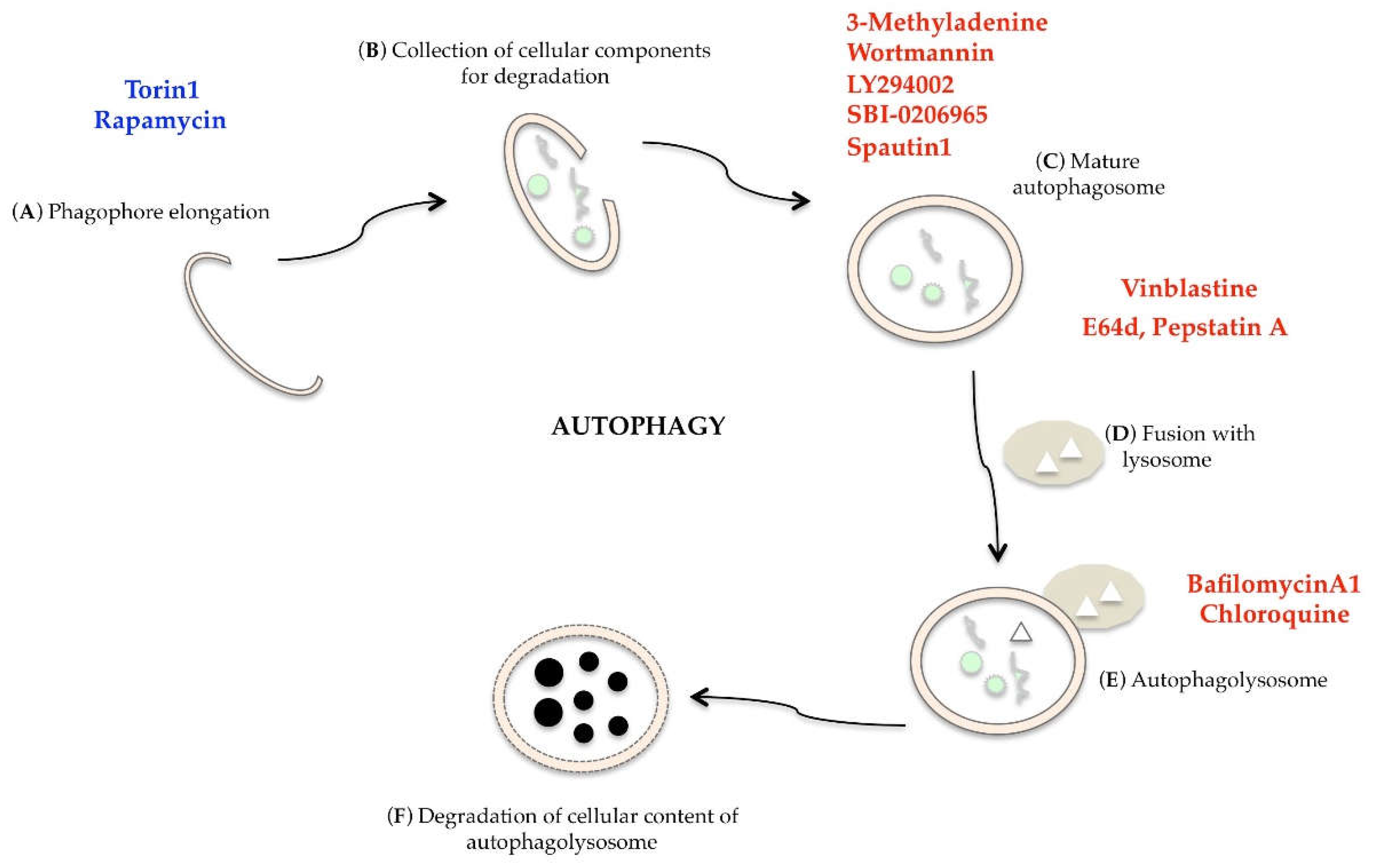
- Yang, Y.P.; Hu, L.F.; Zheng, H.F.; Mao, C.J.; Hu, W.D.; Xiong, K.P.; Wang, F.; Liu, C.F. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol. Sin. 2013, 34, 625–635. [Google Scholar] [CrossRef]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Met. 2014, 19, 373–379. [Google Scholar] [CrossRef]
- Selvakumaran, M.; Amaravadi, R.K.; Vasilevskaya, I.A.; O’Dwyer, P.J. Autophagy inhibition sensitizes colon cancer cells to antiangiogenic and cytotoxic therapy. Clin. Cancer Res. 2013, 19, 2995–3007. [Google Scholar] [CrossRef]
- Wang, P.; Zhu, L.; Sun, D.; Gan, F.; Gao, S.; Yin, Y.; Chen, L. Natural products as modulatory of autophagy with potential clinical prospects. Apoptosis 2017, 22, 325–356. [Google Scholar] [CrossRef]
- Huang, N.; Perl, A. Metabolism as a target for modulation in autoimmune diseases. Trends Immunol. 2018, 39, 562–576. [Google Scholar] [CrossRef]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passeguè, E. Autophagy maintains the metabolism and function of young and old (hematopoietic) stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef]
- Delgoffe, G.M.; Pollizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Xiao, B.; Worley, P.F.; Powell, D.J. The kinase mTor regulates the differantation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 2011, 12, 295–304. [Google Scholar] [CrossRef]
- Caza, T.N.; Fernandez, D.R.; Talaber, G.; Oaks, Z.; Haas, M.; Madaio, M.P.; Lai, Z.; Miklossy, G.; Singh, R.R.; Chudakov, D.M.; et al. HRES/Rab4-mediated depletion of Drp1 impairs mitochondrial homeostasis and represents a target for treatment in SLE. Ann. Rheum. Dis. 2014, 73, 1888–1897. [Google Scholar] [CrossRef]
- Kato, H.; Perl, A. MTORC1 expands Th17 and IL-4+DN T cells and contracts Tregs in SLE. J. Immunol. 2014, 192, 4134–4144. [Google Scholar] [CrossRef]
- Kato, H.; Perl, A. The IL-21-mTOR axis blocks Treg differentiation and function by suppression of autophagy in patients with systemic lupus erythematosus. Arthritis Rheumatol. 2018, 70, 427–438. [Google Scholar] [CrossRef]
- Oaks, Z.; Winans, T.; Caza, T.; Fernandez, D.; Liu, Y.; Landas, S.K.; Banki, K.; Perl, A. Mitochondrial dysfunction in the liver and antiphospholipid antibody production precede disease onset and respond to rapamycin in lupus-prone mice. Arthritis Rheumatol. 2016, 69, 1035–1044. [Google Scholar] [CrossRef]
- Clarke, A.J.; Ellinghaus, U.; Cortini, A.; Stranks, A.; Simon, A.K.; Botto, M.; VyseM, T.J. Autophagy is activated in systemic lupus erythematosus and required for plasmablsst development. Ann. Rheum. Dis. 2015, 74, 912–920. [Google Scholar] [CrossRef]
- Wei, J.; Long, L.; Yang, K.; Guy, C.; Shrestha, S.; Chen, Z.; Wu, C.; Vogel, P.; Neale, G.; Green, D.R.; et al. Autophagy enfornces functional integrity of regulatory T cells by coupling enviromental cues and metabolic homeostasis. Nat. Immunol. 2016, 17, 277–285. [Google Scholar] [CrossRef]
- Patel, D.D.; Kuchroo, V.K. Th17 cell pathway in human immunity: Lessons from genetics and therapeutic interventions. Immunity 2015, 43, 1040–1051. [Google Scholar] [CrossRef]
- Valencia, X.; Yarboro, C.; Illei, G.; Lipsky, P.E. Deficient CD4+CD25 high t regulatory cell function in patients with active systemic lupus erythematosus. J. Immunol. 2007, 178, 2579–2588. [Google Scholar] [CrossRef]
- Bertsias, G.K.; Tektonidou, M.; Amoura, Z.; Aringer, M.; Bajema, I.; Berden, J.H.; Boletis, J.; Cervera, R.; Dorner, T.; Doria, A.; et al. European League Against R, European League Against Rheumatism and European Renal Association-European Dialysis and Transplant Association: Joint European League Against Rheumatism and European Renal Association-European Dialysis and Transplant Association (EULAR/ERA-EDTA) recommendations for the management of adult and paediatric lupus nephritis. Ann. Rheum. Dis. 2012, 71, 1771–1782. [Google Scholar]
- An, N.; Chen, Y.; Wang, C.; Yang, C.; Wu, Z.H.; Xue, J.; Ye, L.; Wang, S.; Liu, H.F.; Pan, Q.; et al. Chloroquine autophagic inhibition rebalances Th17/Treg-mediated immunity and ameliorates systemic lupus erythematosus. Cell. Physiol. Biochem. 2017, 44, 412–422. [Google Scholar] [CrossRef]
- Sehgal, S.N.; Bansbavk, C.C. Rapamycin: In vitro profile of a new immunosoppressive macrolide. Ann. N. Y. Acad. Sci. 1993, 685, 58–67. [Google Scholar] [CrossRef]
- Warner, L.M.; Adams, L.M.; Sehgal, S.N. Rapamycin prolongs survival and arrests pathophysiologic changes in murine systemic lupus erythematosus. Arthritis Rheum. 1994, 3, 289–297. [Google Scholar] [CrossRef]
- Lai, Z.W.; Kelly, R.; Winans, T.; Marchena, I.; Shadakshari, A.; Yu, J.; Dawood, M.; Garcia, R.; Tily, H.; Francis, L.; et al. Sirolimus in patients with clinically active systemic lipus erythematosus resistant to, or intolerant of, conventional medications: A single-arm, open-label, phase 1/2 trial. Lancet 2018, 391, 1186–1196. [Google Scholar] [CrossRef]
- Kiss, E.; Kovacs, L.; Szodoray, P. Malignancies in systemic lupus erythematosus. Autoimmun. Rev. 2010, 9, 195–199. [Google Scholar] [CrossRef]
- Younes, A.; Samad, N. Utility of mTOR inhibition in hematologic malignancies. Oncologist 2011, 16, 730–741. [Google Scholar] [CrossRef]
- de Duve, C.; de Bary, T.; Poole, B.; Trouet, A.; Tulkens, A.; Van Hoof, F. Commentary. Lysosomotropic agents. Biochem. Pharmacol. 1974, 23, 2495–2531. [Google Scholar] [CrossRef]
- Levine, B.; Klionsky, D.J. Autophagy wins the 2016 Nobel Prize in phisiology or medicine: Breakthroughs in baker’s yeast fuel advances in biomedical research. Proc. Natl. Acad. Sci. USA 2017, 114, 201–205. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types | Morphological Features | Biochemical Features | Core Regulators |
|---|---|---|---|
| Apoptosis |
|
| Positive:
|
| Necrosis |
|
| Positive:
|
| Autophagy |
|
| Positive:
|
| Method | Description |
|---|---|
| Optical and electron microscopy | Display vacuoles inside the cytoplasm, their content, their stage of maturation, and the turnover of autophagic compartments |
| GFP-LC3 fluorescence microscopy | Monitor vacuolar/lysosomal localization |
| LC3 western blotting | Monitor autophagic flux with or without autophagic modulators |
| Flow cytometry | Autphagosome quantification by fluorescent probes |
| Western blot | p62 and related LC3 binding protein turnover |
| Kinase assays or western blotting | mTOR, AMPK and ULK1 kinase activity |
| ATG genes silencing | Allows to identify ATG target proteins involved in the phenomenon |
| Combination of Treatment | Type of Tumor | Trial on ClinicalTrials.gov |
|---|---|---|
| Sirolimus or vorinostat + HCQ | Advanced solid tumors | NCT01266057 |
| Single HCQ | Glioblastoma and astrocytoma | NCT02432417 |
| HCQ + Vorinostat | Malignant solid neoplasm | NCT01023737 |
| Cisplatin, etoposide + HCQ | Stage 4 small cell lung cancer | NCT00969306 |
| HCQ + Abraxane + Gemcitabine | Pancreatic adenocarcinoma | NCT01978184 |
| Sorafenib + HCQ | Refractory or relapsed solid tumors | NCT01634893 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Condello, M.; Pellegrini, E.; Caraglia, M.; Meschini, S. Targeting Autophagy to Overcome Human Diseases. Int. J. Mol. Sci. 2019, 20, 725. https://doi.org/10.3390/ijms20030725
Condello M, Pellegrini E, Caraglia M, Meschini S. Targeting Autophagy to Overcome Human Diseases. International Journal of Molecular Sciences. 2019; 20(3):725. https://doi.org/10.3390/ijms20030725
Chicago/Turabian StyleCondello, Maria, Evelin Pellegrini, Michele Caraglia, and Stefania Meschini. 2019. "Targeting Autophagy to Overcome Human Diseases" International Journal of Molecular Sciences 20, no. 3: 725. https://doi.org/10.3390/ijms20030725
APA StyleCondello, M., Pellegrini, E., Caraglia, M., & Meschini, S. (2019). Targeting Autophagy to Overcome Human Diseases. International Journal of Molecular Sciences, 20(3), 725. https://doi.org/10.3390/ijms20030725