Diabetes Impairs Angiogenesis and Induces Endothelial Cell Senescence by Up-Regulating Thrombospondin-CD47-Dependent Signaling

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
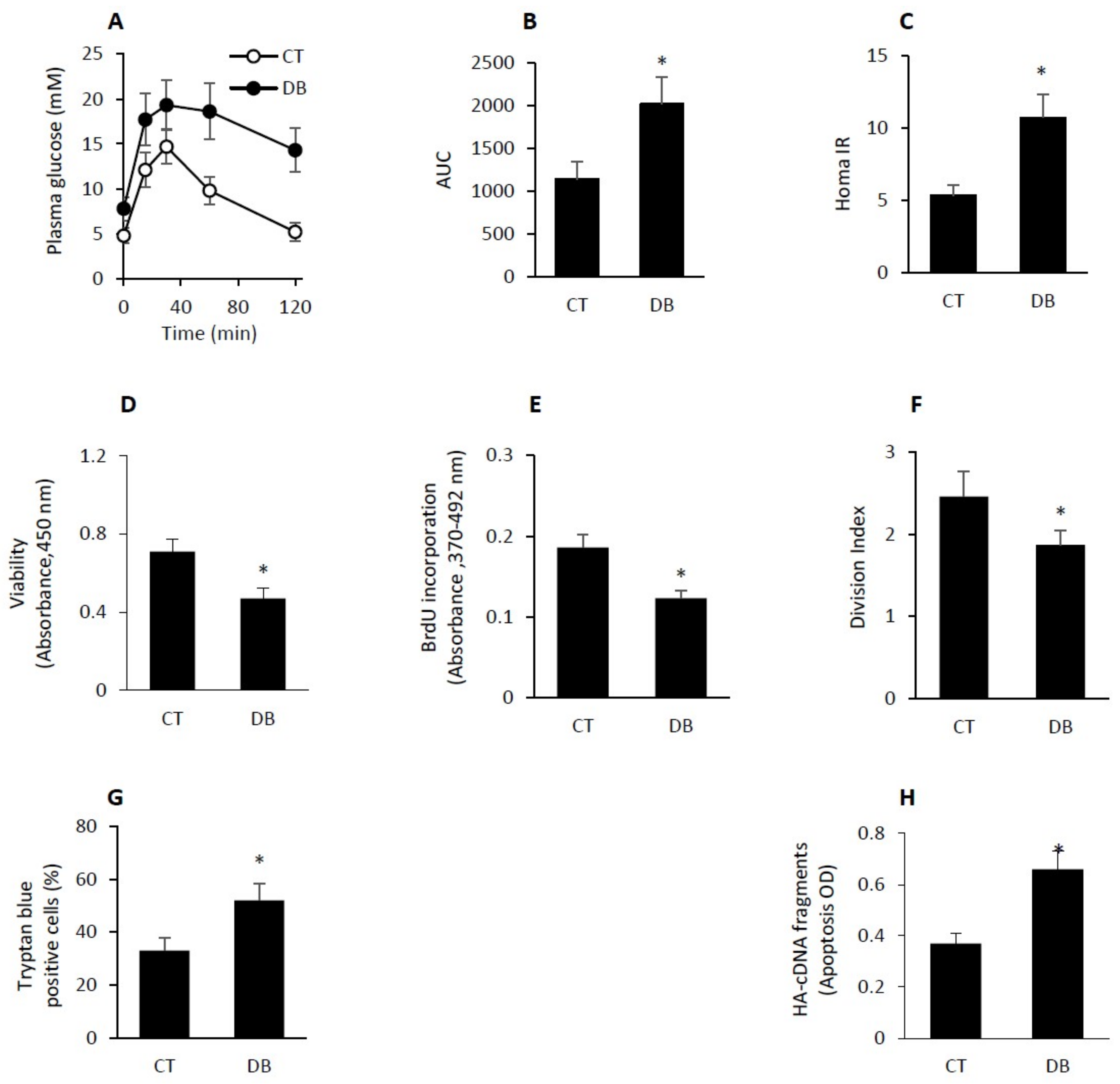
2.1. GK Diabetic W-ECs Showed Low Proliferation Rate, Decreased Survival and Increased Apoptosis
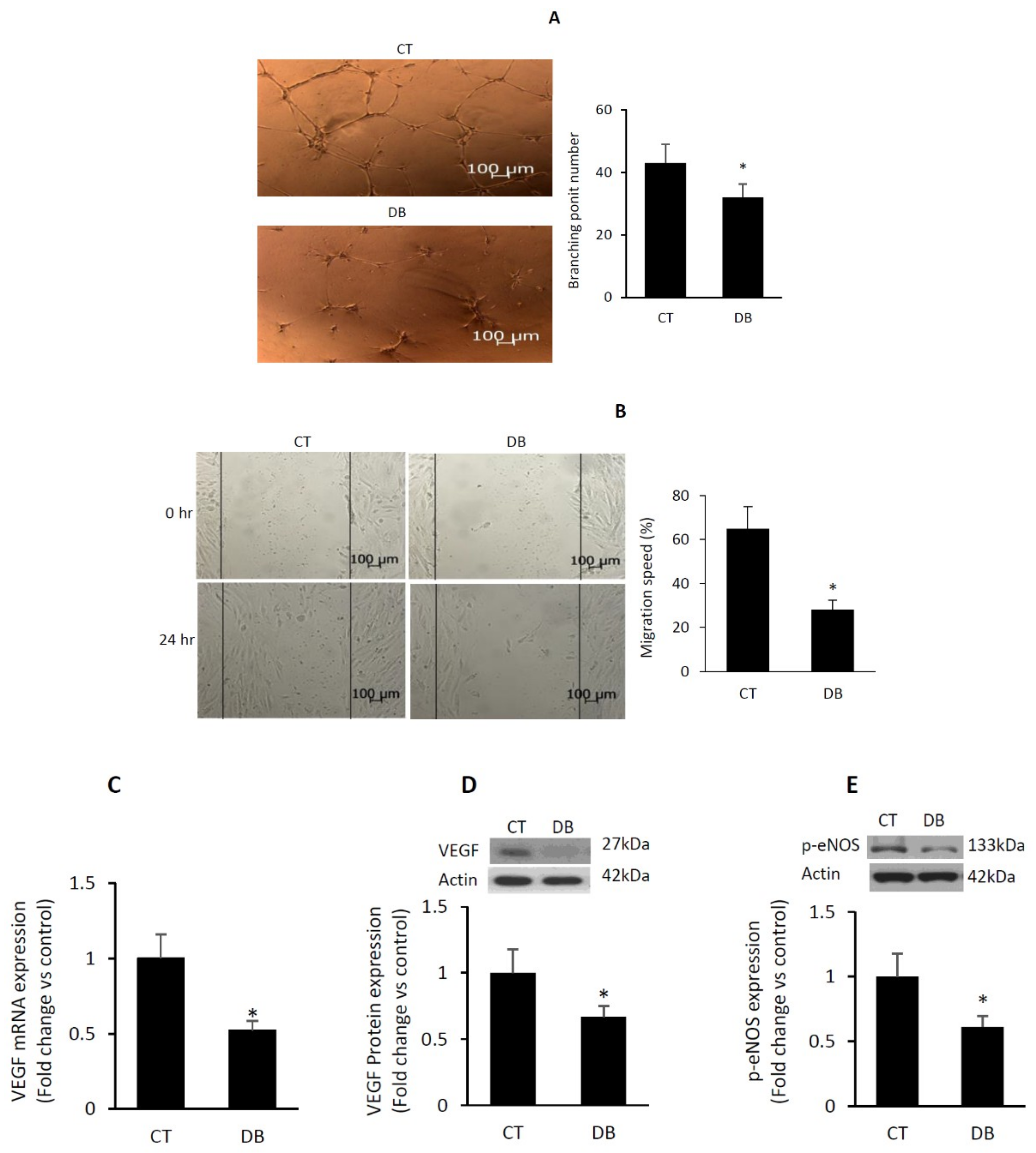
2.2. Diabetes Suppresses Angiogenic Capacity in W-ECs
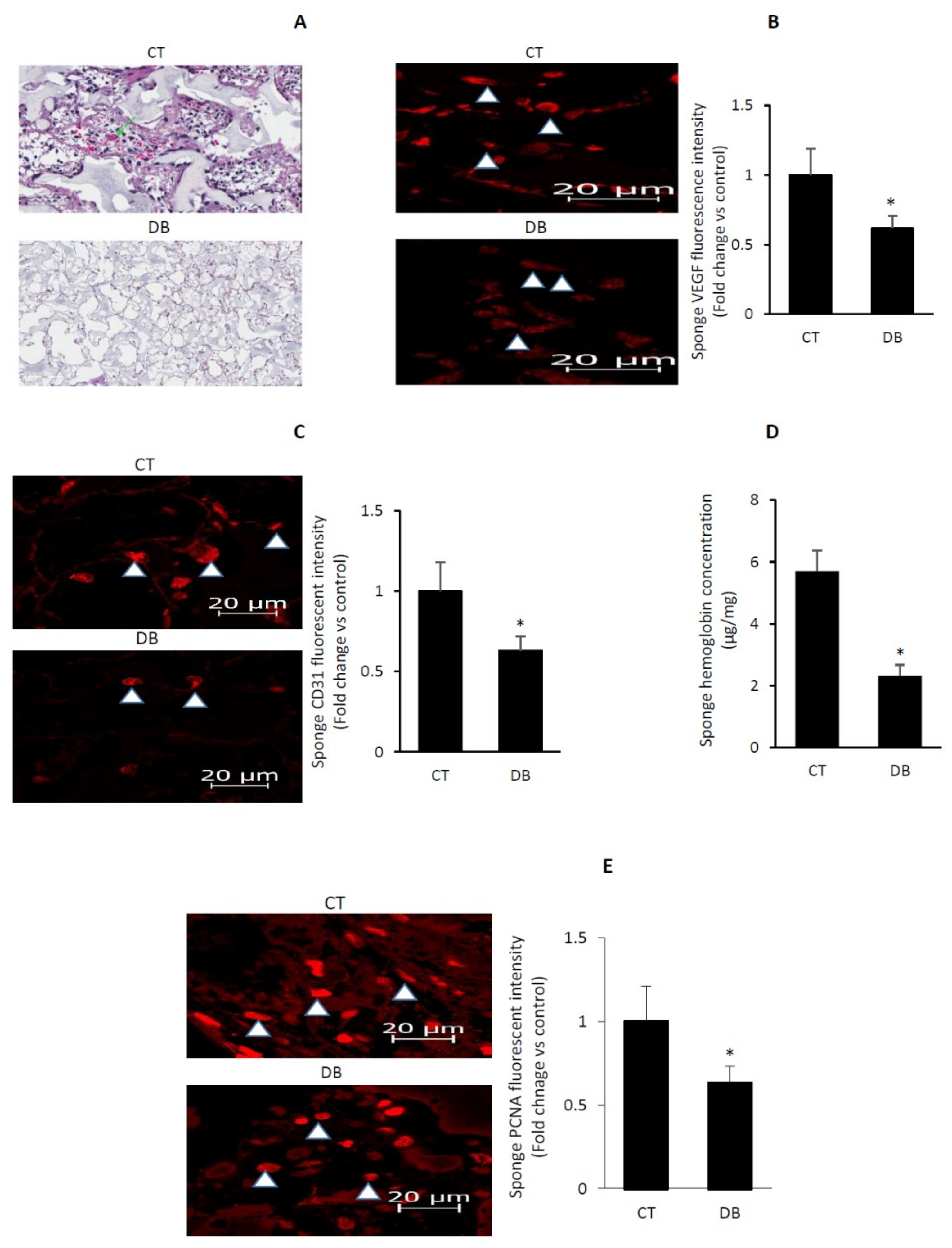
2.3. Diabetes Impairs Angiogenic Activity in PVA Sponge Model of Wound Healing
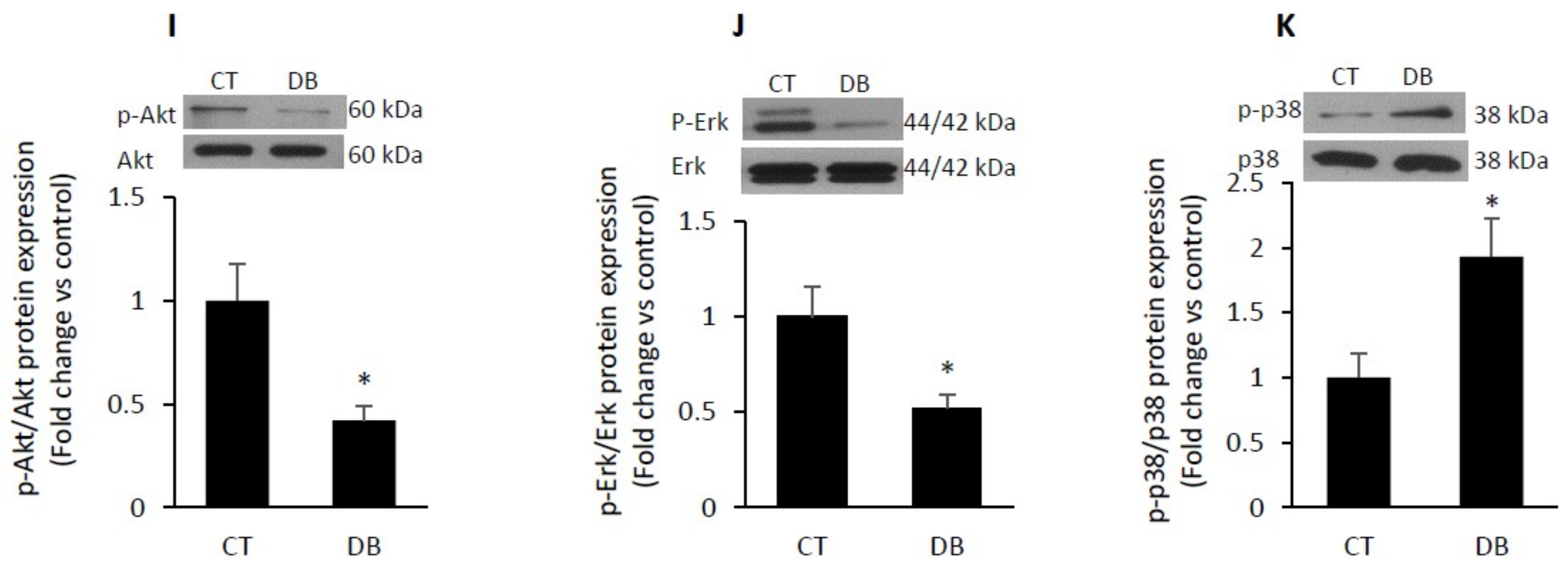
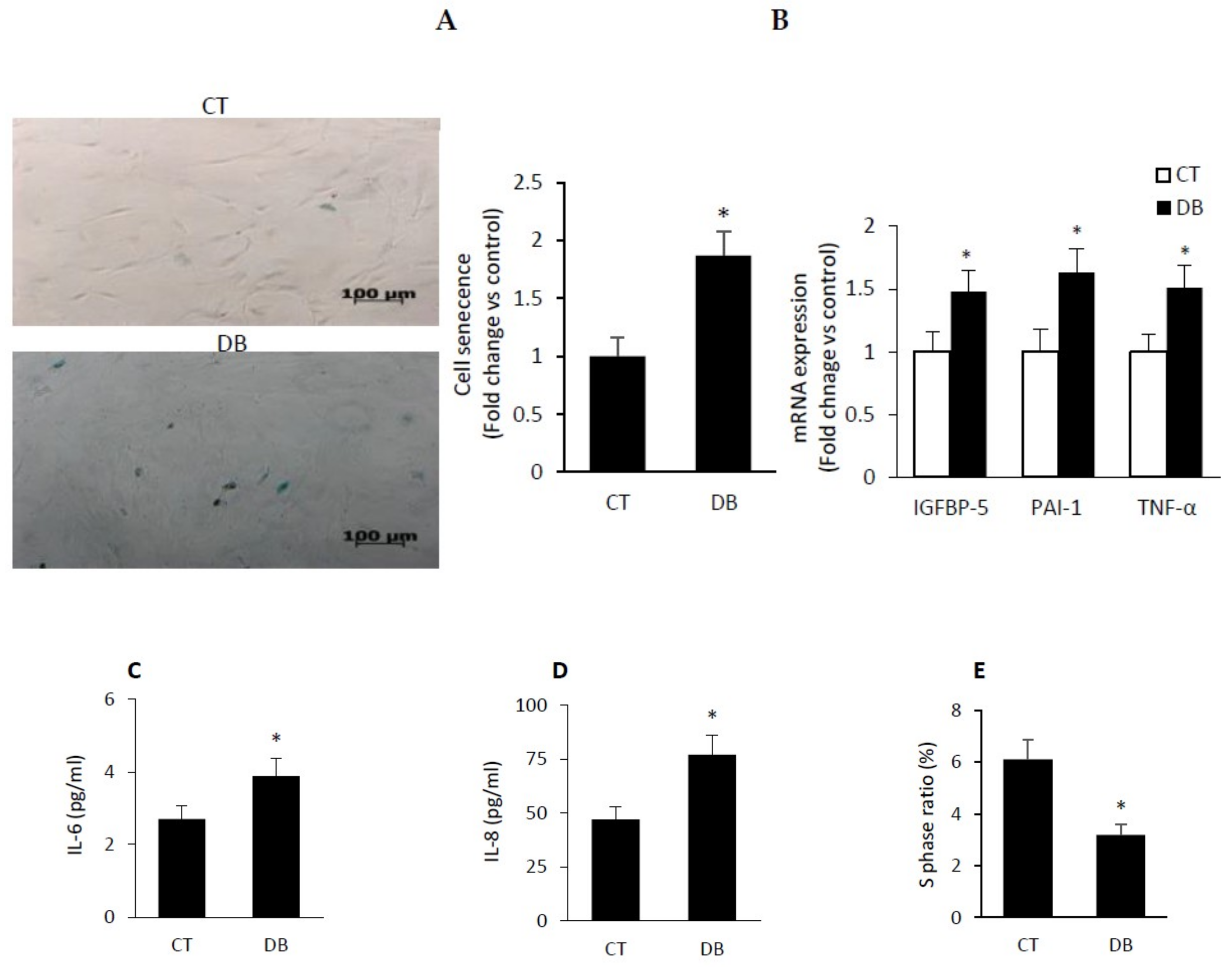
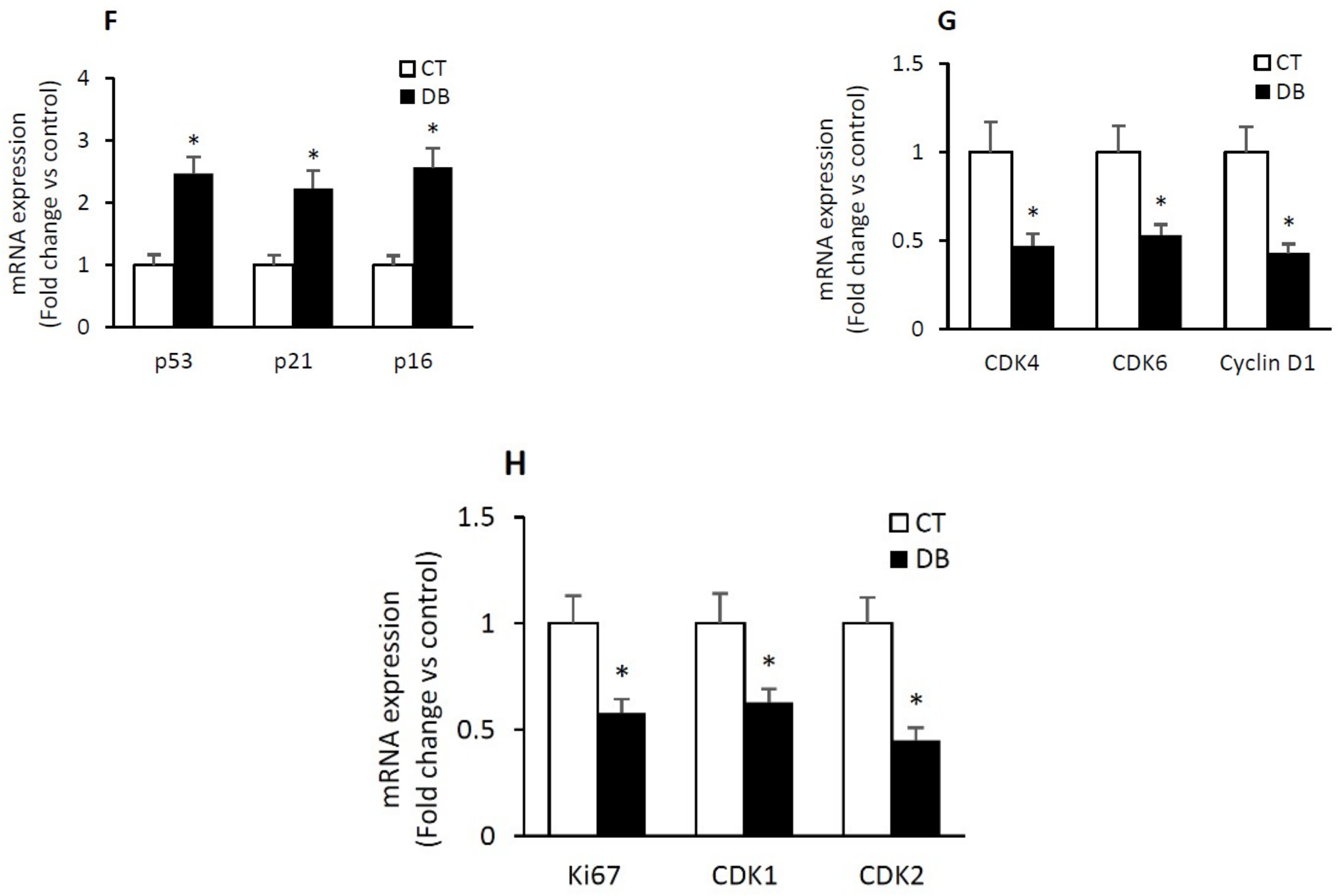
2.4. Diabetic W-ECs Exhibited a Characteristic Feature Consistent with Cellular Senescence and Cell-Cycle Arrest
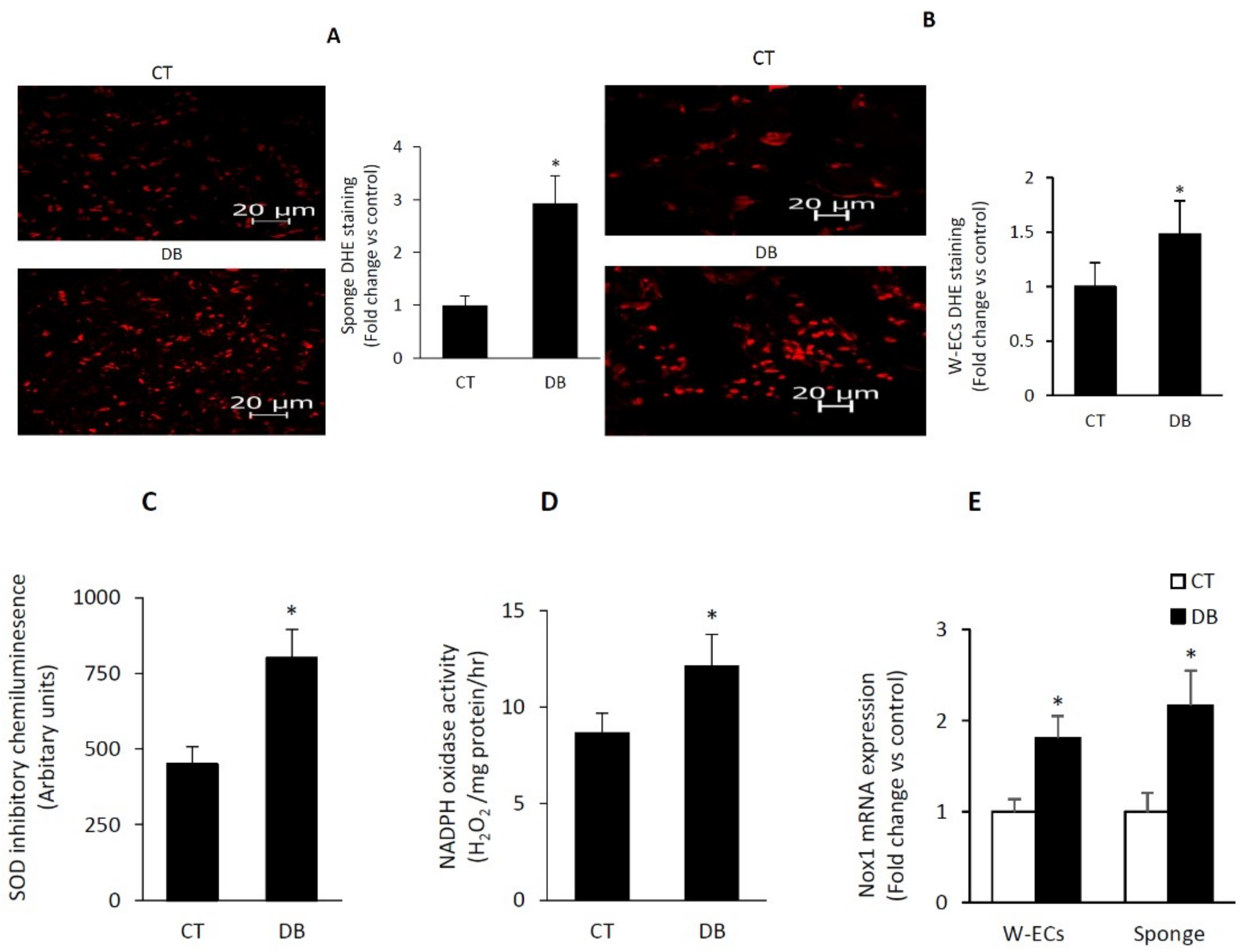
2.5. Diabetes Induces a Heightened State of Oxidative Stress in Cultured W-ECs and In Vivo Sponge Model of Wound Healing
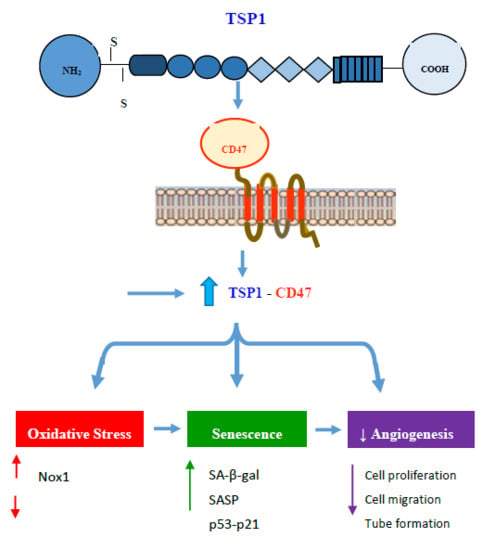
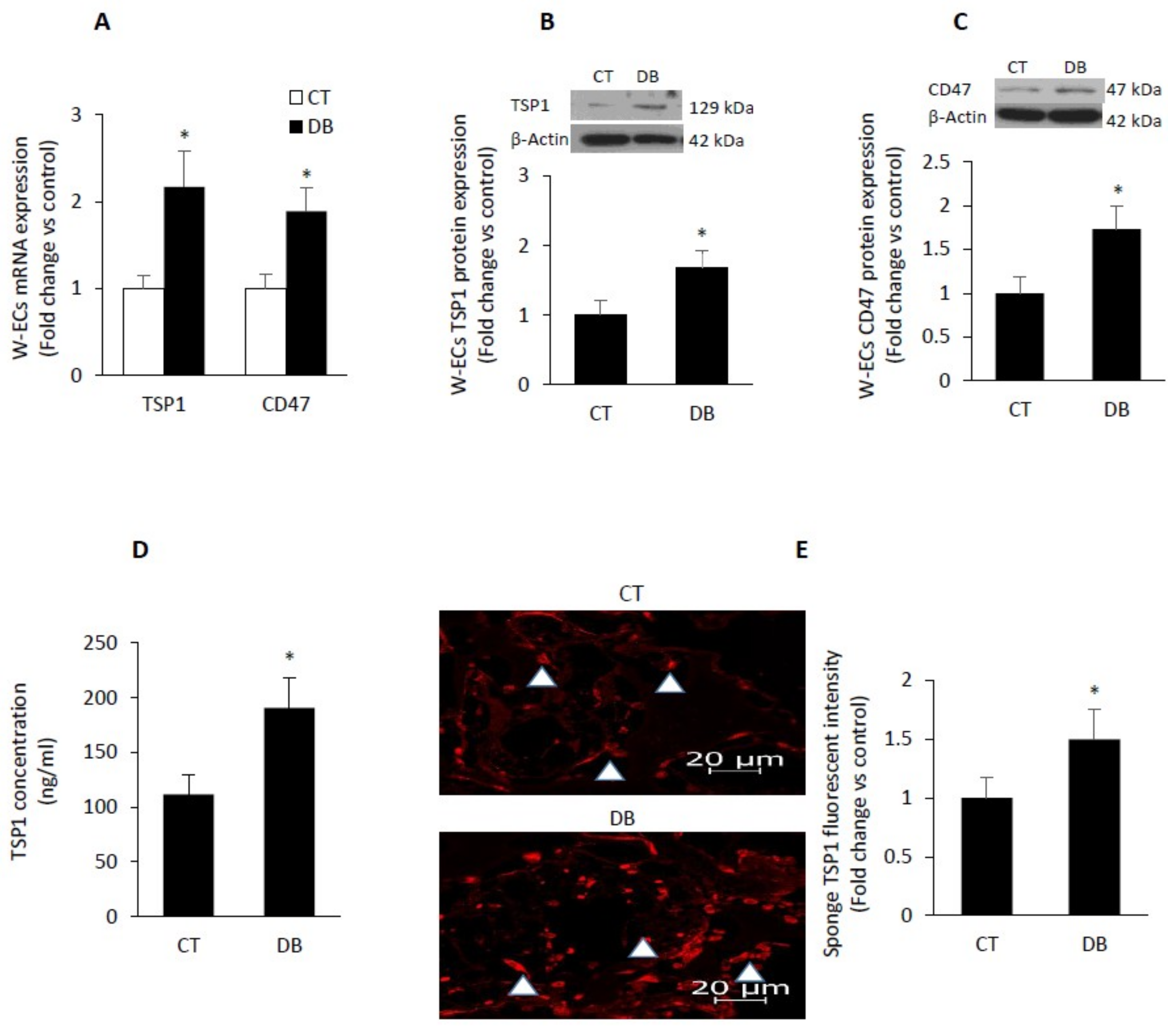
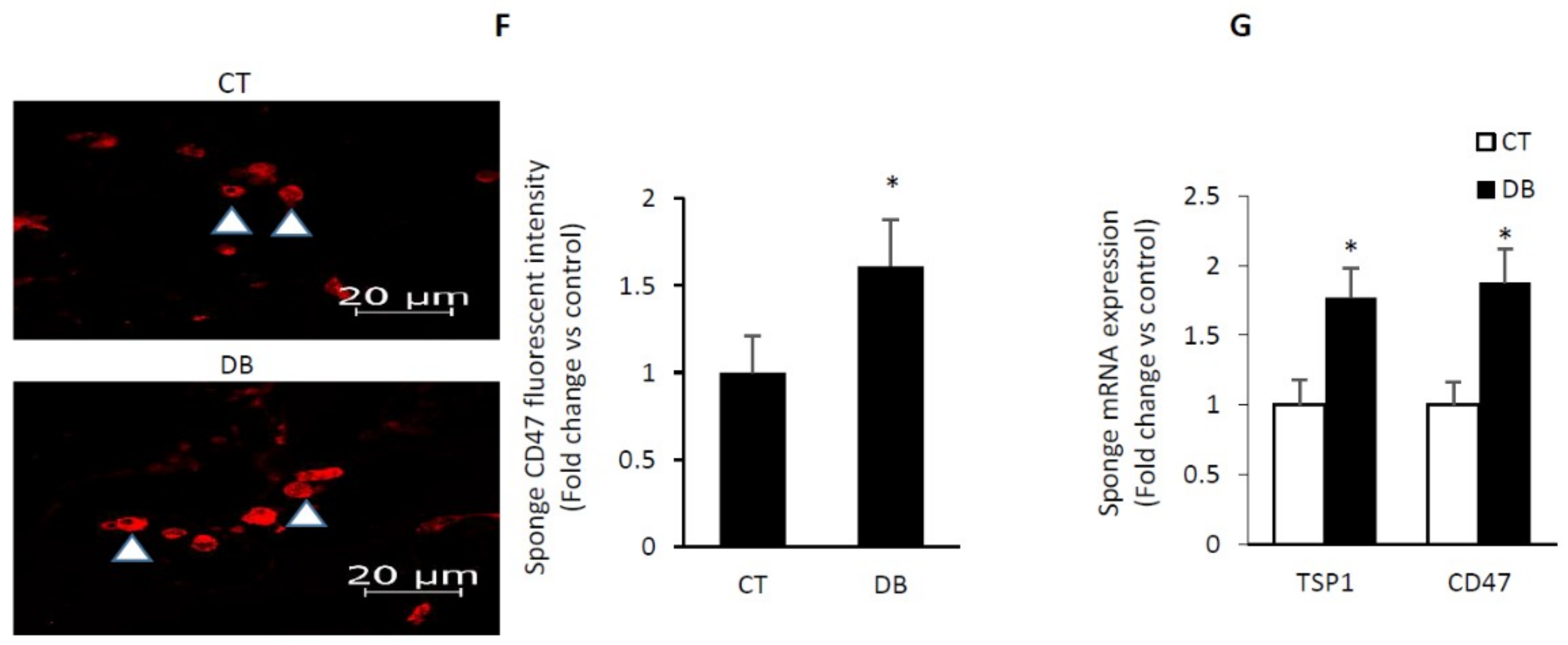
2.6. Diabetes Up-Regulates TSP1-CD47 System in Both Endothelial Cells and in PVA Sponge Model of Wound Healing
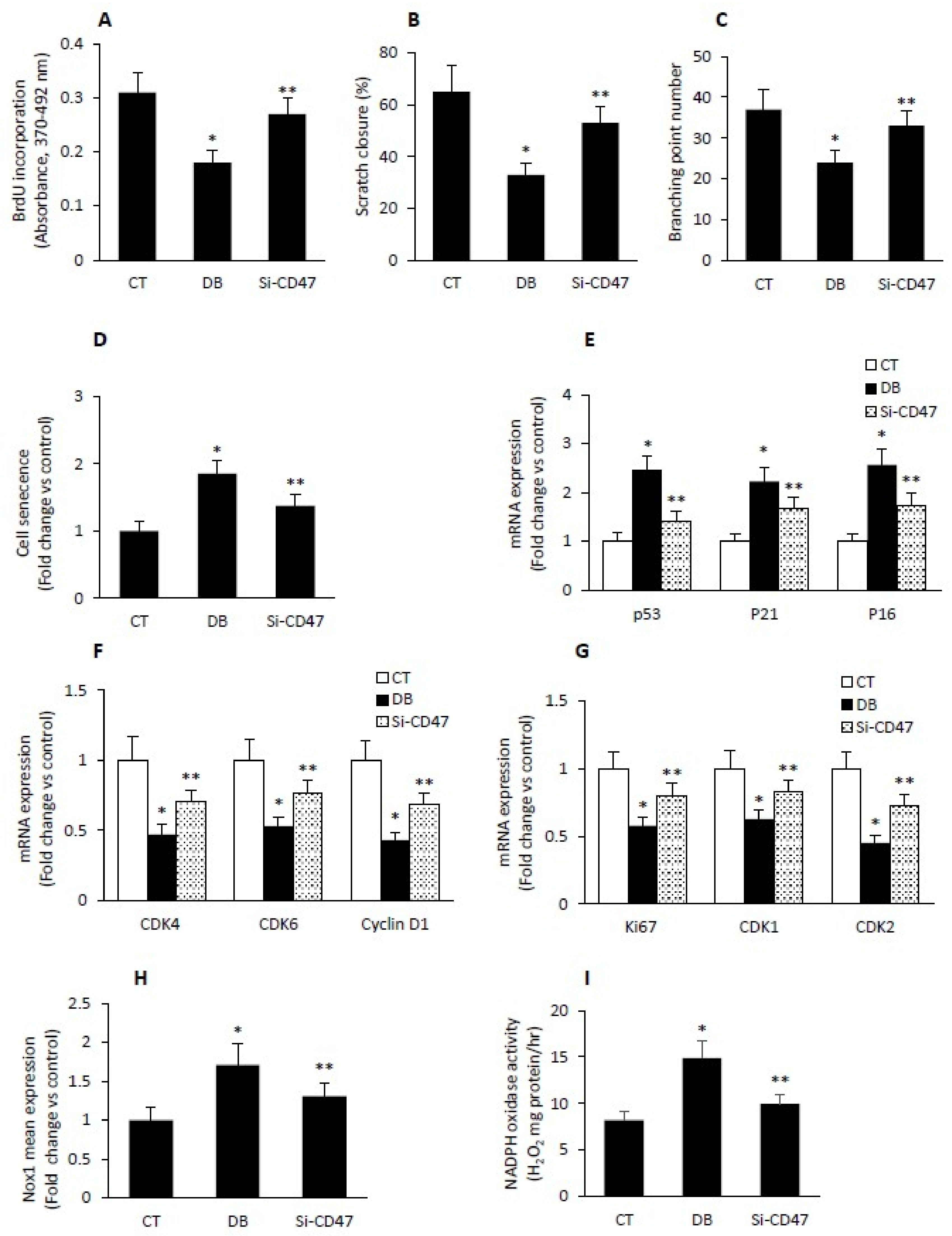
2.7. Diabetes Impairs Angiogenesis and Induces Endothelial Cell Senescence by Up-Regulating TSP1-CD47-Depndent Signaling
3. Discussion
4. Materials and Methods
4.1. Animals and an In Vivo Sponge Model of Angiogenesis
4.2. GTT and MOMA-IR Assessment
4.3. Assessment of, Fibroblast Invasion, Cell Proliferation and Microvascular Density in Sponge Implants
4.4. W-ECs as an In Vitro Model of Angiogenesis
4.5. Assessment of Key W-EC Functions
4.5.1. Cell Proliferation
4.5.2. Wound Healing Assay
4.5.3. Tube Formation Assay
4.6. Assessment of mRNA Expression, Protein Levels and ROS Contents in Sponge Implants and W-ECs
4.6.1. Real-Time PCR for mRNA Quantitation
4.6.2. Western Blot
4.6.3. Immunofluorescence Imaging
4.6.4. ELISA Quantification of TSP1
4.6.5. siRNA Transfection
4.7. Senescence-Associated β-Galactosidase (SA-β-gal) Expression Assay
4.8. ROS Generation and NAD(P)H Oxidase Activity
4.9. Apoptosis Assay
4.10. Statistical Analysis
Funding
Acknowledgments
Conflicts of Interest
References
- Palmer, A.K.; Tchkonia, T.; LeBrasseur, N.K.; Chini, E.N.; Xu, M.; Kirkland, J.L. Cellular Senescence in Type 2 Diabetes: A Therapeutic Opportunity. Diabetes 2015, 64, 2289–2298. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.A.; Creager, M.A.; Libby, P. Diabetes and atherosclerosis: Epidemiology, pathophysiology, and management. Jama 2002, 287, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Brem, H.; Tomic-Canic, M. Cellular and molecular basis of wound healing in diabetes. J. Clin. Investig. 2007, 117, 1219–1222. [Google Scholar] [CrossRef] [PubMed]
- Kannel, W.B.; McGee, D.L. Diabetes and glucose tolerance as risk factors for cardiovascular disease: The Framingham study. Diabetes Care 1979, 2, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Jeffcoate, W.J.; Harding, K.G. Diabetic foot ulcers. Lancet 2003, 361, 1545–1551. [Google Scholar] [CrossRef]
- Bitar, M.S.; Ayed, A.K.; Abdel-Halim, S.M.; Isenovic, E.R.; Al-Mulla, F. Inflammation and apoptosis in aortic tissues of aged type II diabetes: Amelioration with alpha-lipoic acid through phosphatidylinositol 3-kinase/Akt-dependent mechanism. Life Sci. 2010, 86, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Bitar, M.S.; Wahid, S.; Mustafa, S.; Al-Saleh, E.; Dhaunsi, G.S.; Al-Mulla, F. Nitric oxide dynamics and endothelial dysfunction in type II model of genetic diabetes. Eur. J. Pharmacol. 2005, 511, 53–64. [Google Scholar] [CrossRef]
- Tahergorabi, Z.; Khazaei, M. Imbalance of angiogenesis in diabetic complications: The mechanisms. Int. J. Prev. Med. 2012, 3, 827–838. [Google Scholar] [CrossRef]
- Falanga, V. Wound healing and its impairment in the diabetic foot. Lancet 2005, 366, 1736–1743. [Google Scholar] [CrossRef]
- Galiano, R.D.; Tepper, O.M.; Pelo, C.R.; Bhatt, K.A.; Callaghan, M.; Bastidas, N.; Bunting, S.; Steinmetz, H.G.; Gurtner, G.C. Topical vascular endothelial growth factor accelerates diabetic wound healing through increased angiogenesis and by mobilizing and recruiting bone marrow-derived cells. Am. J. Pathol. 2004, 164, 1935–1947. [Google Scholar] [CrossRef]
- Waltenberger, J. VEGF resistance as a molecular basis to explain the angiogenesis paradox in diabetes mellitus. Biochem. Soc. Trans. 2009, 37 Pt 6, 1167–1170. [Google Scholar] [CrossRef]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Counter, C.M.; Avilion, A.A.; LeFeuvre, C.E.; Stewart, N.G.; Greider, C.W.; Harley, C.B.; Bacchetti, S. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992, 11, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, M.T.; Nunez, M.I.; Villalobos, M.; Siles, E.; McMillan, T.J.; Pedraza, V.; Ruiz de Almodovar, J.M. A comparison of p53 and p16 expression in human tumor cells treated with hyperthermia or ionizing radiation. Int. J. Cancer 1997, 72, 307–312. [Google Scholar] [CrossRef]
- Matsui-Hirai, H.; Hayashi, T.; Yamamoto, S.; Ina, K.; Maeda, M.; Kotani, H.; Iguchi, A.; Ignarro, L.J.; Hattori, Y. Dose-dependent modulatory effects of insulin on glucose-induced endothelial senescence in vitro and in vivo: A relationship between telomeres and nitric oxide. J. Pharmacol. Exp. Ther. 2011, 337, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.Y.; Awad, E.M.; Oszwald, A.; Mayr, M.; Yin, X.; Waltenberger, B.; Stuppner, H.; Lipovac, M.; Uhrin, P.; Breuss, J.M. Premature senescence of endothelial cells upon chronic exposure to TNFalpha can be prevented by N-acetyl cysteine and plumericin. Sci. Rep. 2017, 7, 39501. [Google Scholar] [CrossRef] [PubMed]
- Corre, I.; Paris, F.; Huot, J. The p38 pathway, a major pleiotropic cascade that transduces stress and metastatic signals in endothelial cells. Oncotarget 2017, 8, 55684–55714. [Google Scholar] [CrossRef]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Haendeler, J.; Hoffmann, J.; Diehl, J.F.; Vasa, M.; Spyridopoulos, I.; Zeiher, A.M.; Dimmeler, S. Antioxidants inhibit nuclear export of telomerase reverse transcriptase and delay replicative senescence of endothelial cells. Circ. Res. 2004, 94, 768–775. [Google Scholar] [CrossRef]
- Matsushita, H.; Chang, E.; Glassford, A.J.; Cooke, J.P.; Chiu, C.P.; Tsao, P.S. eNOS activity is reduced in senescent human endothelial cells: Preservation by hTERT immortalization. Circ. Res. 2001, 89, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.G.; Zhang, J.; Block, E.R.; Patel, J.M. Senescence-enhanced oxidative stress is associated with deficiency of mitochondrial cytochrome c oxidase in vascular endothelial cells. Mech. Ageing Dev. 2003, 124, 911–919. [Google Scholar] [CrossRef]
- Yoon, H.J.; Cho, S.W.; Ahn, B.W.; Yang, S.Y. Alterations in the activity and expression of endothelial NO synthase in aged human endothelial cells. Mech. Ageing Dev. 2010, 131, 119–123. [Google Scholar] [CrossRef]
- Lakatta, E.G.; Levy, D. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part I: Aging arteries: A “set up” for vascular disease. Circulation 2003, 107, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Rivard, A.; Fabre, J.E.; Silver, M.; Chen, D.; Murohara, T.; Kearney, M.; Magner, M.; Asahara, T.; Isner, J.M. Age-dependent impairment of angiogenesis. Circulation 1999, 99, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Isenberg, J.S.; Popel, A.S. Human expression patterns: Qualitative and quantitative analysis of thrombospondin-1 under physiological and pathological conditions. J. Cell. Mol. Med. 2018, 22, 2086–2097. [Google Scholar] [CrossRef] [PubMed]
- Zaslavsky, A.; Baek, K.H.; Lynch, R.C.; Short, S.; Grillo, J.; Folkman, J.; Italiano, J.E., Jr.; Ryeom, S. Platelet-derived thrombospondin-1 is a critical negative regulator and potential biomarker of angiogenesis. Blood 2010, 115, 4605–4613. [Google Scholar] [CrossRef]
- Isenberg, J.S.; Ridnour, L.A.; Dimitry, J.; Frazier, W.A.; Wink, D.A.; Roberts, D.D. CD47 is necessary for inhibition of nitric oxide-stimulated vascular cell responses by thrombospondin-1. J. Biol. Chem. 2006, 281, 26069–26080. [Google Scholar] [CrossRef]
- Sick, E.; Jeanne, A.; Schneider, C.; Dedieu, S.; Takeda, K.; Martiny, L. CD47 update: A multifaceted actor in the tumour microenvironment of potential therapeutic interest. Br. J. Pharmacol. 2012, 167, 1415–1430. [Google Scholar] [CrossRef]
- Kaur, S.; Soto-Pantoja, D.R.; Stein, E.V.; Liu, C.; Elkahloun, A.G.; Pendrak, M.L.; Nicolae, A.; Singh, S.P.; Nie, Z.; Levens, D.; et al. Thrombospondin-1 signaling through CD47 inhibits self-renewal by regulating c-Myc and other stem cell transcription factors. Sci. Rep. 2013, 3, 1673. [Google Scholar] [CrossRef]
- Gao, Q.; Chen, K.; Gao, L.; Zheng, Y.; Yang, Y.G. Thrombospondin-1 signaling through CD47 inhibits cell cycle progression and induces senescence in endothelial cells. Cell Death Dis. 2016, 7, e2368. [Google Scholar] [CrossRef] [PubMed]
- Drinkwater, S.L.; Smith, A.; Burnand, K.G. What can wound fluids tell us about the venous ulcer microenvironment? Int. J. Low. Extrem. Wounds 2002, 1, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Yagihashi, S.; Tonosaki, A.; Yamada, K.; Kakizaki, M.; Goto, Y. Peripheral neuropathy in selectively-inbred spontaneously diabetic rats: Electrophysiological, morphometrical and freeze-replica studies. Tohoku J. Exp. Med. 1982, 138, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Komatsu, K.; Kurumatani, H. Late onset of diabetic nephropathy in spontaneously diabetic GK rats. Am. J. Nephrol. 2003, 23, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Sone, H.; Kawakami, Y.; Okuda, Y.; Sekine, Y.; Honmura, S.; Matsuo, K.; Segawa, T.; Suzuki, H.; Yamashita, K. Ocular vascular endothelial growth factor levels in diabetic rats are elevated before observable retinal proliferative changes. Diabetologia 1997, 40, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular senescence in cancer and aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef]
- Leong, W.F.; Chau, J.F.; Li, B. p53 Deficiency leads to compensatory up-regulation of p16INK4a. Mol. Cancer Res. 2009, 7, 354–360. [Google Scholar] [CrossRef]
- Kovac, S.; Angelova, P.R.; Holmstrom, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 2015, 1850, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Wingler, K.; Altenhoefer, S.A.; Kleikers, P.W.; Radermacher, K.A.; Kleinschnitz, C.; Schmidt, H.H. VAS2870 is a pan-NADPH oxidase inhibitor. Cell. Mol. Life Sci. 2012, 69, 3159–3160. [Google Scholar] [CrossRef] [PubMed]
- Novelli, E.M.; Little-Ihrig, L.; Jorgensen, D.R.; Shaaban, C.E.; Isenberg, J.S.; Butters, M.A.; Rosano, C. Elevated Plasma Thrombospondin-1 (TSP1) Levels Are Correlated with Oxidative Stress and Worse Cognitive Function in Sickle Cell Disease. Blood 2015, 126, 2184. [Google Scholar]
- Smadja, D.M.; d’Audigier, C.; Bieche, I.; Evrard, S.; Mauge, L.; Dias, J.V.; Labreuche, J.; Laurendeau, I.; Marsac, B.; Dizier, B.; et al. Thrombospondin-1 is a plasmatic marker of peripheral arterial disease that modulates endothelial progenitor cell angiogenic properties. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Gupta, P.; Wild, R.; Ramakrishnan, S.; Hebbel, R.P. Binding and displacement of vascular endothelial growth factor (VEGF) by thrombospondin: Effect on human microvascular endothelial cell proliferation and angiogenesis. Angiogenesis 1999, 3, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Pagano, K.; Torella, R.; Foglieni, C.; Bugatti, A.; Tomaselli, S.; Zetta, L.; Presta, M.; Rusnati, M.; Taraboletti, G.; Colombo, G.; et al. Direct and allosteric inhibition of the FGF2/HSPGs/FGFR1 ternary complex formation by an antiangiogenic, thrombospondin-1-mimic small molecule. PLoS ONE 2012, 7, e36990. [Google Scholar] [CrossRef] [PubMed]
- Ergul, A.; Abdelsaid, M.; Fouda, A.Y.; Fagan, S.C. Cerebral neovascularization in diabetes: Implications for stroke recovery and beyond. J. Cereb. Blood Flow Metab. 2014, 34, 553–563. [Google Scholar] [CrossRef]
- Bitar, M.S.; Al-Mulla, F. Upregulation of CREM/ICER suppresses wound endothelial CRE-HIF-1α-VEGF-dependent signaling and impairs angiogenesis in type 2 diabetes. Dis. Models Mechan. 2015, 8, 65–80. [Google Scholar] [CrossRef]
- Sadoun, E.; Reed, M.J. Impaired angiogenesis in aging is associated with alterations in vessel density, matrix composition, inflammatory response, and growth factor expression. J. Histochem. Cytochem. 2003, 51, 1119–1130. [Google Scholar] [CrossRef]
- Neill, T.; Painter, H.; Buraschi, S.; Owens, R.T.; Lisanti, M.P.; Schaefer, L.; Iozzo, R.V. Decorin antagonizes the angiogenic network: Concurrent inhibition of Met, hypoxia inducible factor 1alpha, vascular endothelial growth factor A, and induction of thrombospondin-1 and TIMP3. J. Biol. Chem. 2012, 287, 5492–5506. [Google Scholar] [CrossRef]
- Bitar, M.S.; Abdel-Halim, S.M.; Al-Mulla, F. Caveolin-1/PTRF upregulation constitutes a mechanism for mediating p53-induced cellular senescence: Implications for evidence-based therapy of delayed wound healing in diabetes. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E951–E963. [Google Scholar] [CrossRef]
- Hampel, B.; Fortschegger, K.; Ressler, S.; Chang, M.W.; Unterluggauer, H.; Breitwieser, A.; Sommergruber, W.; Fitzky, B.; Lepperdinger, G.; Jansen-Durr, P.; et al. Increased expression of extracellular proteins as a hallmark of human endothelial cell in vitro senescence. Exp. Gerontol. 2006, 41, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Yanaka, M.; Honma, T.; Sato, K.; Shinohara, N.; Ito, J.; Tanaka, Y.; Tsuduki, T.; Ikeda, I. Increased monocytic adhesion by senescence in human umbilical vein endothelial cells. Biosci. Biotechnol. Biochem. 2011, 75, 1098–1103. [Google Scholar] [CrossRef] [PubMed]
- Rombouts, C.; Aerts, A.; Quintens, R.; Baselet, B.; El-Saghire, H.; Harms-Ringdahl, M.; Haghdoost, S.; Janssen, A.; Michaux, A.; Yentrapalli, R.; et al. Transcriptomic profiling suggests a role for IGFBP5 in premature senescence of endothelial cells after chronic low dose rate irradiation. Int. J. Radiat. Biol. 2014, 90, 560–574. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 4350965, 22. [Google Scholar] [CrossRef] [PubMed]
- Kurz, D.J.; Decary, S.; Hong, Y.; Trivier, E.; Akhmedov, A.; Erusalimsky, J.D. Chronic oxidative stress compromises telomere integrity and accelerates the onset of senescence in human endothelial cells. J. Cell Sci. 2004, 117 Pt 11, 2417–2426. [Google Scholar] [CrossRef]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Vigneron, A.; Vousden, K.H. p53, ROS and senescence in the control of aging. Aging 2010, 2, 471–474. [Google Scholar] [CrossRef]
- Jimenez, B.; Volpert, O.V.; Reiher, F.; Chang, L.; Munoz, A.; Karin, M.; Bouck, N. c-Jun N-terminal kinase activation is required for the inhibition of neovascularization by thrombospondin-1. Oncogene 2001, 20, 3443–3448. [Google Scholar] [CrossRef]
- Isenberg, J.S.; Jia, Y.; Fukuyama, J.; Switzer, C.H.; Wink, D.A.; Roberts, D.D. Thrombospondin-1 inhibits nitric oxide signaling via CD36 by inhibiting myristic acid uptake. J. Biol. Chem. 2007, 282, 15404–15415. [Google Scholar] [CrossRef]
- Kanda, S.; Shono, T.; Tomasini-Johansson, B.; Klint, P.; Saito, Y. Role of thrombospondin-1-derived peptide, 4N1K, in FGF-2-induced angiogenesis. Exp. Cell Res 1999, 252, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Isenberg, J.S.; Ridnour, L.A.; Perruccio, E.M.; Espey, M.G.; Wink, D.A.; Roberts, D.D. Thrombospondin-1 inhibits endothelial cell responses to nitric oxide in a cGMP-dependent manner. Proc. Natl. Acad. Sci. USA 2005, 102, 13141–13146. [Google Scholar] [CrossRef]
- Meijles, D.N.; Sahoo, S.; Al Ghouleh, I.; Amaral, J.H.; Bienes-Martinez, R.; Knupp, H.E.; Attaran, S.; Sembrat, J.C.; Nouraie, S.M.; Rojas, M.M.; et al. The matricellular protein TSP1 promotes human and mouse endothelial cell senescence through CD47 and Nox1. Sci. Signal. 2017, 10, eaaj1784. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.V.; Benslimane-Ahmim, Z.; Egot, M.; Lokajczyk, A.; Grelac, F.; Galy-Fauroux, I.; Juliano, L.; Le-Bonniec, B.; Takiya, C.M.; Fischer, A.M.; et al. A motif within the N-terminal domain of TSP-1 specifically promotes the proangiogenic activity of endothelial colony-forming cells. Biochem. Pharmacol. 2012, 84, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Cointe, S.; Rheaume, E.; Martel, C.; Blanc-Brude, O.; Dube, E.; Sabatier, F.; Dignat-George, F.; Tardif, J.C.; Bonnefoy, A. Thrombospondin-1-Derived Peptide RFYVVMWK Improves the Adhesive Phenotype of CD34(+) Cells From Atherosclerotic Patients With Type 2 Diabetes. Cell Transplant. 2017, 26, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, R.; Akhtar, N.; Lewis, R.L.; Shinners, B.L. Angiogenesis assays: Problems and pitfalls. Cancer Metast. Rev. 2000, 19, 167–172. [Google Scholar] [CrossRef]
- Bitar, M.S. The GSK-3β/Fyn/Nrf2 pathway in fibroblasts and wounds of type 2 diabetes: On the road to an evidence-based therapy of non-healing wounds. Adipocyte 2012, 1, 161–163. [Google Scholar] [CrossRef]
- Bitar, M.S.; Al-Mulla, F. A defect in Nrf2 signaling constitutes a mechanism for cellular stress hypersensitivity in a genetic rat model of type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E1119–E1129. [Google Scholar] [CrossRef]
- Bitar, M.S. Insulin and glucocorticoid-dependent suppression of the IGF-I system in diabetic wounds. Surgery 2000, 127, 687–695. [Google Scholar] [CrossRef]
- Bradshaw, A.D.; Reed, M.J.; Carbon, J.G.; Pinney, E.; Brekken, R.A.; Sage, E.H. Increased fibrovascular invasion of subcutaneous polyvinyl alcohol sponges in SPARC-null mice. Wound Repair Regen. 2001, 9, 522–530. [Google Scholar] [CrossRef]
- Mather, K. Surrogate measures of insulin resistance: Of rats, mice, and men. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E398–E399. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.G.; Bernasconi, S.; Lostaglio, S.; De Calmanovici, R.W.; Martin-Padura, I.; Breviario, F.; Garlanda, C.; Ramponi, S.; Mantovani, A.; Vecchi, A. A general strategy for isolation of endothelial cells from murine tissues. Characterization of two endothelial cell lines from the murine lung and subcutaneous sponge implants. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1599–1604. [Google Scholar] [CrossRef] [PubMed]
- Al-Mulla, F.; Leibovich, S.J.; Francis, I.M.; Bitar, M.S. Impaired TGF-beta signaling and a defect in resolution of inflammation contribute to delayed wound healing in a female rat model of type 2 diabetes. Mol. Biosyst. 2011, 7, 3006–3020. [Google Scholar] [CrossRef] [PubMed]










© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bitar, M.S. Diabetes Impairs Angiogenesis and Induces Endothelial Cell Senescence by Up-Regulating Thrombospondin-CD47-Dependent Signaling. Int. J. Mol. Sci. 2019, 20, 673. https://doi.org/10.3390/ijms20030673
Bitar MS. Diabetes Impairs Angiogenesis and Induces Endothelial Cell Senescence by Up-Regulating Thrombospondin-CD47-Dependent Signaling. International Journal of Molecular Sciences. 2019; 20(3):673. https://doi.org/10.3390/ijms20030673
Chicago/Turabian StyleBitar, Milad S. 2019. "Diabetes Impairs Angiogenesis and Induces Endothelial Cell Senescence by Up-Regulating Thrombospondin-CD47-Dependent Signaling" International Journal of Molecular Sciences 20, no. 3: 673. https://doi.org/10.3390/ijms20030673
APA StyleBitar, M. S. (2019). Diabetes Impairs Angiogenesis and Induces Endothelial Cell Senescence by Up-Regulating Thrombospondin-CD47-Dependent Signaling. International Journal of Molecular Sciences, 20(3), 673. https://doi.org/10.3390/ijms20030673