Lipid–Protein Interactions in Niemann–Pick Type C Disease: Insights from Molecular Modeling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
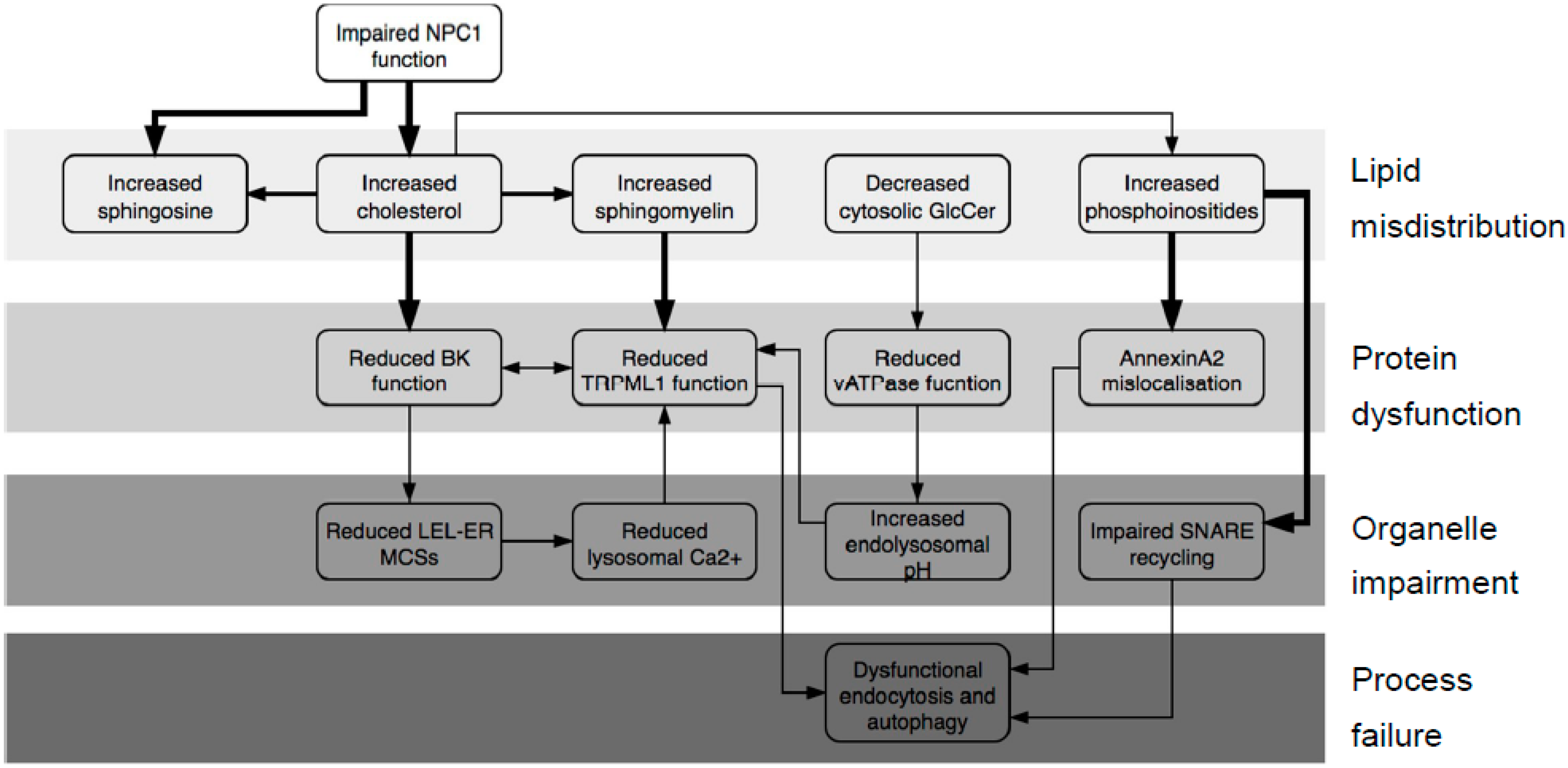
2. Results
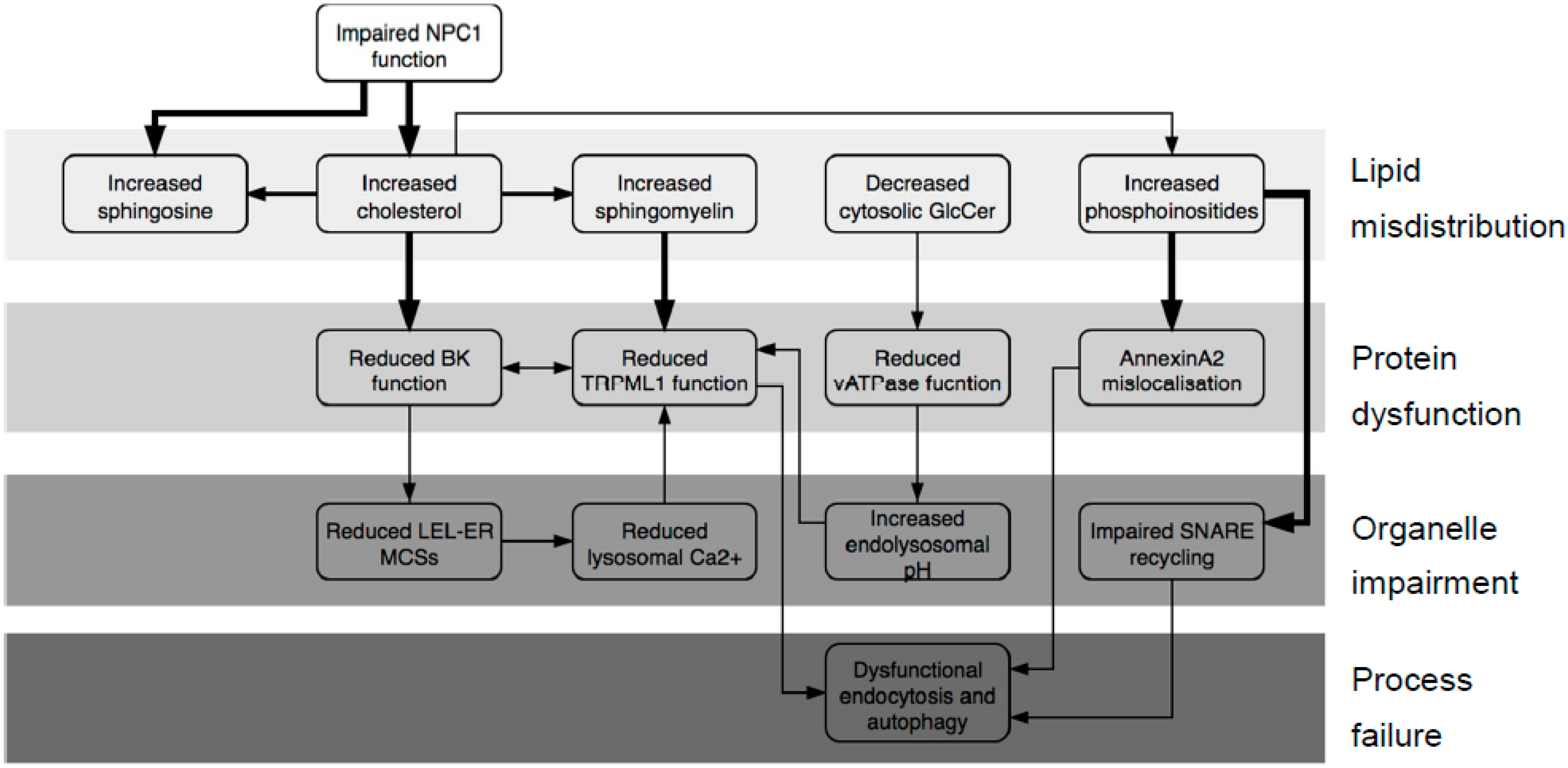
2.1. What Does the NPC System Do?
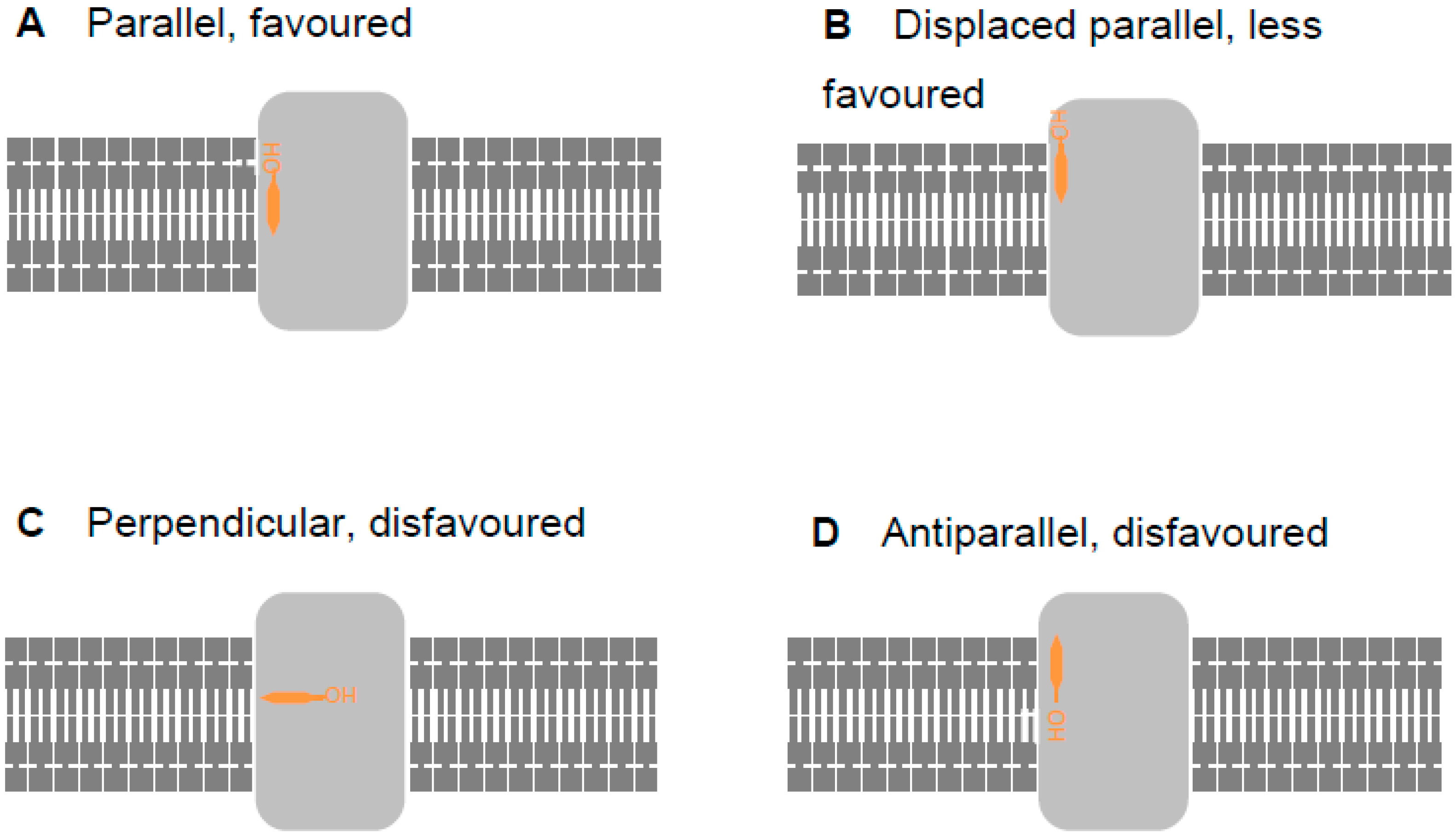
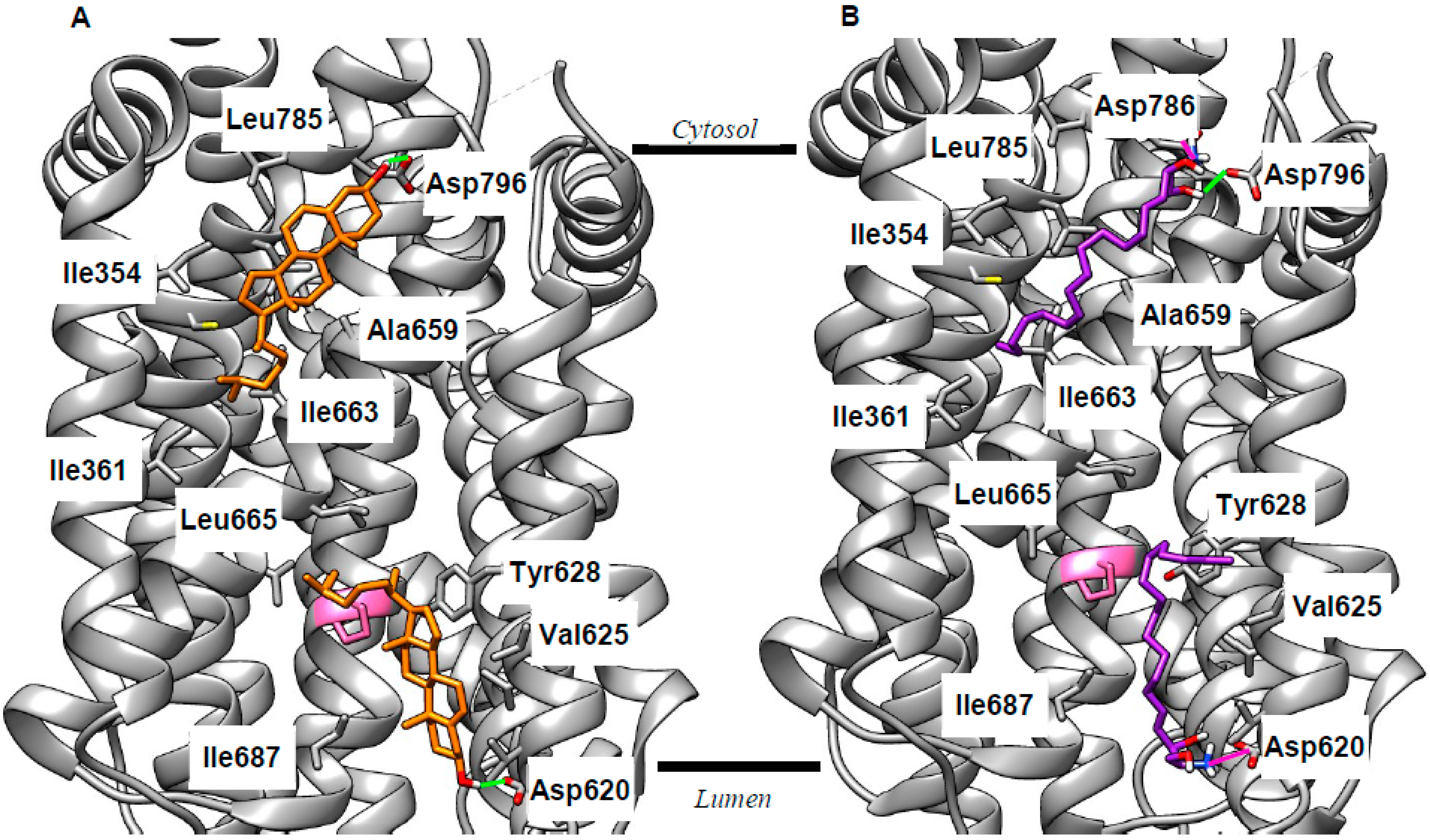
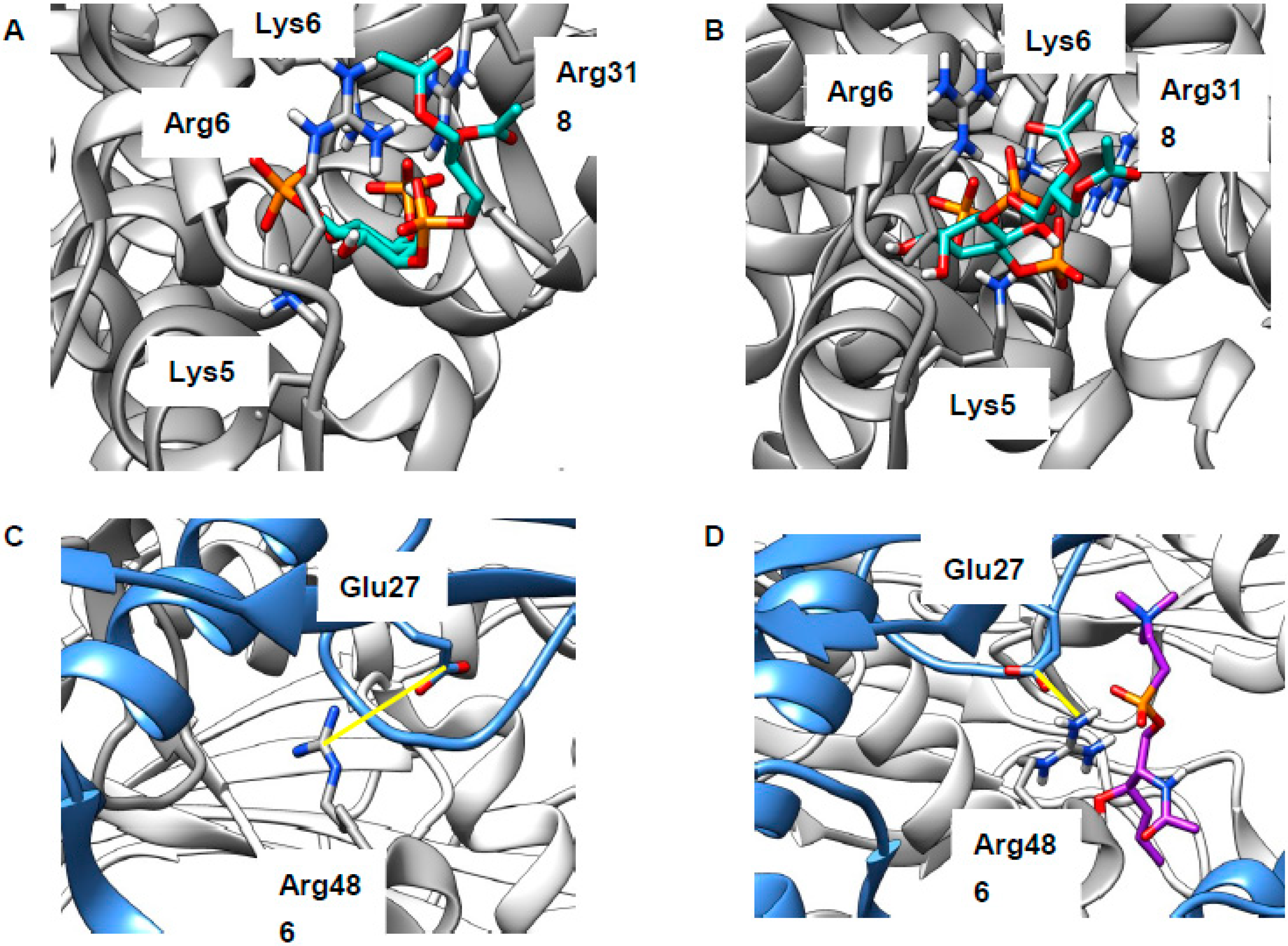
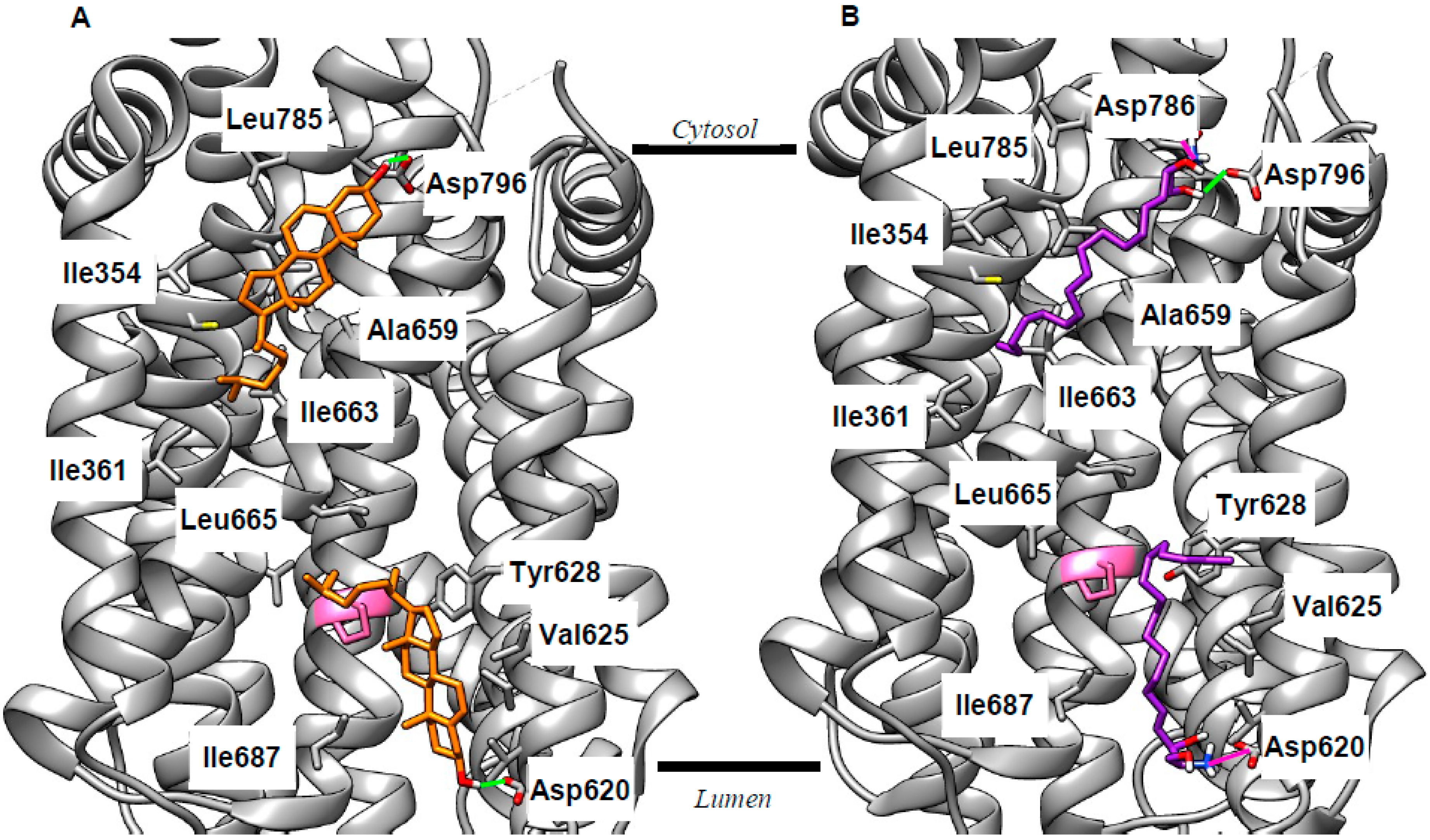
2.2. Revising the BK–Cholesterol Interaction Site
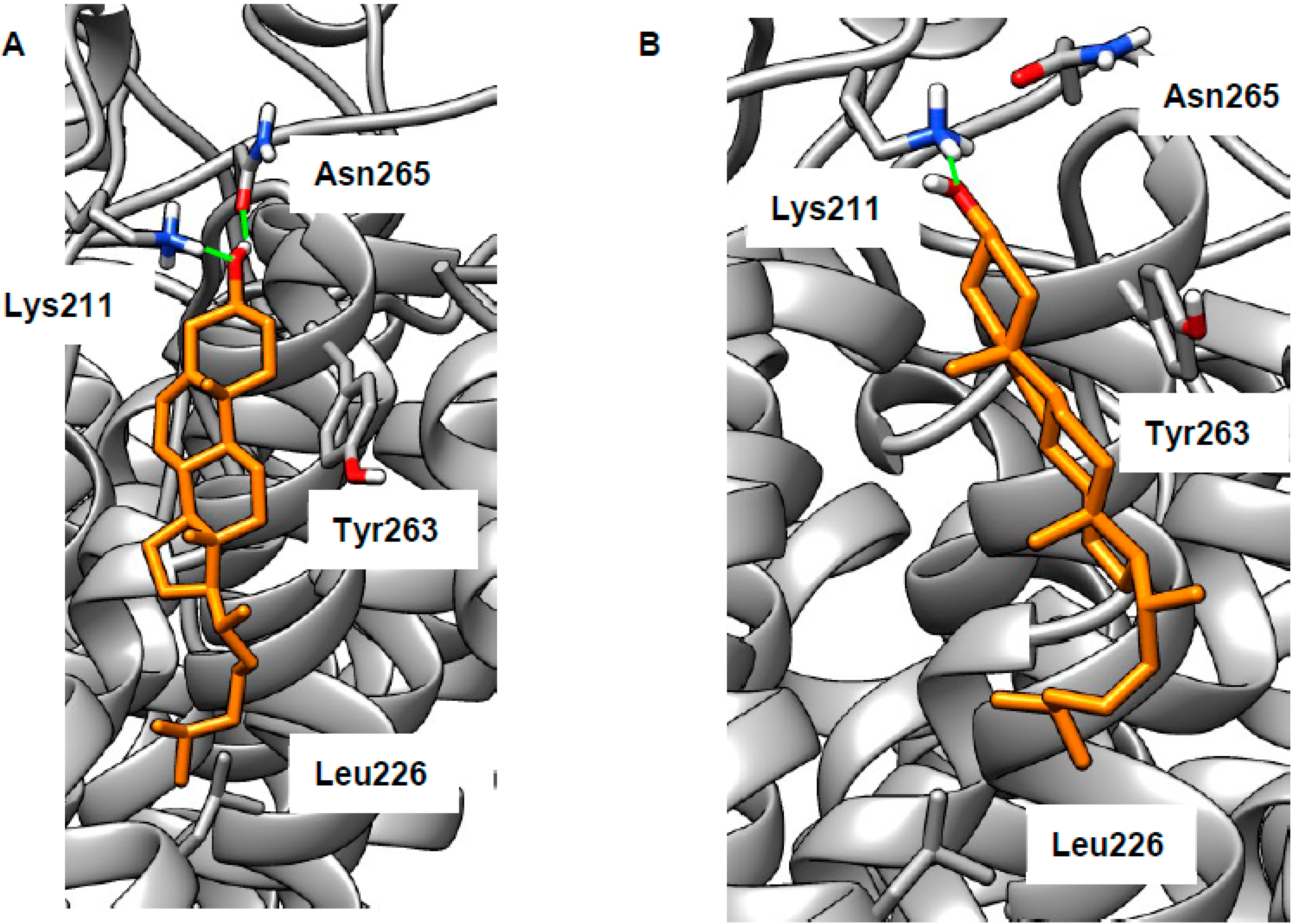
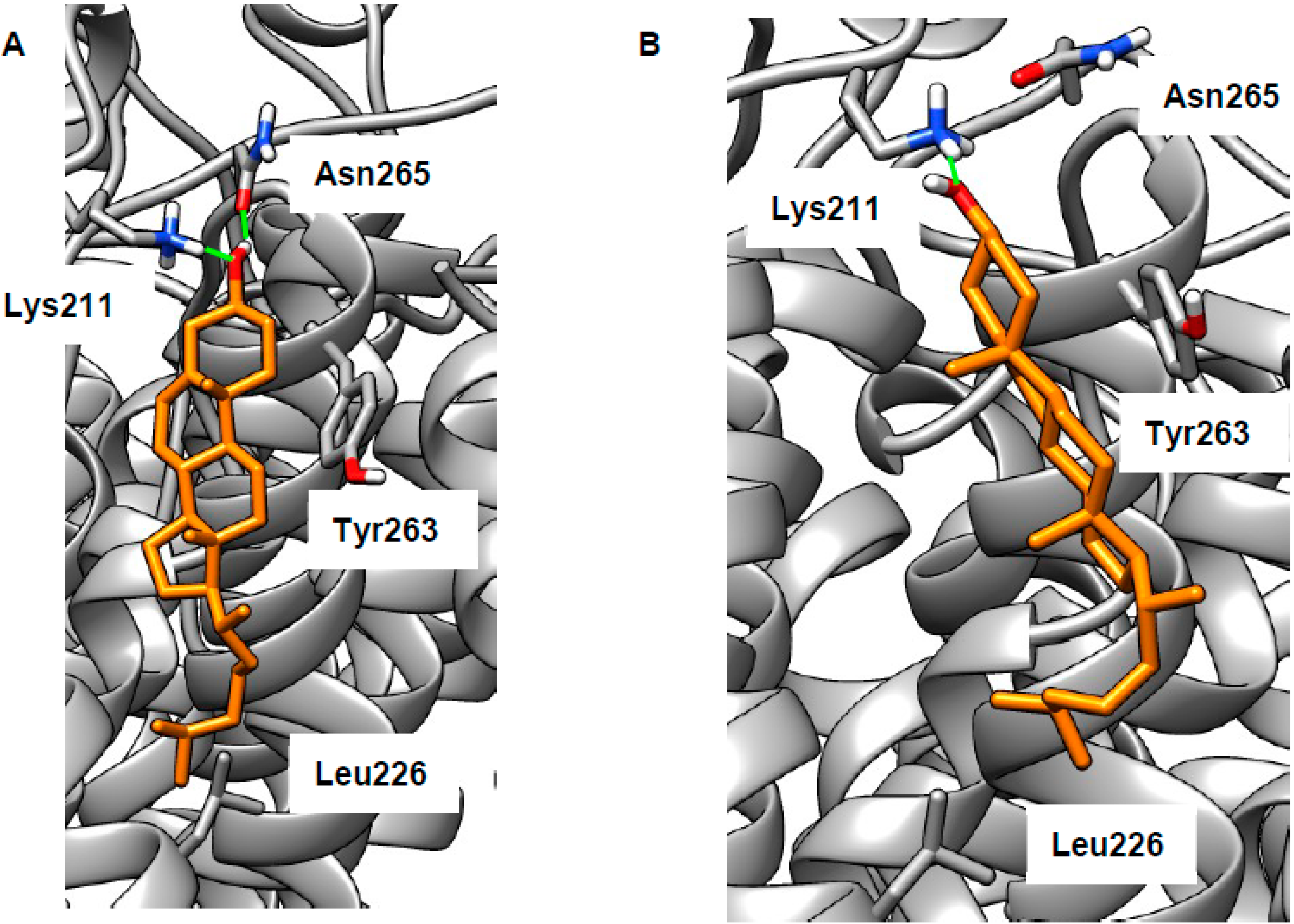
2.3. LEL Ca2+ and TRPML1
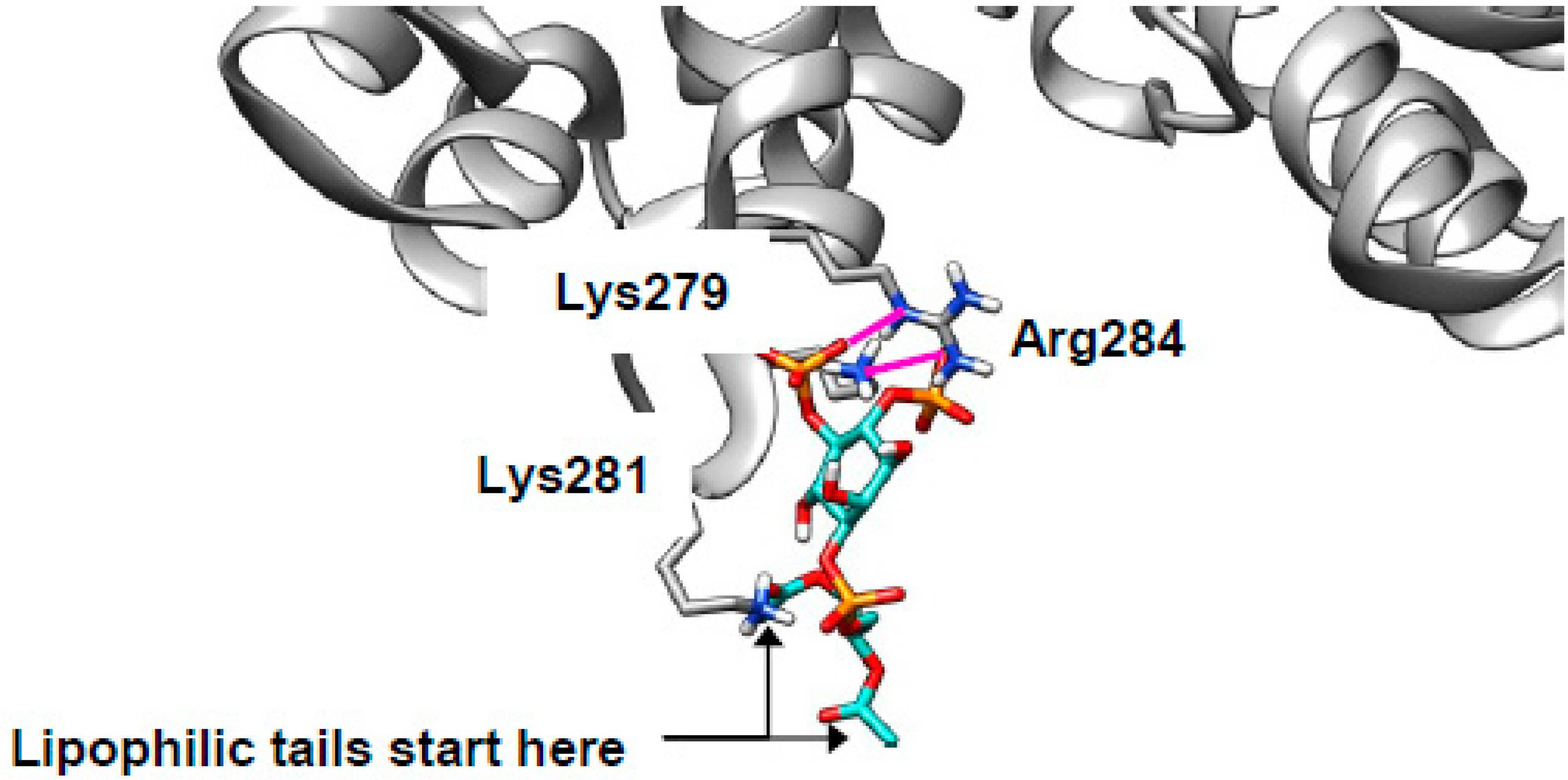
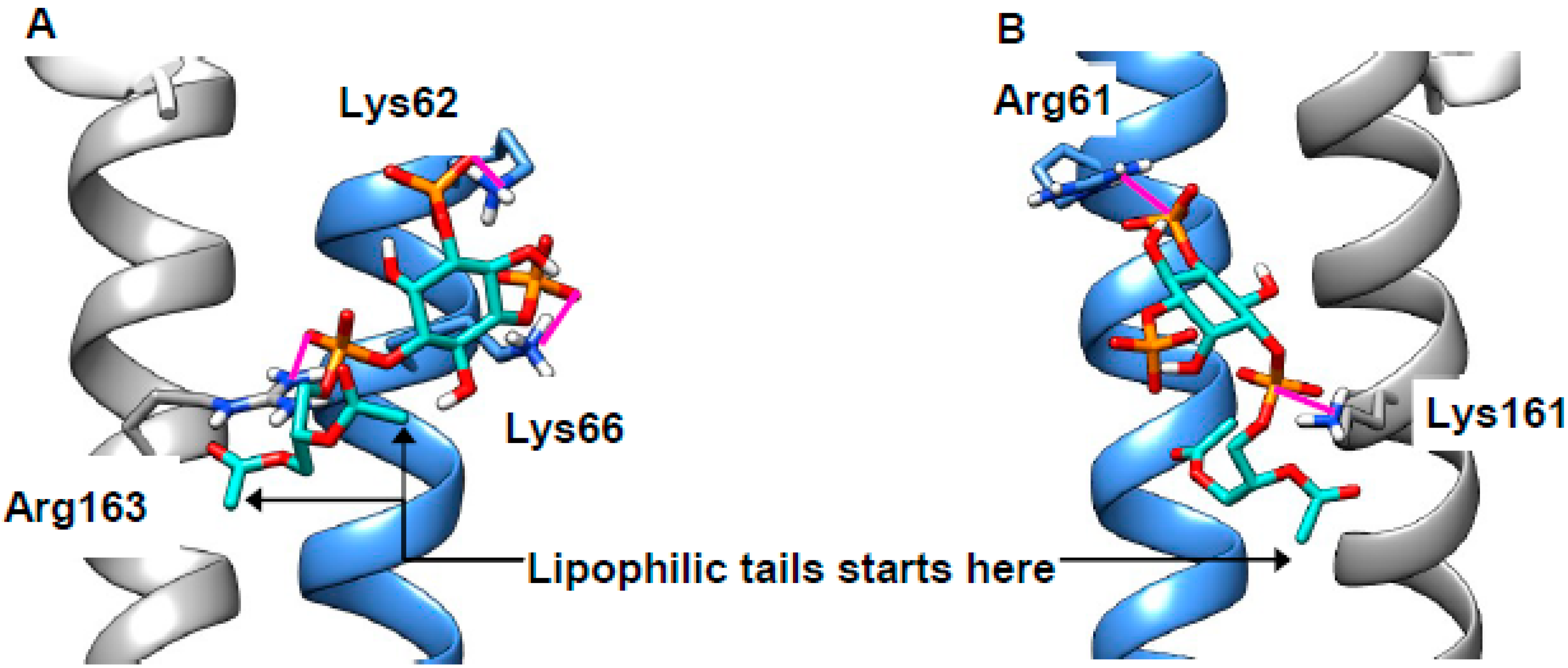
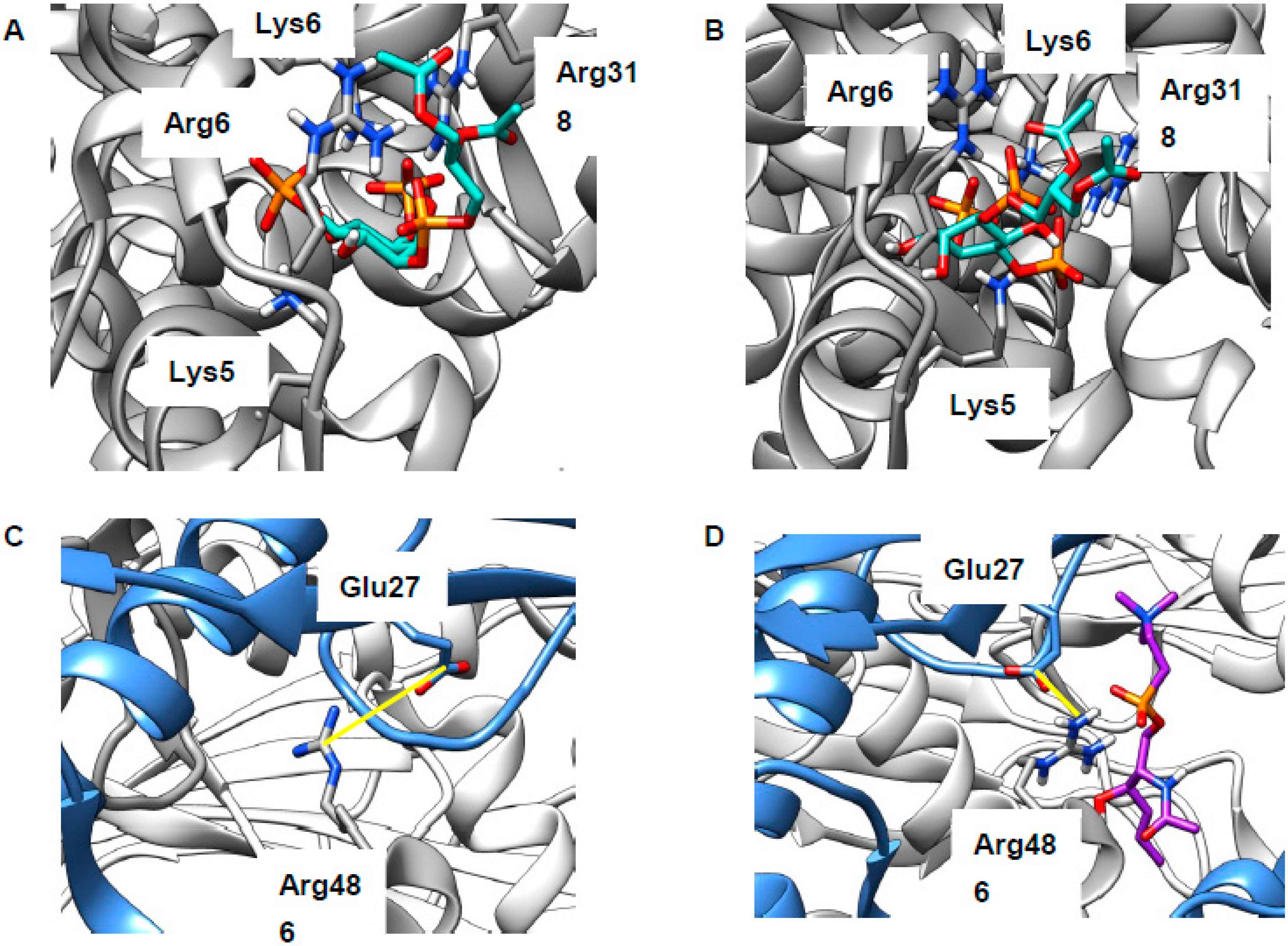
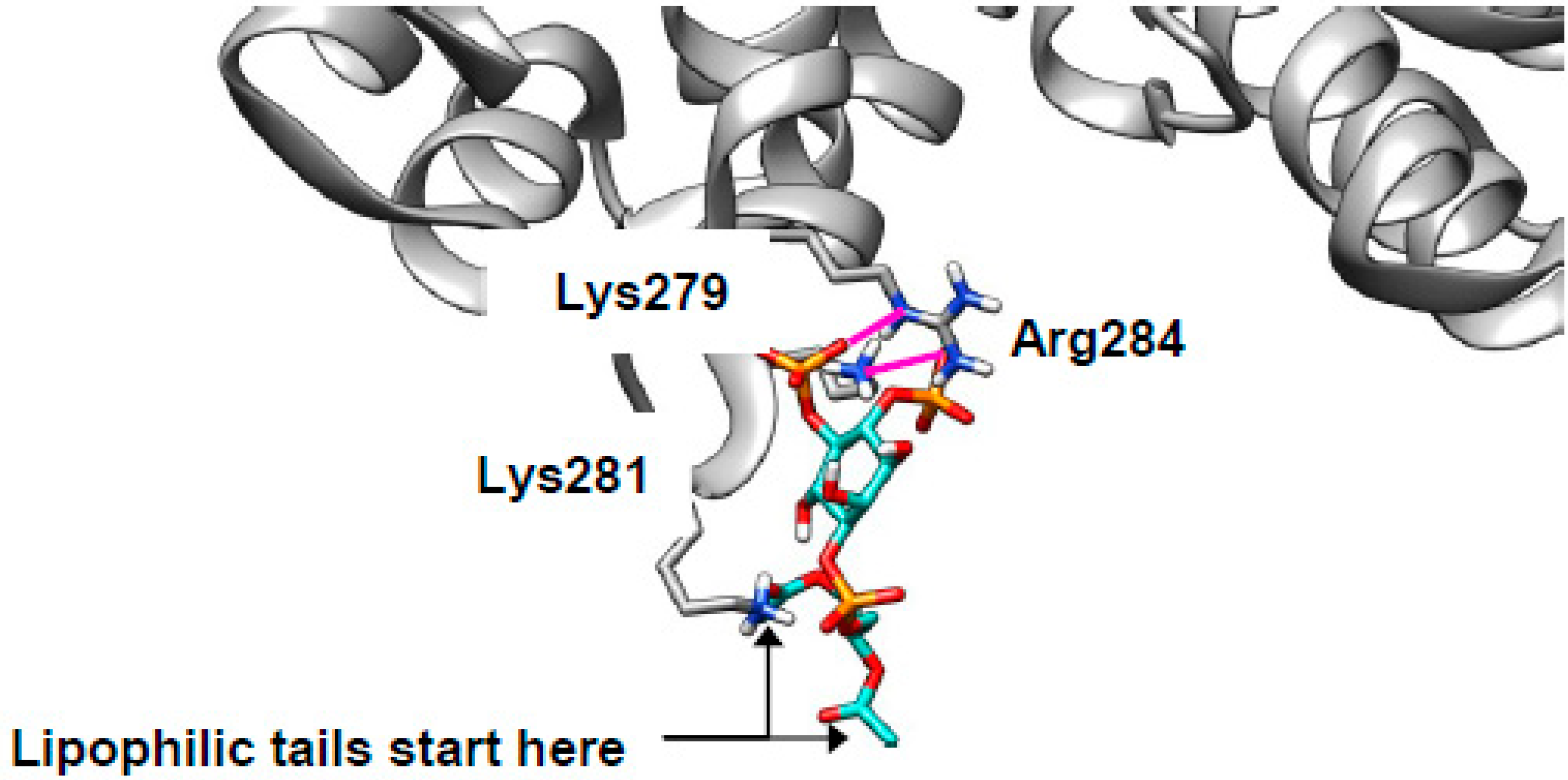
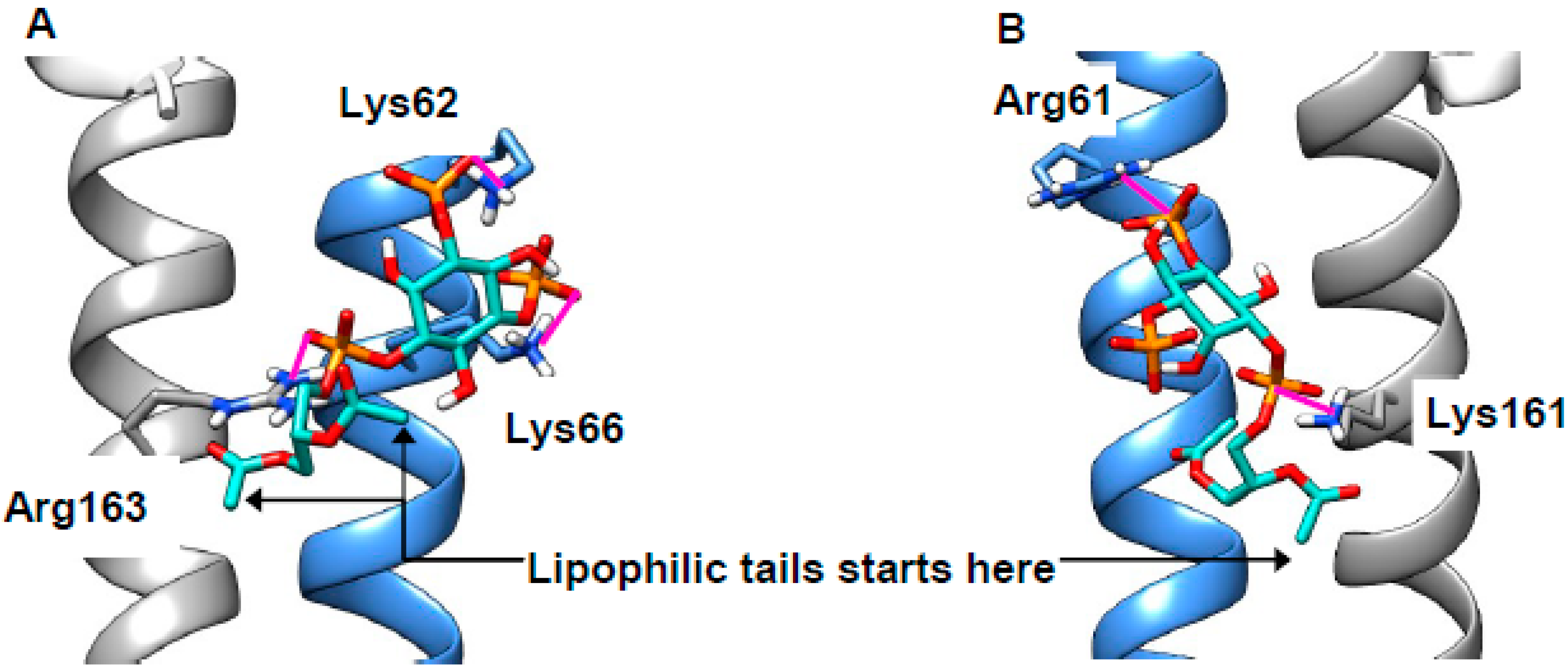
2.4. Cholesterol Clusters Phosphoinositides—Implications for Annexin Localization
2.5. Cholesterol Clusters Phosphoinositides—Implications for SNARE Complex Disassembly
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Anx | Annexin |
| BK | Big potassium |
| CRAC | Cholesterol recognition amino-acid consensus |
| ER | Endoplasmic reticulum |
| LDL | Low-density lipoprotein |
| LEL | Late endolysosome |
| LSD | Lysosomal Storage Disorder |
| MCS | Membrane contact site |
| mTOR | mechanistic Target of Rapamycin |
| NPC | Niemann–Pick type C |
| NPCD | Niemann–Pick type C disease |
| NSF | N-ethylmaleimide Sensitive Fusion |
| NTD | N-terminal domain |
| PI(3,5)P2 | Phosphatidylinositol-3,5-bisphosphate |
| PI(4,5)P2 | Phosphatidylinositol-4,5-bisphosphate |
| RCK | Regulation of conductance by potassium |
| RMS | Root mean square |
| SM | Sphingomyelin |
| SNARE | Soluble NSF protein attachment receptor |
| Sph | Sphingosine |
| SSD | Sterol-sensing domain |
| Stx | Syntaxin |
| TPC | Two-pore channel |
| TRPML | Transient receptor potential mucolipin |
| VAMP | Vesicle-associated membrane protein |
References
- Deffieu, M.S.; Pfeffer, S.R. Niemann–Pick Type C 1 Function Requires Lumenal Domain Residues That Mediate Cholesterol-Dependent NPC2 Binding. Proc. Natl. Acad. Sci. USA 2011, 108, 18932–18936. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Saha, P.; Li, J.; Blobel, G.; Pfeffer, S.R. Clues to the Mechanism of Cholesterol Transfer from the Structure of NPC1 Middle Lumenal Domain Bound to NPC2. Proc. Natl. Acad. Sci. USA 2016, 113, 10079–10084. [Google Scholar] [CrossRef] [PubMed]
- Infante, R.E.; Wang, M.L.; Radhakrishnan, A.; Kwon, H.J.; Brown, M.S.; Goldstein, J.L. NPC2 Facilitates Bidirectional Transfer of Cholesterol between NPC1 and Lipid Bilayers, a Step in Cholesterol Egress from Lysosomes. Proc. Natl. Acad. Sci. USA 2008, 105, 15287–15292. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.L.; Motamed, M.; Infante, R.E.; Abi-Mosleh, L.; Kwon, H.J.; Brown, M.S.; Goldstein, J.L. Identification of Surface Residues on Niemann-Pick C2 Essential for Hydrophobic Handoff of Cholesterol to NPC1 in Lysosomes. Cell Metab. 2010, 12, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.J.; Abi-Mosleh, L.; Wang, M.L.; Deisenhofer, J.; Goldstein, J.L.; Brown, M.S.; Infante, R.E. Structure of N-Terminal Domain of NPC1 Reveals Distinct Subdomains for Binding and Transfer of Cholesterol. Cell 2009, 137, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lu, F.; Trinh, M.N.; Schmiege, P.; Seemann, J.; Wang, J.; Blobel, G. 3.3 Å Structure of Niemann–Pick C1 Protein Reveals Insights into the Function of the C-Terminal Luminal Domain in Cholesterol Transport. Proc. Natl. Acad. Sci. USA 2017, 114, 9116–9121. [Google Scholar] [CrossRef] [PubMed]
- Pfisterer, S.G.; Peränen, J.; Ikonen, E. LDL-cholesterol Transport to the Endoplasmic Reticulum: Current Concepts. Curr. Opin. Lipidol. 2016, 27. [Google Scholar] [CrossRef]
- Steck, T.L.; Lange, Y. How Slow Is the Transbilayer Diffusion (Flip-Flop) of Cholesterol? Biophys. J. 2012, 102, 945–946. [Google Scholar] [CrossRef] [PubMed]
- Abi-Mosleh, L.; Infante, R.E.; Radhakrishnan, A.; Goldstein, J.L.; Brown, M.S. Cyclodextrin Overcomes Deficient Lysosome-to-Endoplasmic Reticulum Transport of Cholesterol in Niemann-Pick Type C Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 19316–19321. [Google Scholar] [CrossRef] [PubMed]
- Frolov, A.; Zielinski, S.E.; Crowley, J.R.; Dudley-Rucker, N.; Schaffer, J.E.; Ory, D.S. NPC1 and NPC2 Regulate Cellular Cholesterol Homeostasis through Generation of Low Density Lipoprotein Cholesterol-Derived Oxysterols. J. Biol. Chem. 2003, 278, 25517–25525. [Google Scholar] [CrossRef]
- Tharkeshwar, A.K.; Trekker, J.; Vermeire, W.; Pauwels, J.; Sannerud, R.; Priestman, D.A.; Te Vruchte, D.; Vints, K.; Baatsen, P.; Decuypere, J.-P.; et al. A Novel Approach to Analyze Lysosomal Dysfunctions through Subcellular Proteomics and Lipidomics: The Case of NPC1 Deficiency. Sci. Rep. 2017, 7, 41408. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Zhong, X.Z.; Zou, Y.; Zhang, Z.; Toro, L.; Dong, X.-P. BK Channels Alleviate Lysosomal Storage Diseases by Providing Positive Feedback Regulation of Lysosomal Ca2+ Release. Dev. Cell 2015, 33, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, X.; Gao, Q.; Lawas, M.; Yu, L.; Cheng, X.; Gu, M.; Sahoo, N.; Li, X.; Li, P.; et al. A Voltage-Dependent K+ Channel in the Lysosome Is Required for Refilling Lysosomal Ca2+ Stores. J. Cell Biol. 2017, 216, 1715–1730. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; O’Connell, R.J.; Feinberg-Zadek, P.L.; Johnston, L.J.; Treistman, S.N. Bilayer Thickness Modulates the Conductance of the BK Channel in Model Membranes. Biophys. J. 2004, 86, 3620–3633. [Google Scholar] [CrossRef] [PubMed]
- Lam, R.S.; Shaw, A.R.; Duszyk, M. Membrane Cholesterol Content Modulates Activation of BK Channels in Colonic Epithelia. Biochim. Biophys. Acta-Biomembr. 2004, 1667, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; O’Connell, R.J.; Jacob, R.F.; Mason, R.P.; Treistman, S.N. Regulation of the Gating of BKCa Channel by Lipid Bilayer Thickness. J. Biol. Chem. 2007, 282, 7276–7286. [Google Scholar] [CrossRef] [PubMed]
- Purcell, E.K.; Liu, L.; Thomas, P.V.; Duncan, R.K. Cholesterol Influences Voltage-Gated Calcium Channels and BK-Type Potassium Channels in Auditory Hair Cells. PLoS ONE 2011, 6, e26289. [Google Scholar] [CrossRef]
- Bukiya, A.N.; Belani, J.D.; Rychnovsky, S.; Dopico, A.M. Specificity of Cholesterol and Analogs to Modulate BK Channels Points to Direct Sterol–channel Protein Interactions. J. Gen. Physiol. 2010, 137, 93–110. [Google Scholar] [CrossRef]
- Yuan, C.; Chen, M.; Covey, D.F.; Johnston, L.J.; Treistman, S.N. Cholesterol Tuning of BK Ethanol Response Is Enantioselective, and Is a Function of Accompanying Lipids. PLoS ONE 2011, 6, e27572. [Google Scholar] [CrossRef]
- Pryor, P.R.; Reimann, F.; Gribble, F.M.; Luzio, J.P. Mucolipin-1 Is a Lysosomal Membrane Protein Required for Intracellular Lactosylceramide Traffic. Traffic 2006, 7, 1388–1398. [Google Scholar] [CrossRef]
- Singh, A.K.; McMillan, J.; Bukiya, A.N.; Burton, B.; Parrill, A.L.; Dopico, A.M. Multiple Cholesterol Recognition/Interaction Amino Acid Consensus (CRAC) Motifs in Cytosolic C Tail of Slo1 Subunit Determine Cholesterol Sensitivity of Ca2+- and Voltage-Gated K+ (BK) Channels. J. Biol. Chem. 2012, 287, 20509–20521. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Hite, R.K.; MacKinnon, R. Cryo-EM Structure of the Open High-Conductance Ca2+-Activated K+ Channel. Nature 2017, 541, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Hite, R.K.; Tao, X.; MacKinnon, R. Structural Basis for Gating the High-Conductance Ca2+-Activated K+ Channel. Nature 2017, 541, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Wang, X.; Li, X.; Zhang, X.; Yao, Z.; Dibble, S.; Dong, X.; Yu, T.; Lieberman, A.P.; Showalter, H.D.; et al. Lipid Storage Disorders Block Lysosomal Trafficking by Inhibiting a TRP Channel and Lysosomal Calcium Release. Nat. Commun. 2012, 3, 731. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, W.K.; Benvin, N.M.; Zhou, X.; Su, D.; Li, H.; Wang, S.; Michailidis, I.E.; Tong, L.; Li, X.; et al. Structural Basis of Dual Ca2+/PH Regulation of the Endolysosomal TRPML1 Channel. Nat. Struct. Mol. Biol. 2017, 24. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, K.; Leung, K.; Krishnan, Y. High Lumenal Chloride in the Lysosome Is Critical for Lysosome Function. Elife 2017, 6, e28862. [Google Scholar] [CrossRef] [PubMed]
- Lafourcade, C.; Sobo, K.; Kieffer-Jaquinod, S.; Garin, J.; van der Goot, F.G. Regulation of the V-ATPase along the Endocytic Pathway Occurs through Reversible Subunit Association and Membrane Localization. PLoS ONE 2008, 3, e2758. [Google Scholar] [CrossRef]
- Wheeler, S.; Haberkant, P.; Sullo, N.; Ferraz, M.J.; Halter, D.; Sprong, H.; Aerts, J.M.F.G.; Sillence, D.J. Cytoplasmic Glucosylceramide Modulates Endolysosomal Function in NPC Disease. Neurobiol. Dis. 2019. In press. [Google Scholar]
- Jiang, Z.; Redfern, R.E.; Isler, Y.; Ross, A.H.; Gericke, A. Cholesterol Stabilizes Fluid Phosphoinositide Domains. Chem. Phys. Lipids 2014, 182, 52–61. [Google Scholar] [CrossRef]
- Picas, L.; Gaits-Iacovoni, F.; Goud, B. The Emerging Role of Phosphoinositide Clustering in Intracellular Trafficking and Signal Transduction [Version 1; Referees: 4 Approved]. F1000Research 2016, 5. [Google Scholar] [CrossRef]
- Mayran, N.; Parton, R.G.; Gruenberg, J. Annexin II Regulates Multivesicular Endosome Biogenesis in the Degradation Pathway of Animal Cells. EMBO J. 2003, 22, 3242–3253. [Google Scholar] [CrossRef] [PubMed]
- te Vruchte, D.; Lloyd-Evans, E.; Veldman, R.J.; Neville, D.C.A.; Dwek, R.A.; Platt, F.M.; van Blitterswijk, W.J.; Sillence, D.J. Accumulation of Glycosphingolipids in Niemann-Pick C Disease Disrupts Endosomal Transport. J. Biol. Chem. 2004, 279, 26167–26175. [Google Scholar] [CrossRef] [PubMed]
- Sargiacomo, M.; Sudol, M.; Tang, Z.; Lisanti, M.P. Signal Transducing Molecules and Glycosyl-Phosphatidylinositol-Linked Proteins Form a Caveolin-Rich Insoluble Complex in MDCK Cells. J. Cell Biol. 1993, 122, 789–807. [Google Scholar] [CrossRef]
- Babiychuk, E.B.; Draeger, A. Annexins in Cell Membrane Dynamics. J. Cell Biol. 2000, 150, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Casey, L.; Pike, L.J. Compartmentalization of Phosphatidylinositol 4,5-Bisphosphate in Low-Density Membrane Domains in the Absence of Caveolin. Biochem. Biophys. Res. Commun. 1998, 245, 684–690. [Google Scholar] [CrossRef] [PubMed]
- Gokhale, N.A.; Abraham, A.; Digman, M.A.; Gratton, E.; Cho, W. Phosphoinositide Specificity of and Mechanism of Lipid Domain Formation by Annexin A2-P11 Heterotetramer. J. Biol. Chem. 2005, 280, 42831–42840. [Google Scholar] [CrossRef] [PubMed]
- Drücker, P.; Pejic, M.; Grill, D.; Galla, H.J.; Gerke, V. Cooperative Binding of Annexin A2 to Cholesterol- and Phosphatidylinositol-4,5-Bisphosphate-Containing Bilayers. Biophys. J. 2014, 107, 2070–2081. [Google Scholar] [CrossRef] [PubMed]
- Pipalia, N.H.; Hao, M.; Mukherjee, S.; Maxfield, F.R. Sterol, Protein and Lipid Trafficking in Chinese Hamster Ovary Cells with Niemann-Pick Type C1 Defect. Traffic 2007, 8, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.; Martinez-Lopez, N.; Otten, E.G.; Carroll, B.; Maetzel, D.; Singh, R.; Sarkar, S.; Korolchuk, V.I. Autophagy, Lipophagy and Lysosomal Lipid Storage Disorders. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2016, 1861, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Fraldi, A.; Annunziata, F.; Lombardi, A.; Kaiser, H.-J.; Medina, D.L.; Spampanato, C.; Fedele, A.O.; Polishchuk, R.; Sorrentino, N.C.; Simons, K.; et al. Lysosomal Fusion and SNARE Function Are Impaired by Cholesterol Accumulation in Lysosomal Storage Disorders. EMBO J. 2010, 29, 3607–3620. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.H.; Tamm, L.K. Molecular Mechanism of Cholesterol- and Polyphosphoinositide-Mediated Syntaxin Clustering. Biochemistry 2011, 50, 9014–9022. [Google Scholar] [CrossRef]
- Murray, D.H.; Tamm, L.K. Clustering of Syntaxin-1A in Model Membranes Is Modulated by Phosphatidylinositol 4,5-Bisphosphate and Cholesterol. Biochemistry 2009, 48, 4617–4625. [Google Scholar] [CrossRef] [PubMed]
- Khelashvili, G.; Galli, A.; Weinstein, H. Phosphatidylinositol 4,5-Biphosphate (PIP(2)) Lipids Regulate the Phosphorylation of Syntaxin N-Terminus by Modulating Both Its Position and Local Structure. Biochemistry 2012, 51, 7685–7698. [Google Scholar] [CrossRef]
- Singer-Lahat, D.; Barak-Broner, N.; Sheinin, A.; Greitzer-Antes, D.; Michaelevski, I.; Lotan, I. The Dual Function of the Polybasic Juxtamembrane Region of Syntaxin 1A in Clamping Spontaneous Release and Stimulating Ca2+-Triggered Release in Neuroendocrine Cells. J. Neurosci. 2018, 38, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Papadopoulos, V. Peripheral-Type Benzodiazepine Receptor Function in Cholesterol Transport. Identification of a Putative Cholesterol Recognition/Interaction Amino Acid Sequence and Consensus Pattern. Endocrinology 1998, 139, 4991–4997. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Barrantes, F.J. How Cholesterol Interacts with Membrane Proteins: An Exploration of Cholesterol-Binding Sites Including CRAC, CARC and Tilted Domains. Front. Physiol. 2013, 4. [Google Scholar] [CrossRef]
- Baier, C.J.; Fantini, J.; Barrantes, F.J. Disclosure of Cholesterol Recognition Motifs in Transmembrane Domains of the Human Nicotinic Acetylcholine Receptor. Sci. Rep. 2011, 1, 69. [Google Scholar] [CrossRef]
- Mahfoud, R.; Garmy, N.; Maresca, M.; Yahi, N.; Puigserver, A.; Fantini, J. Identification of a Common Sphingolipid-Binding Domain in Alzheimer, Prion, and HIV-1 Proteins. J. Biol. Chem. 2002, 277, 11292–11296. [Google Scholar] [CrossRef]
- Contreras, F.-X.; Ernst, A.M.; Haberkant, P.; Bjorkholm, P.; Lindahl, E.; Gonen, B.; Tischer, C.; Elofsson, A.; von Heijne, G.; Thiele, C.; et al. Molecular Recognition of a Single Sphingolipid Species by a Protein/’s Transmembrane Domain. Nature 2012, 481, 525–529. [Google Scholar] [CrossRef]
- Björkholm, P.; Ernst, A.M.; Hacke, M.; Wieland, F.; Brügger, B.; von Heijne, G. Identification of Novel Sphingolipid-Binding Motifs in Mammalian Membrane Proteins. Biochim. Biophys. Acta-Biomembr. 2014, 1838, 2066–2070. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM Database and PPM Web Server: Resources for Positioning of Proteins in Membranes. Nucleic Acids Res. 2012, 40, 370–376. [Google Scholar] [CrossRef]
- Khelashvili, G.; Pabst, G.; Harries, D. Cholesterol Orientation and Tilt Modulus in DMPC Bilayers. J. Phys. Chem. B 2010, 114, 7524–7534. [Google Scholar] [CrossRef]
- Millard, E.E.; Gale, S.E.; Dudley, N.; Zhang, J.; Schaffer, J.E.; Ory, D.S. The Sterol-Sensing Domain of the Niemann-Pick C1 (NPC1) Protein Regulates Trafficking of Low Density Lipoprotein Cholesterol. J. Biol. Chem. 2005, 280, 28581–28590. [Google Scholar] [CrossRef]
- Ohgami, N.; Ko, D.C.; Thomas, M.; Scott, M.P.; Chang, C.C.Y.; Chang, T.-Y. Binding between the Niemann–Pick C1 Protein and a Photoactivatable Cholesterol Analog Requires a Functional Sterol-Sensing Domain. Proc. Natl. Acad. Sci. USA 2004, 101, 12473–12478. [Google Scholar] [CrossRef]
- Castellano, B.M.; Thelen, A.M.; Moldavski, O.; Feltes, M.; van der Welle, R.E.N.; Mydock-McGrane, L.; Jiang, X.; van Eijkeren, R.J.; Davis, O.B.; Louie, S.M.; et al. Lysosomal Cholesterol Activates MTORC1 via an SLC38A9-Niemann-Pick C1 Signaling Complex. Science 2017, 355, 1306–1311. [Google Scholar] [CrossRef]
- Di Scala, C.; Fantini, J.; Yahi, N.; Barrantes, F.J.; Chahinian, H. Anandamide Revisited: How Cholesterol and Ceramides Control Receptor-Dependent and Receptor-Independent Signal Transmission Pathways of a Lipid Neurotransmitter. Biomolecules 2018, 8. [Google Scholar] [CrossRef]
- Fantini, J.; Di Scala, C.; Evans, L.S.; Williamson, P.T.F.; Barrantes, F.J. A Mirror Code for Protein-Cholesterol Interactions in the Two Leaflets of Biological Membranes. Sci. Rep. 2016, 6, 21907. [Google Scholar] [CrossRef]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-Pick Disease Type C1 Is a Sphingosine Storage Disease That Causes Deregulation of Lysosomal Calcium. Nat. Med. 2008, 14, 1247–1255. [Google Scholar] [CrossRef]
- Höglinger, D.; Nadler, A.; Haberkant, P.; Kirkpatrick, J.; Schifferer, M.; Stein, F.; Hauke, S.; Porter, F.D.; Schultz, C. Trifunctional Lipid Probes for Comprehensive Studies of Single Lipid Species in Living Cells. Proc. Natl. Acad. Sci. USA 2017, 114, 1566–1571. [Google Scholar] [CrossRef]
- Höglinger, D.; Haberkant, P.; Aguilera-Romero, A.; Riezman, H.; Porter, F.D.; Platt, F.M.; Galione, A.; Schultz, C. Intracellular Sphingosine Releases Calcium from Lysosomes. Elife 2015, 4, e10616. [Google Scholar] [CrossRef]
- Schonauer, S.; Körschen, H.G.; Penno, A.; Rennhack, A.; Breiden, B.; Sandhoff, K.; Gutbrod, K.; Dörmann, P.; Raju, D.N.; Haberkant, P.; et al. Identification of a Feedback Loop Involving Beta-Glucosidase 2 and Its Product Sphingosine Sheds Light on the Molecular Mechanisms in Gaucher Disease. J. Biol. Chem. 2017. [Google Scholar] [CrossRef]
- Ridley, C.M.; Thur, K.E.; Shanahan, J.; Thillaiappan, N.B.; Shen, A.; Uhl, K.; Walden, C.M.; Rahim, A.A.; Waddington, S.N.; Platt, F.M.; et al. Beta-Glucosidase 2 (GBA2) Activity and Imino Sugar Pharmacology. J. Biol. Chem. 2013. [Google Scholar] [CrossRef]
- Sasaki, H.; Arai, H.; Cocco, M.J.; White, S.H. PH Dependence of Sphingosine Aggregation. Biophys. J. 2009, 96, 2727–2733. [Google Scholar] [CrossRef]
- Hanson, M.A.; Cherezov, V.; Griffith, M.T.; Roth, C.B.; Jaakola, V.-P.; Chien, E.Y.T.; Velasquez, J.; Kuhn, P.; Stevens, R.C. A Specific Cholesterol Binding Site Is Established by the 2.8 Å Structure of the Human Β2-Adrenergic Receptor. Structure 2008, 16, 897–905. [Google Scholar] [CrossRef]
- Xu, M.; Liu, K.; Swaroop, M.; Porter, F.D.; Sidhu, R.; Finkes, S.; Ory, D.S.; Marugan, J.J.; Xiao, J.; Southall, N.; et al. D-Tocopherol Reduces Lipid Accumulation in Niemann-Pick Type C1 and Wolman Cholesterol Storage Disorders. J. Biol. Chem. 2012, 287, 39349–39360. [Google Scholar] [CrossRef]
- Garrity, A.G.; Wang, W.; Collier, C.M.D.; Levey, S.A.; Gao, Q.; Xu, H. The Endoplasmic Reticulum, Not the PH Gradient, Drives Calcium Refilling of Lysosomes. Elife 2016, 5, e15887. [Google Scholar] [CrossRef]
- Zhong, X.Z.; Sun, X.; Cao, Q.; Dong, G.; Schiffmann, R.; Dong, X.P. BK Channel Agonist Represents a Potential Therapeutic Approach for Lysosomal Storage Diseases. Sci. Rep. 2016, 6, 33684. [Google Scholar] [CrossRef]
- Schmiege, P.; Fine, M.; Blobel, G.; Li, X. Human TRPML1 Channel Structures in Open and Closed Conformations. Nature 2017, 550, 366. [Google Scholar] [CrossRef]
- Fine, M.; Schmiege, P.; Li, X. Structural Basis for PtdInsP2-Mediated Human TRPML1 Regulation. Nat. Commun. 2018, 9, 4192. [Google Scholar] [CrossRef]
- Hirschi, M.; Herzik, M.A., Jr.; Wie, J.; Suo, Y.; Borschel, W.F.; Ren, D.; Lander, G.C.; Lee, S.-Y. Cryo-Electron Microscopy Structure of the Lysosomal Calcium-Permeable Channel TRPML3. Nature 2017, 550, 411. [Google Scholar] [CrossRef]
- Venkatachalam, K.; Kiselyov, K. Chapter 25—TRPML1-Dependent Processes as Therapeutic Targets. In TRP Channels as Therapeutic Targets; Szallasi, A., Ed.; Academic Press: Boston, MA, USA, 2015; pp. 469–482. [Google Scholar]
- Vergarajauregui, S.; Connelly, P.S.; Daniels, M.P.; Puertollano, R. Autophagic Dysfunction in Mucolipidosis Type IV Patients. Hum. Mol. Genet. 2008, 17, 2723–2737. [Google Scholar] [CrossRef]
- Choudhury, A.; Dominguez, M.; Puri, V.; Sharma, D.K.; Narita, K.; Wheatley, C.L.; Marks, D.L.; Pagano, R.E. Rab Proteins Mediate Golgi Transport of Caveola-Internalized Glycosphingolipids and Correct Lipid Trafficking in Niemann-Pick C Cells. J. Clin. Investig. 2002, 109, 1541–1550. [Google Scholar] [CrossRef]
- Harvald, E.B.; Olsen, A.S.B.; Faergeman, N.J. Autophagy in the Light of Sphingolipid Metabolism. Apoptosis 2015, 20, 658–670. [Google Scholar] [CrossRef]
- Ruas, M.; Rietdorf, K.; Arredouani, A.; Davis, L.C.; Lloyd-Evans, E.; Koegel, H.; Funnell, T.M.; Morgan, A.J.; Ward, J.A.; Watanabe, K.; et al. Purified TPC Isoforms Form NAADP Receptors with Distinct Roles for Ca2+ Signaling and Endolysosomal Trafficking. Curr. Biol. 2010, 20, 703–709. [Google Scholar] [CrossRef]
- Lin, P.-H.; Duann, P.; Komazaki, S.; Park, K.H.; Li, H.; Sun, M.; Sermersheim, M.; Gumpper, K.; Parrington, J.; Galione, A.; et al. Lysosomal Two-Pore Channel Subtype 2 (TPC2) Regulates Skeletal Muscle Autophagic Signaling. J. Biol. Chem. 2015, 290, 3377–3389. [Google Scholar] [CrossRef]
- Cao, Q.; Zhong, X.Z.; Zou, Y.; Murrell-Lagnado, R.; Zhu, M.X.; Dong, X.-P. Calcium Release through P2X4 Activates Calmodulin to Promote Endolysosomal Membrane Fusion. J. Cell Biol. 2015, 209, 879–894. [Google Scholar] [CrossRef]
- Tian, X.; Gala, U.; Zhang, Y.; Shang, W.; Nagarkar Jaiswal, S.; di Ronza, A.; Jaiswal, M.; Yamamoto, S.; Sandoval, H.; Duraine, L.; et al. A Voltage-Gated Calcium Channel Regulates Lysosomal Fusion with Endosomes and Autophagosomes and Is Required for Neuronal Homeostasis. PLOS Biol. 2015, 13, e1002103. [Google Scholar] [CrossRef]
- Reddy, J.V.; Ganley, I.G.; Pfeffer, S.R. Clues to Neuro-Degeneration in Niemann-Pick Type C Disease from Global Gene Expression Profiling. PLoS ONE 2006, 1, e19. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Zhang, F.; Kemp, M.M.; Linhardt, R.J.; Waisman, D.M.; Head, J.F.; Seaton, B.A. Crystallographic Analysis of Calcium-Dependent Heparin Binding to Annexin A2. J. Biol. Chem. 2006, 281, 31689–31695. [Google Scholar] [CrossRef]
- Mullock, B.M.; Smith, C.W.; Ihrke, G.; Bright, N.A.; Lindsay, M.; Parkinson, E.J.; Brooks, D.A.; Parton, R.G.; James, D.E.; Luzio, J.P.; et al. Syntaxin 7 Is Localized to Late Endosome Compartments, Associates with Vamp 8, and Is Required for Late Endosome–Lysosome Fusion. Mol. Biol. Cell 2000, 11, 3137–3153. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.; Weber, G.; Wahl, M.C.; Jahn, R. Helical Extension of the Neuronal SNARE Complex into the Membrane. Nature 2009, 460, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The Hairpin-Type Tail-Anchored SNARE Syntaxin 17 Targets to Autophagosomes for Fusion with Endosomes/Lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Takáts, S.; Nagy, P.; Varga, Á.; Pircs, K.; Kárpáti, M.; Varga, K.; Kovács, A.L.; Hegedűs, K.; Juhász, G. Autophagosomal Syntaxin17-Dependent Lysosomal Degradation Maintains Neuronal Function in Drosophila. J. Cell Biol. 2013, 201, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Furuta, N.; Fujita, N.; Noda, T.; Yoshimori, T.; Amano, A. Combinational Soluble N-Ethylmaleimide-Sensitive Factor Attachment Protein Receptor Proteins VAMP8 and Vti1b Mediate Fusion of Antimicrobial and Canonical Autophagosomes with Lysosomes. Mol. Biol. Cell 2010, 21, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Rosenhouse-Dantsker, A.; Mehta, D.; Levitan, I. Regulation of Ion Channels by Membrane Lipids. In Comprehensive Physiology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011. [Google Scholar]
- Dong, X.; Shen, D.; Wang, X.; Dawson, T.; Li, X.; Zhang, Q.; Cheng, X.; Zhang, Y.; Weisman, L.S.; Delling, M.; et al. PI(3,5)P2 Controls Membrane Trafficking by Direct Activation of Mucolipin Ca2+ Release Channels in the Endolysosome. Nat. Commun. 2010, 1, 38. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, X.; Xu, H. Phosphoinositide Isoforms Determine Compartment-Specific Ion Channel Activity. Proc. Natl. Acad. Sci. USA 2012, 109, 11384–11389. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, X.; Dong, X.; Samie, M.; Li, X.; Cheng, X.; Goschka, A.; Shen, D.; Zhou, Y.; Harlow, J.; et al. TPC Proteins Are Phosphoinositide- Activated Sodium-Selective Ion Channels in Endosomes and Lysosomes. Cell 2012, 151, 372–383. [Google Scholar] [CrossRef]
- Domon, M.M.; Besson, F.; Bandorowicz-Pikula, J.; Pikula, S. Annexin A6 Is Recruited into Lipid Rafts of Niemann–Pick Type C Disease Fibroblasts in a Ca2+-Dependent Manner. Biochem. Biophys. Res. Commun. 2011, 405, 192–196. [Google Scholar] [CrossRef]
- Ganley, I.G.; Pfeffer, S.R. Cholesterol Accumulation Sequesters Rab9 and Disrupts Late Endosome Function in NPC1-Deficient Cells. J. Biol. Chem. 2006, 281, 17890–17899. [Google Scholar] [CrossRef]
- Kaptzan, T.; West, S.A.; Holicky, E.L.; Wheatley, C.L.; Marks, D.L.; Wang, T.; Peake, K.B.; Vance, J.; Walkley, S.U.; Pagano, R.E. Development of a Rab9 Transgenic Mouse and Its Ability to Increase the Lifespan of a Murine Model of Niemann-Pick Type C Disease. Am. J. Pathol. 2009, 174, 14–20. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A Multiple Sequence Alignment Editor and Analysis Workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL Workspace: A Web-Based Environment for Protein Structure Homology Modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the Estimation of the Absolute Quality of Individual Protein Structure Models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Studer, G.; Biasini, M.; Schwede, T. Assessing the Local Structural Quality of Transmembrane Protein Models Using Statistical Potentials (QMEANBrane). Bioinformatics 2014, 30, i505–i511. [Google Scholar] [CrossRef]
- Ionescu, C.-M.; Sehnal, D.; Falginella, F.L.; Pant, P.; Pravda, L.; Bouchal, T.; Svobodová Vařeková, R.; Geidl, S.; Koča, J. AtomicChargeCalculator: Interactive Web-Based Calculation of Atomic Charges in Large Biomolecular Complexes and Drug-like Molecules. J. Cheminform. 2015, 7, 50. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A Programming Language for Software Integration and Development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wheeler, S.; Schmid, R.; Sillence, D.J. Lipid–Protein Interactions in Niemann–Pick Type C Disease: Insights from Molecular Modeling. Int. J. Mol. Sci. 2019, 20, 717. https://doi.org/10.3390/ijms20030717
Wheeler S, Schmid R, Sillence DJ. Lipid–Protein Interactions in Niemann–Pick Type C Disease: Insights from Molecular Modeling. International Journal of Molecular Sciences. 2019; 20(3):717. https://doi.org/10.3390/ijms20030717
Chicago/Turabian StyleWheeler, Simon, Ralf Schmid, and Dan J Sillence. 2019. "Lipid–Protein Interactions in Niemann–Pick Type C Disease: Insights from Molecular Modeling" International Journal of Molecular Sciences 20, no. 3: 717. https://doi.org/10.3390/ijms20030717
APA StyleWheeler, S., Schmid, R., & Sillence, D. J. (2019). Lipid–Protein Interactions in Niemann–Pick Type C Disease: Insights from Molecular Modeling. International Journal of Molecular Sciences, 20(3), 717. https://doi.org/10.3390/ijms20030717