Current Coverage of the mTOR Pathway by Next-Generation Sequencing Oncology Panels


Abstract
:1. Introduction
1.1. mTOR Pathway
1.2. mTOR Signaling in Cancer
1.3. Next-Generation-Sequencing
2. Summary and Comparison of Oncological NGS Panels and Their Coverage of the mTOR Pathway
Oncological NGS Gene Panels
3. Discussion and Conclusions
Supplementary Materials
Conflicts of Interest
References
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Schalm, S.S.; Fingar, D.C.; Sabatini, D.M.; Blenis, J. TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr. Biol. 2003, 13, 797–806. [Google Scholar] [CrossRef]
- Nojima, H.; Tokunaga, C.; Eguchi, S.; Oshiro, N.; Hidayat, S.; Yoshino, K.; Hara, K.; Tanaka, N.; Avruch, J.; Yonezawa, K. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J. Biol. Chem. 2003, 278, 15461–15464. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell 2006, 11, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Harris, T.E.; Roth, R.A.; Lawrence, J.C., Jr. PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J. Biol. Chem. 2007, 282, 20036–20044. [Google Scholar] [CrossRef]
- Frias, M.A.; Thoreen, C.C.; Jaffe, J.D.; Schroder, W.; Sculley, T.; Carr, S.A.; Sabatini, D.M. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr. Biol. 2006, 16, 1865–1870. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.Y.; Kim, D.H.; Jun, C.B.; Kim, Y.M.; Haar, E.V.; Lee, S.I.; Hegg, J.W.; Bandhakavi, S.; Griffin, T.J. PRR5, a novel component of mTOR complex 2, regulates platelet-derived growth factor receptor beta expression and signaling. J. Biol. Chem. 2007, 282, 25604–25612. [Google Scholar] [CrossRef] [PubMed]
- Thedieck, K.; Polak, P.; Kim, M.L.; Molle, K.D.; Cohen, A.; Jeno, P.; Arrieumerlou, C.; Hall, M.N. PRAS40 and PRR5-like protein are new mTOR interactors that regulate apoptosis. PLoS ONE 2007, 2, e1217. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Elis, W.; Menon, S.; Qin, W.; Klekota, J.; Asara, J.M.; Finan, P.M.; Kwiatkowski, D.J.; Murphy, L.O.; Manning, B.D. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol. Cell 2012, 47, 535–546. [Google Scholar] [CrossRef]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef]
- Vander Haar, E.; Lee, S.I.; Bandhakavi, S.; Griffin, T.J.; Kim, D.H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.F.; Kuo, H.P.; Chen, C.T.; Hsu, J.M.; Chou, C.K.; Wei, Y.; Sun, H.L.; Li, L.Y.; Ping, B.; Huang, W.C.; et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 2007, 130, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef]
- Sancak, Y.; Bar–Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef]
- Bar-Peled, L.; Schweitzer, L.D.; Zoncu, R.; Sabatini, D.M. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 2012, 150, 1196–1208. [Google Scholar] [CrossRef]
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Blenis, J.; Cantley, L.C.; Toker, A.; et al. PtdIns(3,4,5)P3-Dependent Activation of the mTORC2 Kinase Complex. Cancer Discov. 2015, 5, 1194–1209. [Google Scholar] [CrossRef]
- Yang, G.; Murashige, D.S.; Humphrey, S.J.; James, D.E. A Positive Feedback Loop between Akt and mTORC2 via SIN1 Phosphorylation. Cell Rep. 2015, 12, 937–943. [Google Scholar] [CrossRef]
- Hsu, P.P.; Kang, S.A.; Rameseder, J.; Zhang, Y.; Ottina, K.A.; Lim, D.; Peterson, T.R.; Choi, Y.; Gray, N.S.; Yaffe, M.B.; et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 2011, 332, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef]
- Duvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef]
- Holz, M.K.; Ballif, B.A.; Gygi, S.P.; Blenis, J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 2005, 123, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Dorrello, N.V.; Peschiaroli, A.; Guardavaccaro, D.; Colburn, N.H.; Sherman, N.E.; Pagano, M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 2006, 314, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Brunn, G.J.; Hudson, C.C.; Sekulic, A.; Williams, J.M.; Hosoi, H.; Houghton, P.J.; Lawrence, J.C., Jr.; Abraham, R.T. Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science 1997, 277, 99–101. [Google Scholar] [CrossRef]
- Gingras, A.C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar] [CrossRef]
- Richter, J.D.; Sonenberg, N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 2005, 433, 477–480. [Google Scholar] [CrossRef]
- Spilka, R.; Ernst, C.; Mehta, A.K.; Haybaeck, J. Eukaryotic translation initiation factors in cancer development and progression. Cancer Lett. 2013, 340, 9–21. [Google Scholar] [CrossRef]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sahra, I.; Howell, J.J.; Asara, J.M.; Manning, B.D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013, 339, 1323–1328. [Google Scholar] [CrossRef] [PubMed]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Gan, X.; Wang, J.; Wang, C.; Sommer, E.; Kozasa, T.; Srinivasula, S.; Alessi, D.; Offermanns, S.; Simon, M.I.; Wu, D. PRR5L degradation promotes mTORC2-mediated PKC-delta phosphorylation and cell migration downstream of Galpha12. Nat. Cell Biol. 2012, 14, 686–696. [Google Scholar] [CrossRef]
- Li, X.; Gao, T. mTORC2 phosphorylates protein kinase Czeta to regulate its stability and activity. EMBO Rep. 2014, 15, 191–198. [Google Scholar]
- Thomanetz, V.; Angliker, N.; Cloetta, D.; Lustenberger, R.M.; Schweighauser, M.; Oliveri, F.; Suzuki, N.; Ruegg, M.A. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J. Cell Biol. 2013, 201, 293–308. [Google Scholar] [CrossRef]
- Garcia-Martinez, J.M.; Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J. 2008, 416, 375–385. [Google Scholar] [CrossRef]
- Fumarola, C.; Bonelli, M.A.; Petronini, P.G.; Alfieri, R.R. Targeting PI3K/AKT/mTOR pathway in non small cell lung cancer. Biochem. Pharmacol. 2014, 90, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Knight, S.D.; Adams, N.D.; Burgess, J.L.; Chaudhari, A.M.; Darcy, M.G.; Donatelli, C.A.; Luengo, J.I.; Newlander, K.A.; Parrish, C.A.; Ridgers, L.H.; et al. Discovery of GSK2126458, a Highly Potent Inhibitor of PI3K and the Mammalian Target of Rapamycin. ACS Med. Chem. Lett. 2010, 1, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Populo, H.; Lopes, J.M.; Soares, P. The mTOR signalling pathway in human cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef] [PubMed]
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR Pathways in Cancer and Autophagy. Cancers 2018, 10, 18. [Google Scholar] [CrossRef]
- Ilagan, E.; Manning, B.D. Emerging role of mTOR in the response to cancer therapeutics. Trends Cancer 2016, 2, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Iyer, G.; Hanrahan, A.J.; Milowsky, M.I.; Al-Ahmadie, H.; Scott, S.N.; Janakiraman, M.; Pirun, M.; Sander, C.; Socci, N.D.; Ostrovnaya, I.; et al. Genome sequencing identifies a basis for everolimus sensitivity. Science 2012, 338, 221. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Grabiner, B.C.; Van Allen, E.M.; Amin-Mansour, A.; Taylor-Weiner, A.; Rosenberg, M.; Gray, N.; Barletta, J.A.; Guo, Y.; Swanson, S.J.; et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N. Engl. J. Med. 2014, 371, 1426–1433. [Google Scholar] [CrossRef]
- Grabiner, B.C.; Nardi, V.; Birsoy, K.; Possemato, R.; Shen, K.; Sinha, S.; Jordan, A.; Beck, A.H.; Sabatini, D.M. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 2014, 4, 554–563. [Google Scholar] [CrossRef]
- Wagle, N.; Grabiner, B.C.; Van Allen, E.M.; Hodis, E.; Jacobus, S.; Supko, J.G.; Stewart, M.; Choueiri, T.K.; Gandhi, L.; Cleary, J.M.; et al. Activating mTOR mutations in a patient with an extraordinary response on a phase I trial of everolimus and pazopanib. Cancer Discov. 2014, 4, 546–553. [Google Scholar] [CrossRef]
- Bissler, J.J.; McCormack, F.X.; Young, L.R.; Elwing, J.M.; Chuck, G.; Leonard, J.M.; Schmithorst, V.J.; Laor, T.; Brody, A.S.; Bean, J.; et al. Sirolimus for Angiomyolipoma in Tuberous Sclerosis Complex or Lymphangioleiomyomatosis. N. Engl. J. Med. 2008, 358, 140–151. [Google Scholar] [CrossRef]
- Krueger, D.A.; Care, M.M.; Holland, K.; Agricola, K.; Tudor, C.; Mangeshkar, P.; Wilson, K.A.; Byars, A.; Sahmoud, T.; Franz, D.N. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N. Engl. J. Med. 2010, 363, 1801–1811. [Google Scholar] [CrossRef] [PubMed]
- Franz, D.N.; Leonard, J.; Tudor, C.; Chuck, G.; Care, M.; Sethuraman, G.; Dinopoulos, A.; Thomas, G.; Crone, K.R. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann. Neurol. 2006, 59, 490–498. [Google Scholar] [CrossRef]
- Davies, D.M.; de Vries, P.J.; Johnson, S.R.; McCartney, D.L.; Cox, J.A.; Serra, A.L.; Watson, P.C.; Howe, C.J.; Doyle, T.; Pointon, K.; et al. Sirolimus therapy for angiomyolipoma in tuberous sclerosis and sporadic lymphangioleiomyomatosis: A phase 2 trial. Clin. Cancer Res. 2011, 17, 4071–4081. [Google Scholar] [CrossRef] [PubMed]
- Klümpen, H.J.; Queiroz, K.C.; Spek, C.A.; van Noesel, C.J.; Brink, H.C.; de Leng, W.W.; de Wilde, R.F.; Mathus-Vliegen, E.M.; Offerhaus, G.J.A.; Alleman, M.A.; et al. mTOR Inhibitor Treatment of Pancreatic Cancer in a Patient With Peutz-Jeghers Syndrome. J. Clin. Oncol. 2011, 29, e150–e153. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.J.; Malinowska-Kolodziej, I.; Morgan, J.A.; Qin, W.; Fletcher, C.D.; Vena, N.; Ligon, A.H.; Antonescu, C.R.; Ramaiya, N.H.; Demetri, G.D.; et al. Clinical Activity of mTOR Inhibition With Sirolimus in Malignant Perivascular Epithelioid Cell Tumors: Targeting the Pathogenic Activation of mTORC1 in Tumors. J. Clin. Oncol. 2010, 28, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Zheng, X.S. Toward rapamycin analog (rapalog)-based precision cancer therapy. Acta Pharmacol. Sin. 2015, 36, 1163–1169. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One Drug, Many Effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef]
- Kelsey, I.; Manning, B.D. mTORC1 status dictates tumor response to targeted therapeutics. Sci. Signal. 2013, 6, pe31. [Google Scholar] [CrossRef]
- White, E.; DiPaola, R.S. The double–edged sword of autophagy modulation in cancer. Clin. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain–terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef]
- Muzzey, D.; Evans, E.A.; Lieber, C. Understanding the Basics of NGS: From Mechanism to Variant Calling. Curr. Genet. Med. Rep. 2015, 3, 158–165. [Google Scholar] [CrossRef]
- Ku, C.S.; Roukos, D.H. From next–generation sequencing to nanopore sequencing technology: paving the way to personalized genomic medicine. Exp. Rev. Med. Devices 2013, 10, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lahens, N.F.; Ricciotti, E.; Smirnova, O.; Toorens, E.; Kim, E.J.; Baruzzo, G.; Hayer, K.E.; Ganguly, T.; Schug, J.; Grant, G.R. A comparison of Illumina and Ion Torrent sequencing platforms in the context of differential gene expression. BMC Genom. 2017, 18, 602. [Google Scholar] [CrossRef]
- Kamps, R.; Brandao, R.D.; Bosch, B.J.; Paulussen, A.D.; Xanthoulea, S.; Blok, M.J.; Romano, A. Next–Generation Sequencing in Oncology: Genetic Diagnosis, Risk Prediction and Cancer Classification. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- mTOR Pathway. Available online: http://www.addgene.org/cancer/mtor–pathway/#gene–list (accessed on 20 December 2018).
- Del Vecchio, F.; Mastroiaco, V.; Di Marco, A.; Compagnoni, C.; Capece, D.; Zazzeroni, F.; Capalbo, C.; Alesse, E.; Tessitore, A. Next–generation sequencing: Recent applications to the analysis of colorectal cancer. J. Transl. Med. 2017, 15, 246. [Google Scholar] [CrossRef] [PubMed]
- Landolt, L.; Marti, H.P.; Beisland, C.; Flatberg, A.; Eikrem, O.S. RNA extraction for RNA sequencing of archival renal tissues. Scand. J. Clin. Lab. Investig. 2016, 76, 426–434. [Google Scholar] [CrossRef] [PubMed]
- AmpliSeq for Illumina Focus Panel Data Sheet. Available online: https://science–docs.illumina.com/documents/LibraryPrep/ampliseq–focus–panel–data–sheet–770–2017–027/Content/Source/Library–Prep/AmpliSeq/focus–panel/ampliseq–focus–panel–data–sheet.htm (accessed on 20 December 2018).
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: somatic cancer genetics at high–resolution. Nucleic Acids Res 2017, 45, D777–D783. [Google Scholar] [CrossRef]
- COSMIC—Catalogue of Somatic Mutations in Cancer. Available online: https://cancer.sanger.ac.uk/cosmic (accessed on 24 January 2019).
- Hyman, D.M.; Smyth, L.M.; Donoghue, M.T.A.; Westin, S.N.; Bedard, P.L.; Dean, E.J.; Bando, H.; El–Khoueiry, A.B.; Perez–Fidalgo, J.A.; Mita, A.; et al. AKT Inhibition in Solid Tumors with AKT1 Mutations. J. Clin. Oncol. 2017, 35, 2251–2259. [Google Scholar] [CrossRef]
- Comtesse, N.; Keller, A.; Diesinger, I.; Bauer, C.; Kayser, K.; Huwer, H.; Lenhof, H.P.; Meese, E. Frequent overexpression of the genes FXR1, CLAPM1 and EFI4G located on amplicon 3q26-27 in squamous cell carcinoma of the lung. Int. J. Cancer 2007, 120, 2538–2544. [Google Scholar] [CrossRef]
- Cheng, F.; Zhao, J.; Hanker, A.B.; Brewer, M.R.; Arteaga, C.L.; Zhao, Z. Transcriptome- and proteome-oriented identification of dysregulated eIF4G, STAT3 and Hippo pathways altered by PIK3CA H1047R in HER2/ER-positive breast cancer. Breast Cancer Res. Treat. 2016, 160, 457–474. [Google Scholar] [CrossRef]
- Sato, T.; Nakashima, A.; Guo, L.; Coffman, K.; Tamanoi, F. Single amino-acid changes that confer constitutive activation of mTOR are discovered in human cancer. Oncogene 2010, 29, 2746–2752. [Google Scholar] [CrossRef] [PubMed]
- Philpott, C.; Tovell, H.; Frayling, I.M.; Cooper, D.N.; Upadhyaya, M. The NF1 somatic mutational landscape in sporadic human cancers. Hum. Genom. 2017, 11, 13. [Google Scholar] [CrossRef] [PubMed]
- Kiuru, M.; Busam, K.J. The NF1 gene in tumor syndromes and melanoma. Lab. Investig. 2017, 97, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Sawa, K.; Koh, Y.; Kawaguchi, T.; Kambayashi, S.; Asai, K.; Mitsuoka, S.; Kimura, T.; Yoshimura, N.; Yoshimoto, N.; Kubo, A.; et al. PIK3CA mutation as a distinctive genetic feature of non-small cell lung cancer with chronic obstructive pulmonary disease: A comprehensive mutational analysis from a multi-institutional cohort. Lung Cancer 2017, 112, 96–101. [Google Scholar] [CrossRef]
- Mei, Z.B.; Duan, C.Y.; Li, C.B.; Cui, L.; Ogino, S. Prognostic role of tumor PIK3CA mutation in colorectal cancer: A systematic review and meta-analysis. Ann. Oncol. 2016, 27, 1836–1848. [Google Scholar] [CrossRef]
- Dirican, E.; Akkiprik, M.; Ozer, A. Mutation distribution and clinical correlations of PIK3CA gene mutations in breast cancer. Tumour Biol. 2016, 37, 7033–7045. [Google Scholar] [CrossRef]
- Lim, S.M.; Park, H.S.; Kim, S.; Ali, S.M.; Greenbowe, J.R.; Yang, I.S.; Kwon, N.J.; Lee, J.L.; Ryu, M.H.; Ahn, J.H.; et al. Next-generation sequencing reveals somatic mutations that conver exceptional response to everolimus. Oncotarget 2016, 7, 10547–10556. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Morrison, C.; Wang, L.; Xiong, D.; Vedell, P.; Cui, P.; Hua, X.; Ding, F.; Lu, Y.; James, M.; et al. Identification of somatic mutations in non-small cell lung carcinomas using whole-exome sequencing. Carcinogenesis 2012, 33, 1270–1276. [Google Scholar] [CrossRef]
- Cheung, L.W.; Mills, G.B. Targeting therapeutic liabilities engendered by PIK3R1 mutations for cancer treatment. Pharmacogenomics 2016, 17, 297–307. [Google Scholar] [PubMed]
- Isakov, N. Protein Kinase C (PKC) isoforms in cancer, tumor promotion and tumor suppression. Semin. Cancer Biol. 2018, 48, 36–52. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.N.; Briggs, J.M. Structural mutation analysis of PTEN and its genotype-phenotype correlations in endometriosis and cancer. Proteins 2016, 84, 1625–1643. [Google Scholar] [PubMed]
- Malaney, P.; Uversky, V.N.; Dave, V. PTEN proteoforms in biology and disease. Cell. Mol. Life Sci. 2017, 74, 2783–2794. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Rashid, A.; Churi, C.; Kar, S.; Zuo, M.; Eterovic, A.K.; Nogueras–Gonzalez, G.M.; Janku, F.; Shroff, R.T.; Aloia, T.A.; et al. Molecular characterization of gallbladder cancer using somatic mutation profiling. Hum. Pathol. 2014, 45, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Kamp, W.M.; Wang, P.Y.; Hwang, P.M. TP53 mutation, mitochondria and cancer. Curr. Opin. Genet. Dev. 2016, 38, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Te Raa, G.D.; Kater, A.P. TP53 dysfunction in CLL: Implications for prognosis and treatment. Best Pract. Res. Clin. Haematol. 2016, 29, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.W.H.; Chan, L.K.; Chiu, Y.T.; Xu, I.M.J.; Poon, R.T.P.; Cheung, T.T.; Tang, C.N.; Tang, V.W.L.; Lo, I.L.O.; Lam, P.W.Y.; et al. TSC1/2 mutations define a molecular subset of HCC with aggressive behaviour and treatment implication. Gut 2017, 66, 1496–1506. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Dai, J.; Xu, T.; Yu, S.; Yu, H.; Tang, H.; Yan, J.; Wu, X.; Yu, J.; Chi, Z.; et al. Analysis of TSC1 mutation spectrum in mucosal melanoma. J. Cancer Res. Clin. Oncol. 2018, 144, 257–267. [Google Scholar] [CrossRef]
- Martin, K.R.; Zhou, W.; Bowman, M.J.; Shih, J.; Au, K.S.; Dittenhafer-Reed, K.E.; Sisson, K.A.; Koeman, J.; Weisenberger, D.J.; Cottingham, S.L.; et al. The genomic landscape of tuberous sclerosis complex. Nat. Commun. 2017, 8, 15816. [Google Scholar] [CrossRef]
- Kim, W.Y.; Kaelin, W.G. Role of VHL gene mutation in human cancer. J. Clin. Oncol. 2004, 22, 4991–5004. [Google Scholar] [CrossRef]
- Xu, J.; Pham, C.G.; Albanese, S.K.; Dong, Y.; Oyama, T.; Lee, C.H.; Rodrik–Outmezguine, V.; Yao, Z.; Han, S.; Chen, D.; et al. Mechanistically distinct cancer-associated mTOR activation clusters predict sensitivity to rapamycin. J. Clin. Investig. 2016, 126, 3526–3540. [Google Scholar] [CrossRef]
- Golob–Schwarzl, N.; Schweiger, C.; Koller, C.; Krassnig, S.; Gogg–Kamerer, M.; Gantenbein, N.; Toeglhofer, A.M.; Wodlej, C.; Bergler, H.; Pertschy, B.; et al. Separation of low and high grade colon and rectum carcinoma by eukaryotic translation initiation factors 1, 5 and 6. Oncotarget 2017, 8, 101224–101243. [Google Scholar] [CrossRef] [PubMed]
- Spilka, R.; Laimer, K.; Bachmann, F.; Spizzo, G.; Vogetseder, A.; Wieser, M.; Muller, H.; Haybaeck, J.; Obrist, P. Overexpression of eIF3a in Squamous Cell Carcinoma of the Oral Cavity and Its Putative Relation to Chemotherapy Response. J. Oncol. 2012, 2012, 901956. [Google Scholar] [CrossRef] [PubMed]
- Spilka, R.; Ernst, C.; Bergler, H.; Rainer, J.; Flechsig, S.; Vogetseder, A.; Lederer, E.; Benesch, M.; Brunner, A.; Geley, S.; et al. eIF3a is over–expressed in urinary bladder cancer and influences its phenotype independent of translation initiation. Cell. Oncol. (Dordrecht) 2014, 37, 253–267. [Google Scholar] [CrossRef] [PubMed]
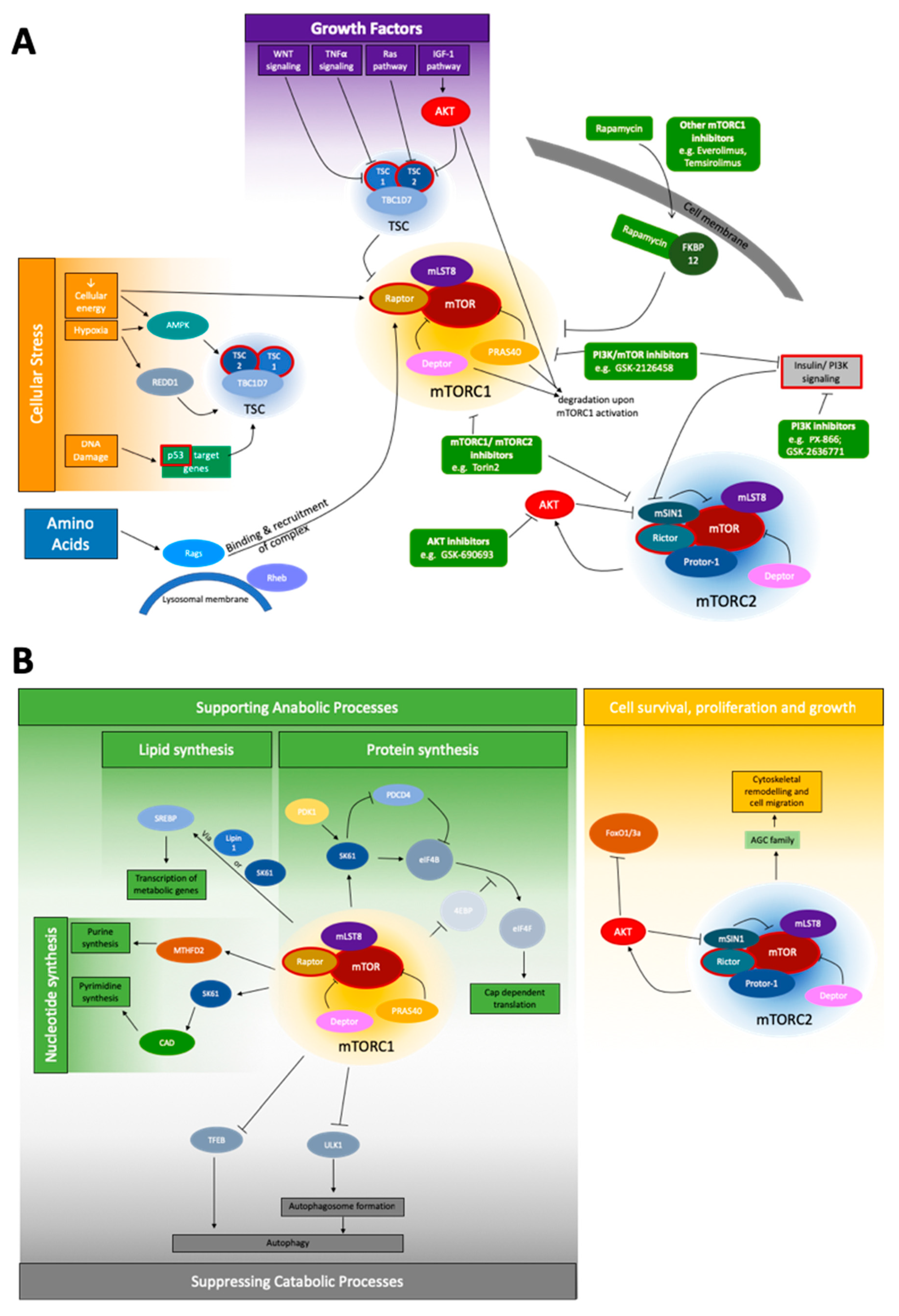

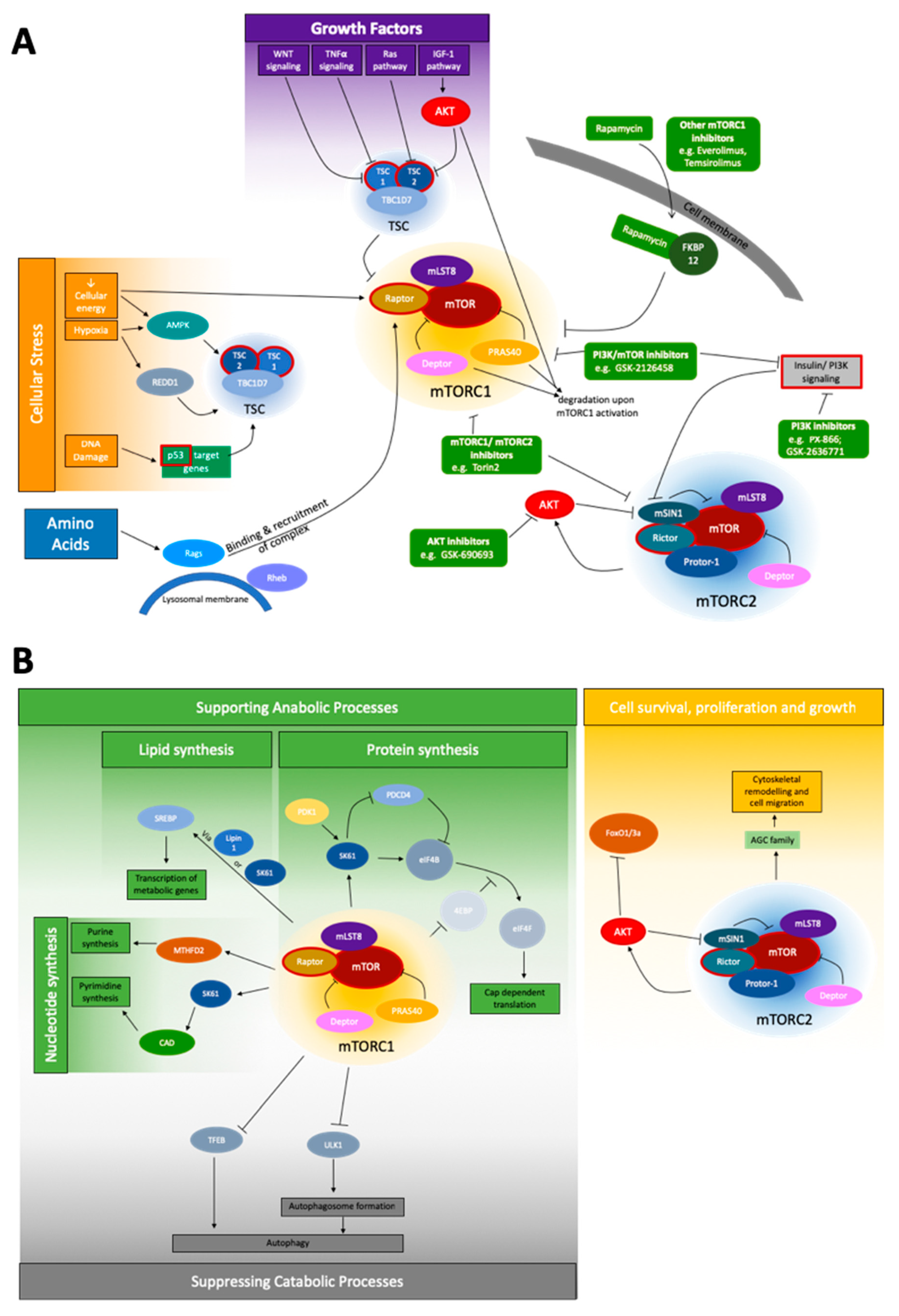{kind=link}
| Abbreviation | Full Name | Function | ↑↓ |
|---|---|---|---|
| mTORC 1 | Stimulating/Inhibiting Signal | ||
| mTOR | mechanistic target of rapamycin | Serine-threonine kinase | - |
| Raptor | regulatory-associated protein of mTOR | Localization of mTOR, substrate recruitment to mTOR [1,3,4] | ↑ |
| mLST8 | mammalian lethal with Sec13 protein 8 | Stabilizing kinase loop [5]; not essential to TORC1 function [6] | - |
| PRAS40 | proline rich AKT substrate 40 kDa | Inhibitory [7]; inhibits substrate binding, phosphorylated by active mTORC1 [9] | ↓ |
| Deptor | DEP-domain-containing mTOR-interacting protein | Inhibitory [8], phosphorylated by active mTORC1 | ↓ |
| mTORC 2 | Stimulating/Inhibiting Signal | ||
| mTOR | mechanistic target of rapamycin | serine-threonine kinase of the phosphoinositide 3-kinase (PI3K)-related family | |
| mLST8 | mammalian lethal with Sec13 protein 8 | Essential for stability and function of mTORC2 [6] | ↑ |
| Deptor | DEP-domain-containing mTOR-interacting protein | Inhibitory [8] | ↓ |
| Rictor | rapamycin-insensitive companion of mTOR | Stabilization [10,11]; shown to interact with Protor-1 [12,13] | ↑ |
| mSIN1 | mammalian stress-activated protein kinase interacting protein | Stabilization [10,11], phosphoinositide-binding PH domain: critical for insulin dependent mTORC2 function, inhibits mTORC2 function in absence of insulin [1] | ↑/↓ |
| Protor-1 | protein observed with Rictor-1 | shown to interact with Rictor [12,13] | |
| mTORC 1 Upstream | Stimulating/Inhibiting Signal | ||
| - | rapamycin | Enters cell and binds FKBP12 [2]; when bound inhibits mTORC 1, but not all functions [15] | ↓ |
| FKBP12 | FK506-binding protein 12 kDa | Is bound by rapamycin, interacts with FBD on mTOR [2]; when bound inhibits mTORC 1, but not all functions [15]; cannot acutely inhibit mTORC2 [2] | - |
| TSC | tuberous sclerosis complex | Consists of TC1, TC2, TBC1D7, negatively regulates mTORC1 via inactivation of Rheb [17], phosphorylated by AKT (mTORC2 independent) [6] | ↓ |
| Rheb | Ras homolog enriched in brain | Stimulates mTOCR1 activity when active [7,16,18] | ↑ |
| IGF-1 pathway | insulin/insulin like growth factor 1 pathway | Causes AKT dependent phosphorylation of TSC2 [1,19] | ↑ |
| Ras pathway | Rat Sarcoma Pathway | Causes TSC2 phosphorylation via ERK and p90rsk [1,16,20] | ↑ |
| AKT | AKT serine/threonine kinase | Phosphorylates TSC2 [1,19]; key effector protein of the insulin/PI3K signaling pathway, and can be activated by mTORC2 [45]; promotes dissociation of PRAS40 from mTORC1. [7,9,21] | ↑ |
| - | Wnt | Inhibits TSC1 [22] | ↑ |
| TNFα | tumor necrosis factor α | Inhibits TSC1 [23] | ↑ |
| AMPK | 5’-AMP-activated protein kinase | Inhibits mTORC1 (in response to reduced cellular energy or hypoxia) by phosphorylating Raptor and activation of TSC2 [1,22,24]; activator of autophagy, activates ULK1 [44] | ↓ |
| REDD1 | regulated in development and DNA damage responses 1 | Activates TSC in response to hypoxia [24] | ↓ |
| - | p53 target genes | Increase TSC activity upon DNA damage [25] | ↓ |
| mTORC 2 Upstream | Stimulating/Inhibiting Signal | ||
| Rapamycin | Enters cell and binds FKBP12 [2]; | ↓ | |
| FKBP12 | FKBP prolyl isomerase | Is bound by rapamycin, interacts with FBD on mTOR cannot acutely inhibit mTORC2 [2]; chronic treatment can inhibit mTORC2 [14] | - |
| PIP3 | Phosphatidylinositol (3,4,5)-trisphosphate | PI3K generated PIP3 binds to PH domain o mSIN1 and relieves inhibition of mTORC2 [29] | ↑ |
| AKT | AKT serine/threonine kinase | Phosphorylates mSIN1, positive feedback loop [30] | ↑ |
| mTORC1 | mammalian target of rapamycin complex 1 | Negative feedback loop between mTORC1 and insulin/PI3K signaling [1,31] | ↓ |
| mTORC1 downstream | Stimulating/Inhibiting Signal | ||
| SREBP | sterol responsive element binding protein | Activated by low sterol levels, in control of expression of metabolic genes, can be activated by mTORC1 independently via S6K1 or Lipin1 [1,32]; expression by mTORC1 increases PPP [1] | ↑ |
| S6K1 | p70S6 Kinase 1 | Can activate SREBP [33,34], when phosphorylated by mTORC1 can be activated by PDK1, promotes mRNA translation intitation [35]; promotes degradation of PDCD4 [36]; phosphorylates CAD (catalyzes first steps in de-novo pyrimidine synthesis) [42] | ↑ |
| - | Lipin1 | Inhibits SREBP in absence of mTORC1, activates when mTORC1 is present [33,34] | ↑ |
| 4EBP | eukaryotic initiation factor 4E binding protein | Inhibits translation by binding eIF4E → prevents assembly of eIF4F complex; when phosphorylated by mTORC1 → dissociates from eIF4e → allows assembly [37,38,39] | ↑ |
| eIF4B | eukaryotic translation initiation factor 4B | Positive regulator of the 5’-cap binding eIF4F complex, activated by S6K1 [35], inhibitor: PDCD4 [36] | ↑ |
| eIF4F complex | eukaryotic translation initiation factor 4F | Positively regulated by eIF4B, 5’-cap binding complex, | ↑ |
| MTHFD2 | methylenetetrahydrofolate dehydrogenase | Controls the mitochondrial tetrahydrofolate cycle, expression increased bymTORC1 induction of purine synthesis [41] | ↑ |
| HIF1α | hypoxia inducible factor 1 | Translation increased by mTORC1 → expression of glycolytic enzymes [33] | ↑ |
| TFEB | Transcription factor EB | expression of genes for autophagy and lysosomal biogenesis, when phosphorylated by mTORC1 →cannot translocate to nucleus [43] | ↓ |
| ULK1 | unc-51 like autophagy activating kinase 1 | Drives autophagosome formation, when phosphorylated by mTORC1 → no interaction with AMPK → no activation [44] | ↓ |
| AMPK | AMP-activated protein kinase | Inhibits mTORC1 (in response to reduced cellular energy or hypoxia) by phosphorylating Raptor and activation of TSC2 [1,22]; activator of autophagy, activates ULK1 [44] | ↓ |
| mTORC2 downstream | Stimulating/Inhibiting Signal | ||
| AKT | AKT serine/threonine kinase | Phosphorylated by mTORC2 [1]; phosphorylates TSC2; key effector protein of the insulin/PI3K signaling pathway [45]; activation by mTORC2 not crucial for the phosphorylation of all, but some of its substrates [6,11] | - |
| FoxO1/3a | Forkhead box protein O1 | TFs, phosphorylated by AKT (mTORC2 dependent) [11] | - |
| GSK3ß | Glycogen synthase kinase 3β | Metabolic regulator, phosphorylated by AKT [6] | - |
| - | AGC (PKA/PKB/PKC) Family | Several members phosphorylated by mTORC2 for regulation of proliferation, survival and cytoskeleton [1,46,47,48,49] | - |
| Panel Name | Number of Genes Covered | mTOR Relevant Genes Covered |
|---|---|---|
| Foundation One | 305 | AKT1/2/3; CCND1; GSK3B; MDM2; MTOR; NF1; PDK1; PIK3C2; PIK3CA/B; PIK3R1; PTEN; RICTOR, RPTOR; SGK1; TNFAIP3; TP53; TSC1/2; VHL |
| Agilent ClearSeq Comprehensive Cancer Panel | 150 | AKT1/2/3; NF1; MTOR; PIK3R1; PIK3CA; PTEN; TP53; VHL |
| Qiagen Human Cancer Predisposition GeneRead DNAseq Targeted Panel V2 | 143 | AKT1; NF1; PIK3CA; PTEN; TP53; TSC1/2; VHL |
| Integrated DNA Technologies (IDT) xGen Pan-Cancer Panel | 127 | AKT1; CCND1; EIF4A2; MTOR; NF1; PIK3CA; PIK3CG; PIK3R1; PTEN; TP53; VHL |
| Archer VariantPlex Solid Tumor | 67 | AKT1; CCND1; MDM2; PIK3CA; PIK3R1; PTEN; TP53; VHL |
| Swift Biosciences Accel-Amplicon 56G Oncology Panel v2 | 56 | AKT1; PIK3CA; PTEN; TP53; TSC1; VHL |
| NEBNExt Direct Cancer HotSpot Panel | 50 | AKT1; PIK3CA; PTEN; TP53; VHL |
| AmpliSeq Cancer Hotspot Panel v2 | 49 | AKT1; PIK3CA; PTEN; TP53; VHL |
| TruSeq Amplicon Cancer Panel | 48 | AKT1; PIK3CA; PTEN; TP53; VHL |
| AmpliSeq for Illumina Focus Panel | 40 | AKT1; CCND1; MTOR; PIK3CA |
| Archer VariantPlex Comprehensive Thyroid and Lung Kit | 31 | AKT1; CCND1; MDM2; PIK3CA; PTEN; TP53 |
| TruSight Tumor 26 | 26 | AKT1; PIK3CA; PTEN; TP53 |
| Agilent SureMASTR Tumor Hotspot | 25 | AKT; PIK3CA; PTEN |
| Qiagen Human Clinically Relevant Tumor GeneRead DNAseq Targeted Panel V2 | 24 | AKT1; PIK3CA; PTEN; TP53 |
| Asuragen QuantideX NGS DNA Hotspot 21 Kit | 21 | AKT1/2; PIK3CA |
| TruSight Tumor 15 | 15 | AKT1; PIK3CA; TP53 |
| Qiagen Human Tumor Actionable Mutations GeneRead DNAseq Targeted Panel v2 | 8 | - |
| Gene | Frequency of Mutation in Cancer |
|---|---|
| 4E-BP | <0.1% |
| AKT1 | 1.1% |
| AKT2 | 0.4% |
| AKT3 | 0.5% |
| CCND1 | 0.3% |
| Deptor | 0.3% |
| eIF3a | 0.8% |
| eIF3b | 0.4% |
| eIF3c | <0.1% |
| eIF3d | 0.3% |
| eIF3e | 0.3% |
| eIF3f | 0.2% |
| eIF3g | 0.2% |
| eIF3h | 0.2% |
| eIF3i | 0.2% |
| eIF3j | 0.1% |
| eIF3k | 0.1% |
| eIF3l | 0.3% |
| eIF3m | 0.2% |
| eIF4a | 0.3% |
| eIF4b | 0.3% |
| eIF4E | 0.2% |
| eIF4g | 1.0% |
| eIF4h | 0.2% |
| FOXO | 0.4% |
| GBL/mLST8 | 0.2% |
| GSK3A | 0.2% |
| GSK3B | 0.4% |
| HIF1α | 0.5% |
| LKB1 | 0.5% |
| MDM2 | 0.4% |
| mSin1 = MAPKAP1 | 0.2% |
| MTHFD2 | 0.1% |
| mTOR | 2.1% |
| NF1 | 3.8% |
| PDK1 | 0.2% |
| PIK3CA | 9.7% |
| PIK3CB | 0.7% |
| PIK3CD | 0.7% |
| PIK3CG | 1.6% |
| PIK3R1 | 1.4% |
| PIK3R2 | 0.5% |
| PIK3R3 | 0.3% |
| PIK3R4 | 0.7% |
| PIK3R5 | 0.7% |
| PIK3R6 | 0.6% |
| PIP3 | 0.4% |
| PKC alpha | 0.4% |
| PKC beta | 1.0% |
| PKC delta | 0.4% |
| PKC epsilon | 0.5% |
| PKC eta | 0.5% |
| PKC gamma | 0.7% |
| PKC iota | 0.4% |
| PKC theta | 0.7% |
| PKC zeta | 0.4% |
| PRAS40 = AKT1S1 | 0.2% |
| Protor = PRR5 | 0.3% |
| PTEN | 5.0% |
| Raptor | 1.0% |
| REDD1 = DDIT4 | 0.1% |
| Rheb | 0.1% |
| Rictor | 1.0% |
| RRAGA | 0.1% |
| RRAGB | 0.2% |
| RRAGC | 0.2% |
| RRAGD | 0.2% |
| S6K | 0.2% |
| SGK | 0.4% |
| SREBP | 0.5% |
| TFEB | 0.3% |
| TNFα | 0.3% |
| TP53 | 25.2% |
| TSC1 | 1.2% |
| TSC2 | 1.7% |
| ULK1 | 0.7% |
| VHL | 4.5% |
| Wnt | 0.3% |
| Gene | Frequency of Mutation in Cancer | Most Common Genetic Mutations | Tissue | Reference |
|---|---|---|---|---|
| AKT1 | 1.1% | E17K, Q79K, L52R | breast, skin, urinary tract | [80,81] |
| eIF4g | 1.0% | T436fs * 86; K643R | colon, lung (overexpression w/o genetic mutation) | [80,82,83] |
| mTOR | 2.1% | S2215Y, S2215F, E1799K, T1977K, L1460P | colon, endometrium, skin, kidney | [80,84] |
| NF1 | 3.8% | R2450 *, R440 *, R1534 * | skin, soft tissue, urinary tract, lung, colon | [80,85,86] |
| PIK3CA | 9.7% | H1047R, E545K, E542K, H1047L, Q546K, R88Q, N345K, C420L | breast, endometrium, urinary tract, colon | [80,87,88,89] |
| PIK3CG | 1.6% | V759I, V165I, R472C, E267K, A84V | skin, colon, lung | [80,90,91] |
| PIK3R1 | 1.4% | N564D, R348 *, K567E, G376R | breast, endometrium, prostate, leukemia | [80,92] |
| PKC beta | 1.0% | D427N, D630N, E533K | lung, skin, colon | [80,93] |
| PTEN | 5.0% | R130G, R130Q, R233 *, R130 * | breast, endometrium, prostate, leukemia | [80,94,95] |
| Raptor | 1.0% | R718C, R139H, Q1264fs * 4, T1121M | various | [80] |
| Rictor | 1.0% | S1101L, R401C | lung, breast | [80,96] |
| TP53 | 25.2% | R175H, R248Q, R273H, R282W, R213 *, G245S, R249S, Y220C, R196 *, R342 * | solid cancer, leukemia, lymphoma, melanoma | [80,97,98] |
| TSC1 | 1.2% | M322T, P1143L | skin, urinary tract, liver | [80,99,100,101] |
| TSC2 | 1.7% | F690fs * 8, R1417fs * 59, S1364fs * 50, K1638 * | liver, breast | [80,101] |
| VHL | 4.5% | kidney, neuroendocrine tumors | R161 *, L89H, S65 * | [80,102,103] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seeboeck, R.; Sarne, V.; Haybaeck, J. Current Coverage of the mTOR Pathway by Next-Generation Sequencing Oncology Panels. Int. J. Mol. Sci. 2019, 20, 690. https://doi.org/10.3390/ijms20030690
Seeboeck R, Sarne V, Haybaeck J. Current Coverage of the mTOR Pathway by Next-Generation Sequencing Oncology Panels. International Journal of Molecular Sciences. 2019; 20(3):690. https://doi.org/10.3390/ijms20030690
Chicago/Turabian StyleSeeboeck, Rita, Victoria Sarne, and Johannes Haybaeck. 2019. "Current Coverage of the mTOR Pathway by Next-Generation Sequencing Oncology Panels" International Journal of Molecular Sciences 20, no. 3: 690. https://doi.org/10.3390/ijms20030690
APA StyleSeeboeck, R., Sarne, V., & Haybaeck, J. (2019). Current Coverage of the mTOR Pathway by Next-Generation Sequencing Oncology Panels. International Journal of Molecular Sciences, 20(3), 690. https://doi.org/10.3390/ijms20030690