Molecular Simulation of Naphthalene, Phenanthrene, and Pyrene Adsorption on MCM-41

 ,
, 
Abstract
1. Introduction
2. Results and Discussion
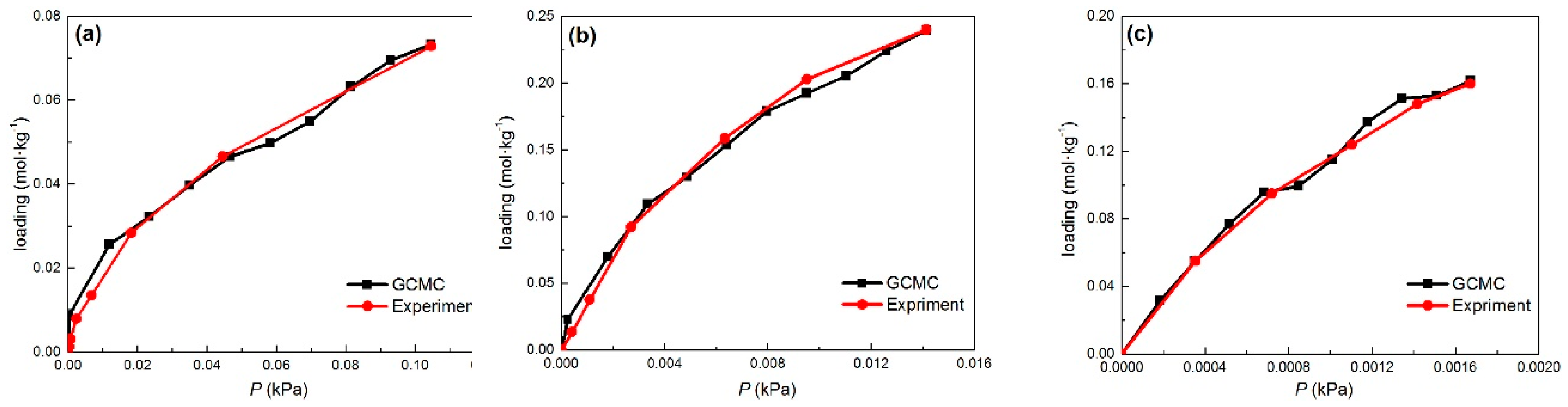
2.1. Adsorption Equlibrium
2.2. Adsorption State
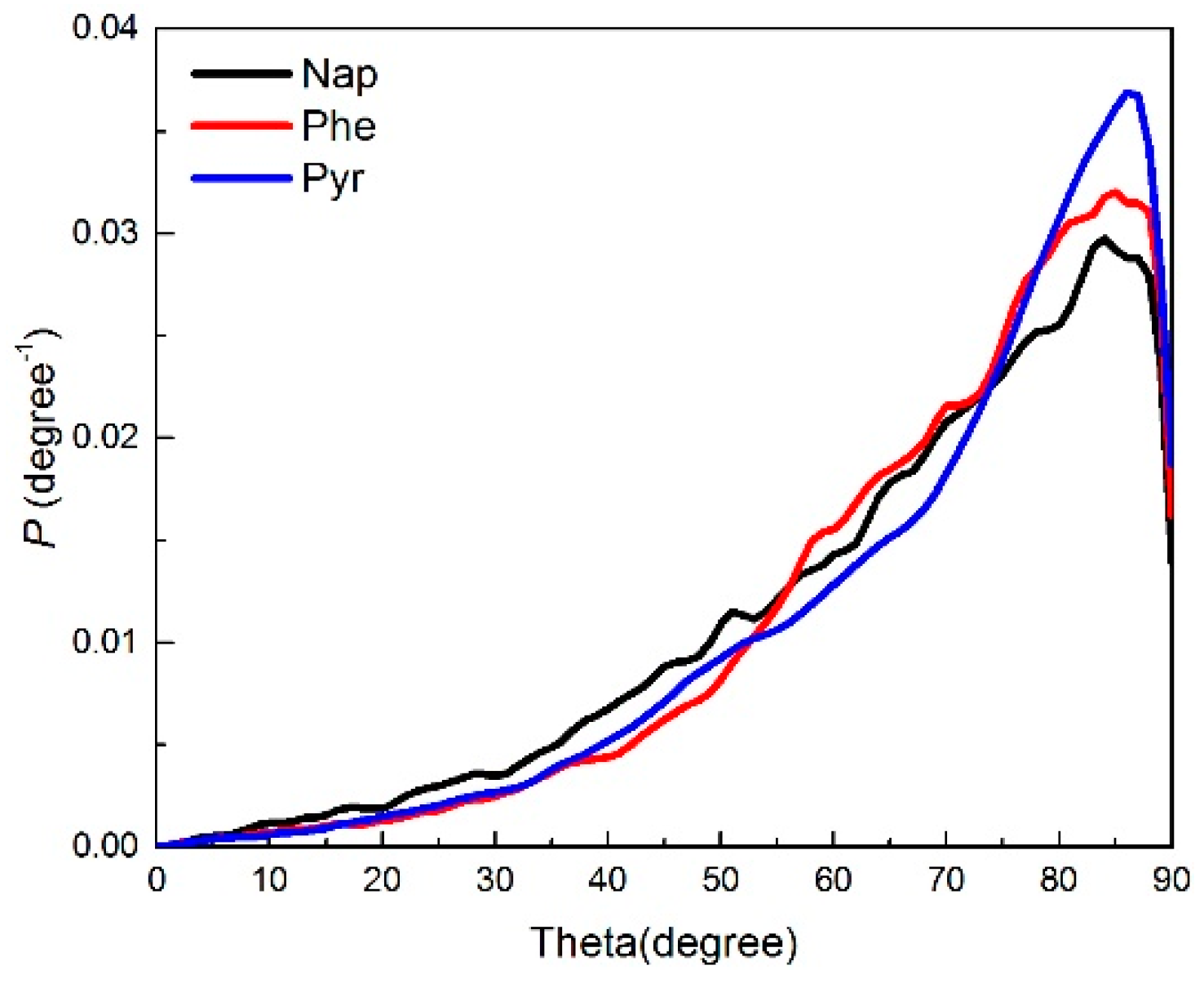
2.2.1. Angle Distribution
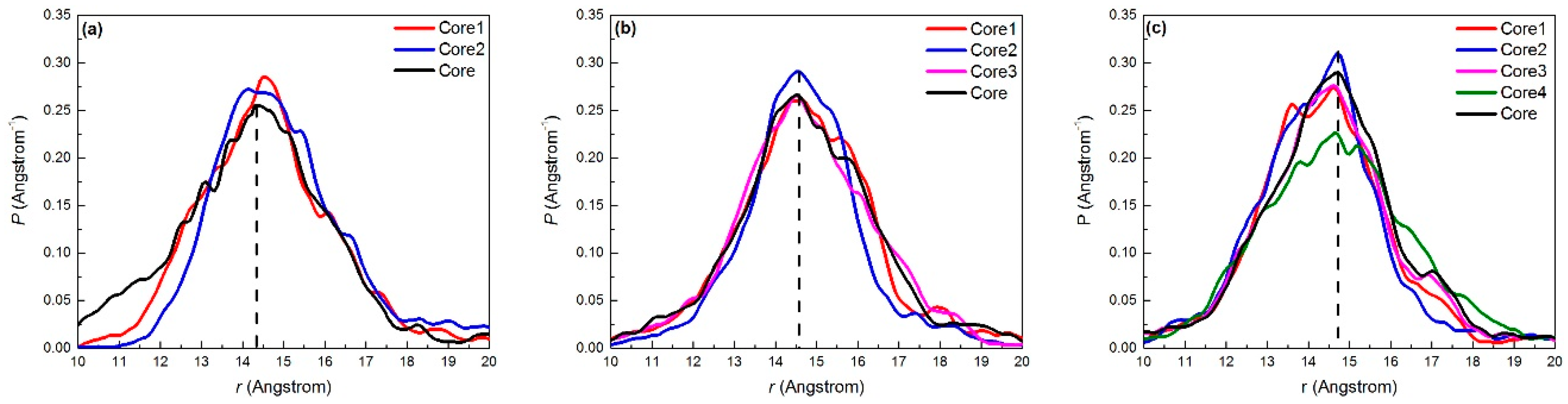
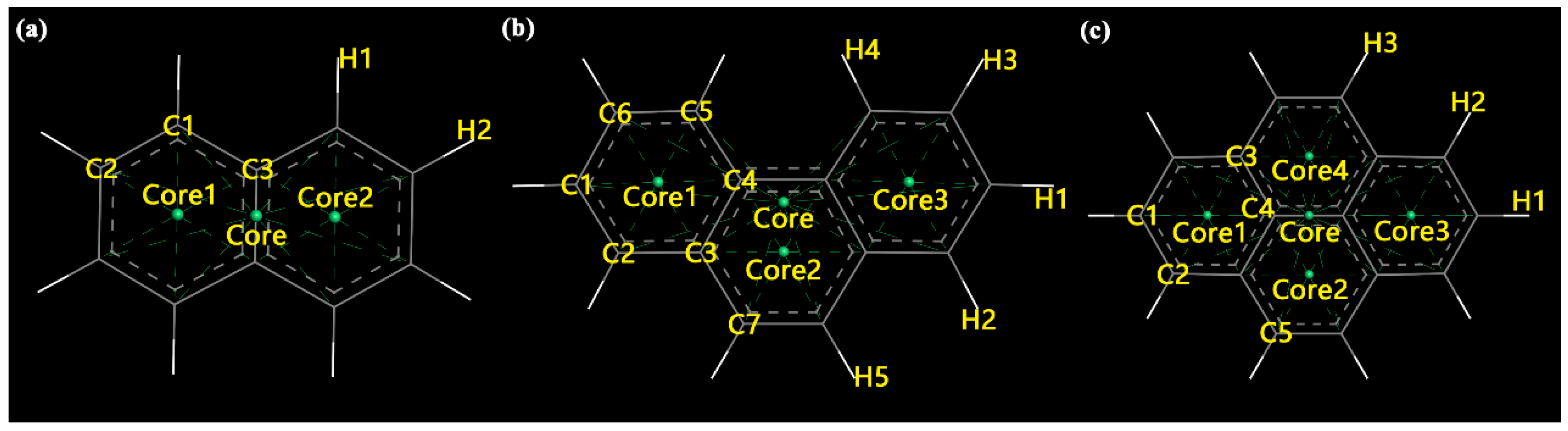
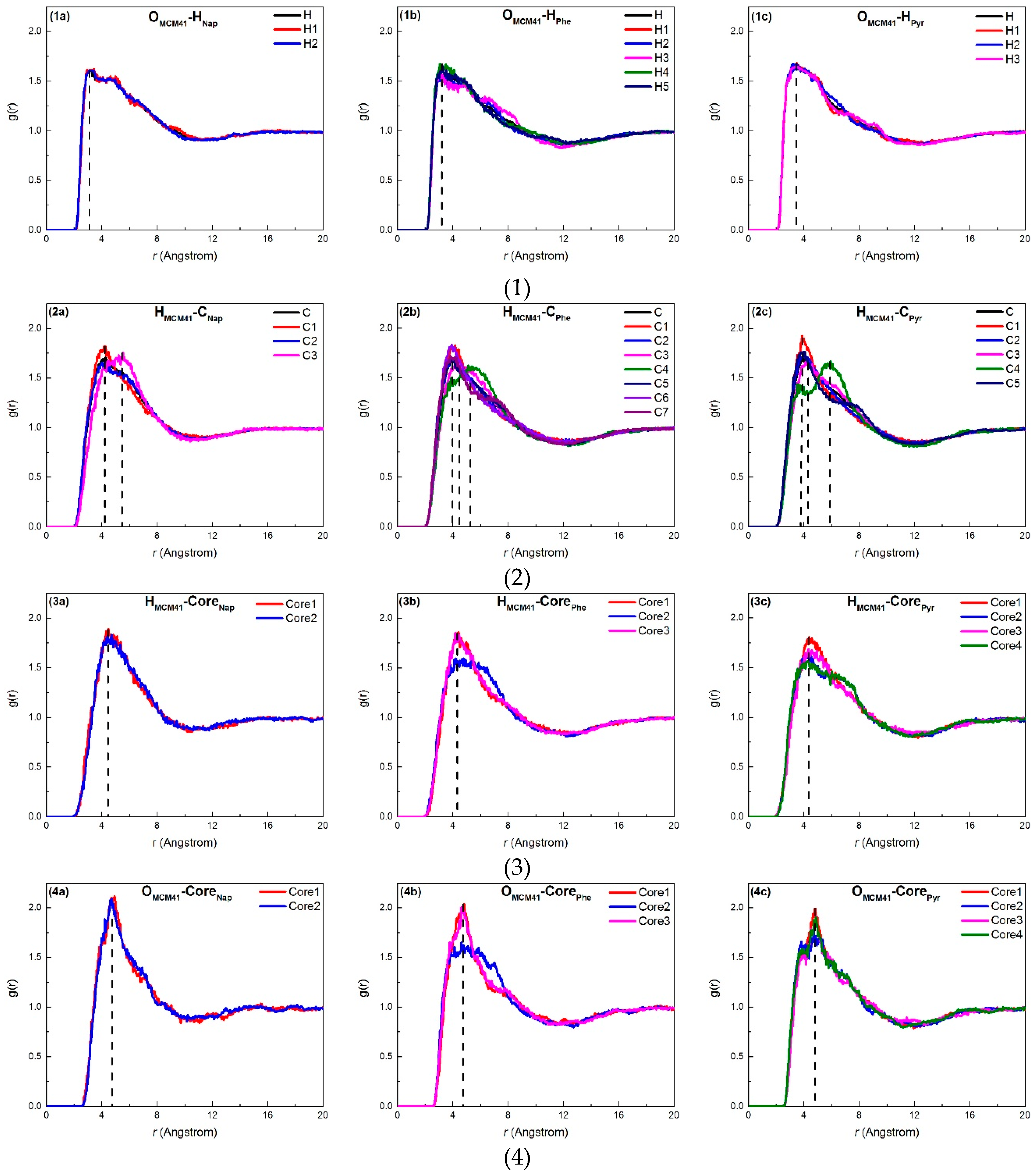
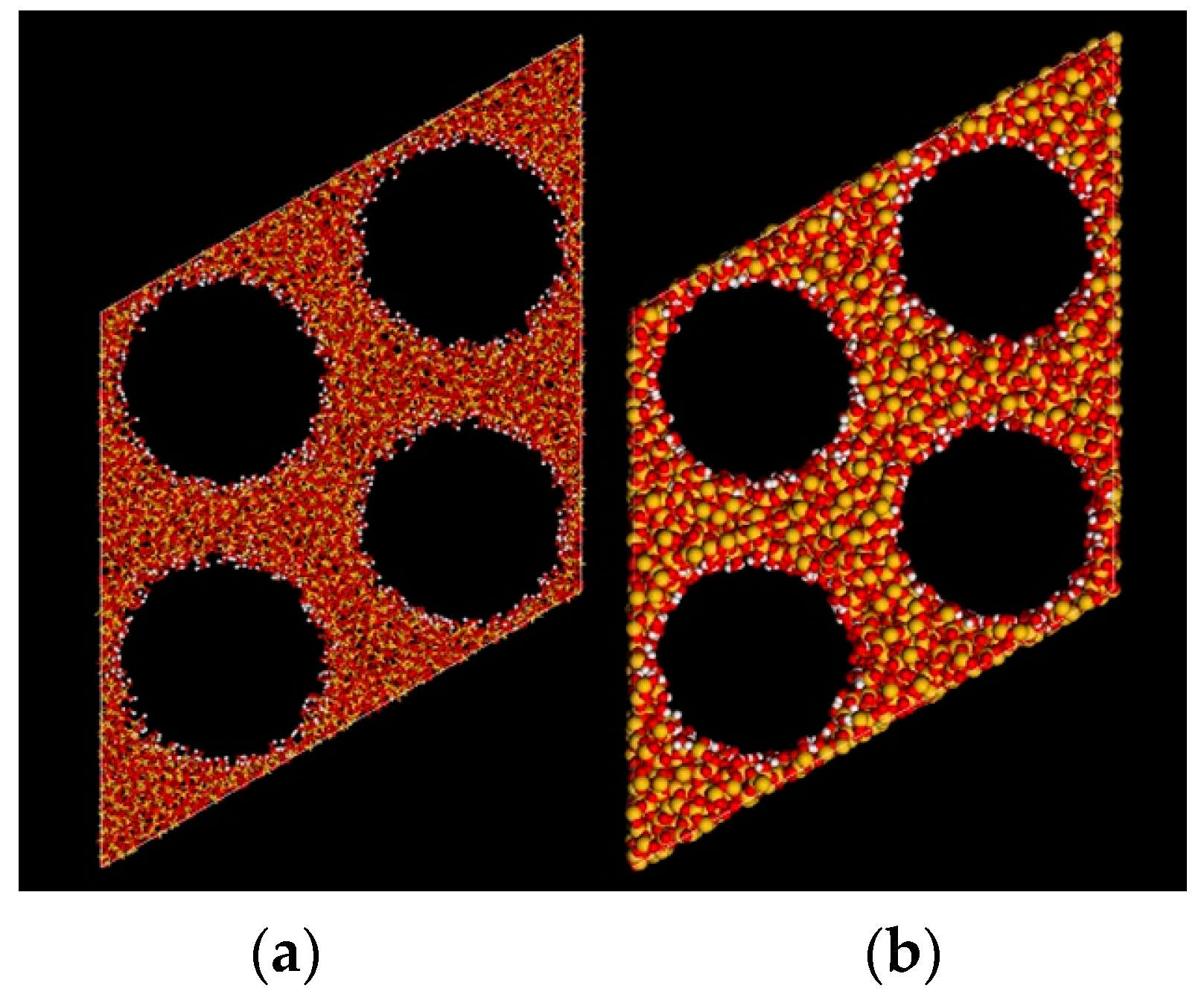
2.2.2. Density Profile
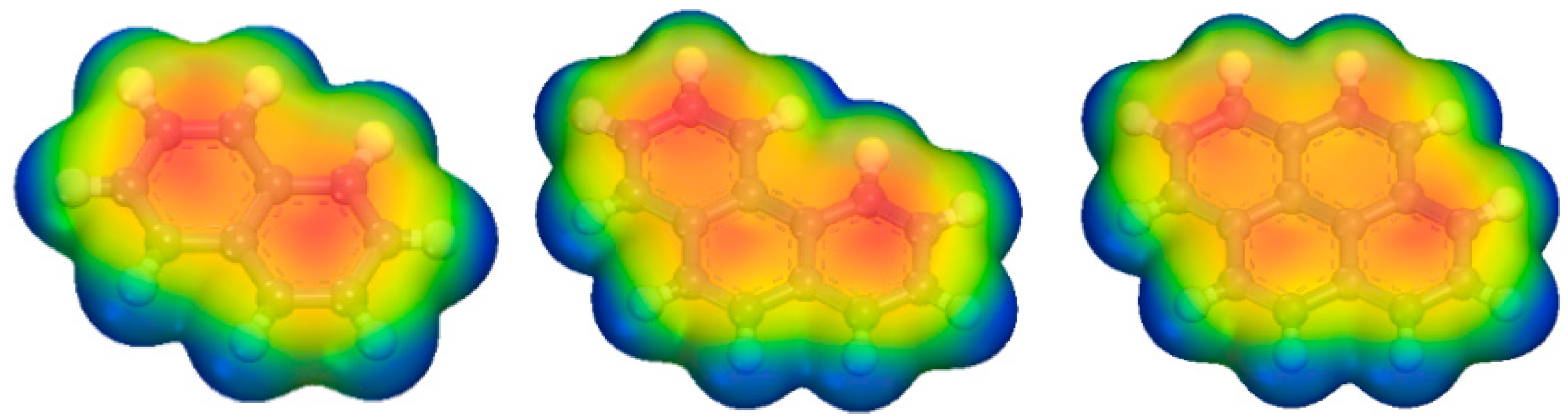
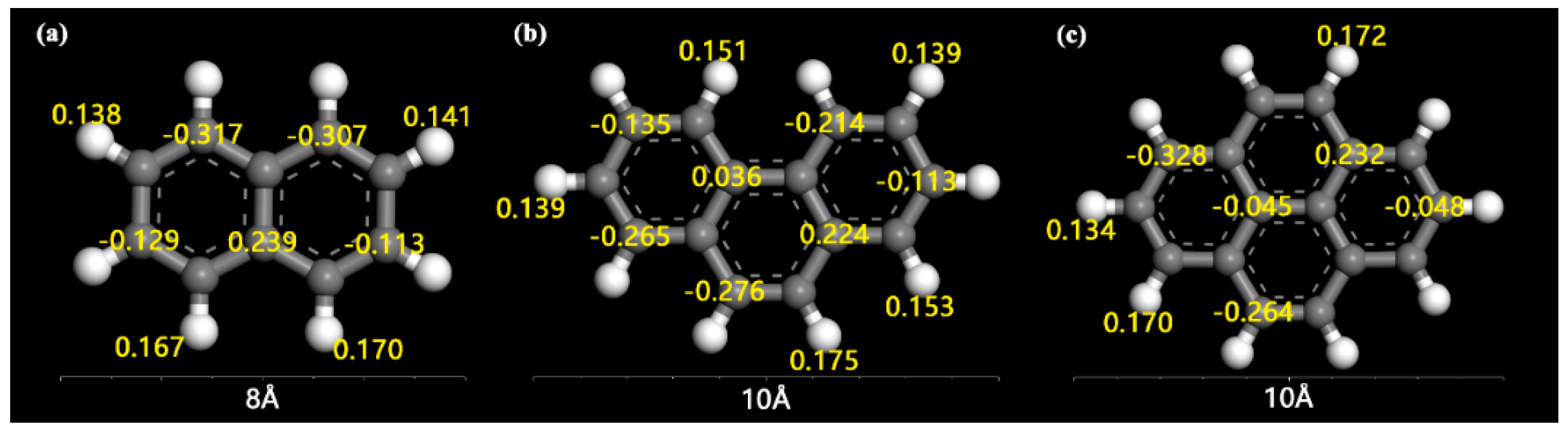
2.3. Adsorption Interactions
3. Methods and Parameters
3.1. Model Structures
3.2. Calculation Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Nap | Naphthalene |
| Phe | Phenanthrene |
| Pyr | Pyrene |
| GCMC | Grand Canonical Monte Carlo |
References
- Casarett, L.J.; Doull, J. Toxicology: The Basic Science of Poisons; McGraw-Hill: New York, NY, USA, 1996. [Google Scholar]
- Li, Z.; Liu, Y.; Yang, X.; Xing, Y.; Wang, Z.; Yang, Q.; Yang, R.T. Desorption kinetics of naphthalene and acenaphthene over two activated carbons via thermogravimetric analysis. Energy Fuels 2015, 29, 5303–5310. [Google Scholar] [CrossRef]
- Cheng, X.; Kan, A.T.; Tomson, M.B. Naphthalene adsorption and desorption from aqueous C60 fullerene. J. Chem. Eng. Data 2004, 49, 675–683. [Google Scholar] [CrossRef]
- Yang, K.U.N.; Wu, W.; Jing, Q.; Zhu, L. Aqueous adsorption of aniline, phenol, and their substitutes by multi-walled carbon nanotubes. Environ. Sci. Technol. 2008, 42, 7931–7936. [Google Scholar] [CrossRef] [PubMed]
- Owabor, C.N.; Ogbeide, S.E.; Susu, A.A. Adsorption and desorption kinetics of naphthalene, anthracene, and pyrene in soil matrix. Pet. Sci. Technol. 2010, 28, 504. [Google Scholar] [CrossRef]
- Long, A.S.; Lemieux, C.L.; Gagné, R.; Lambert, I.B.; White, P.A. Genetic Toxicity of Complex Mixtures of Polycyclic Aromatic Hydrocarbons: Evaluating Dose-Additivity in a Transgenic Mouse Model. Environ. Sci. Technol. 2017, 51, 8138–8148. [Google Scholar] [CrossRef] [PubMed]
- Liamin, M.; Le Mentec, H.; Evrard, B.; Huc, L.; Chalmel, F.; Boutet-Robinet, E.; Le Ferrec, E.; Sparfel, L. Genome-Wide Transcriptional and Functional Analysis of Human T Lymphocytes Treated with Benzo [α] pyrene. Int. J. Mol. Sci. 2018, 19, 3626. [Google Scholar] [CrossRef]
- Libalova, H.; Rossner, P.; Vrbova, K.; Brzicova, T.; Sikorova, J.; Vojtisek-Lom, M.; Beranek, V.; Klema, J.; Ciganek, M.; Neca, J.; et al. Comparative analysis of toxic responses of organic extracts from diesel and selected alternative fuels engine emissions in human lung BEAS-2B cells. Int. J. Mol. Sci. 2016, 17, 1833. [Google Scholar] [CrossRef]
- Mastral, A.M.; Callen, M.S. A review on polycyclic aromatic hydrocarbon (PAH) emissions from energy generation. Environ. Sci. Technol. 2000, 34, 3051–3057. [Google Scholar] [CrossRef]
- Keith, L.; Telliard, W. ES&T special report: Priority pollutants: Ia perspective view. Environ. Sci. Technol. 1979, 13, 416–423. [Google Scholar]
- Albuquerque, M.; Coutinho, M.; Borrego, C. Long-term monitoring and seasonal analysis of polycyclic aromatic hydrocarbons (PAHs) measured over a decade in the ambient air of Porto, Portugal. Sci. Total Environ. 2016, 543, 439–448. [Google Scholar] [CrossRef]
- Dowaidar, A.M.; El-Shahawi, M.S.; Ashour, I. Adsorption of Polycyclic Aromatic Hydrocarbons onto Activated Carbon from Non-Aqueous Media: 1. The Influence of the Organic Solvent Polarity. Sep. Sci. Technol. 2007, 42, 3609–3622. [Google Scholar] [CrossRef]
- Mastral, A.M.; Callen, M.; Murillo, R. Assessment of PAH emissions as a function of coal combustion variables. Fuel 1996, 75, 1533–1536. [Google Scholar] [CrossRef]
- Zhou, H.C.; Zhong, Z.P.; Jin, B.S.; Huang, Y.J.; Xiao, R. Experimental study on the removal of PAHs using in-duct activated carbon injection. Chemosphere 2005, 59, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, Y.; Yang, X.; Xing, Y.; Tsai, C.; Wang, Z.; Yang, Q.; Yang, R.T. Desorption of polycyclic aromatic hydrocarbons on mesoporous sorbents: Thermogravimetric experiments and kinetics study. Ind. Eng. Chem. Res. 2016, 55, 1183–1191. [Google Scholar] [CrossRef]
- Araújo, R.S.; Azevedo, D.C.S.; Cavalcante, C.L., Jr.; Jiménez-López, A.; Rodríguez-Castellón, E. Adsorption of polycyclic aromatic hydrocarbons (PAHs) from isooctane solutions by mesoporous molecular sieves: Influence of the surface acidity. Microporous Mesoporous Mater. 2008, 108, 213–222. [Google Scholar] [CrossRef]
- Shang, J.; Li, G.; Singh, R.; Gu, Q.; Nairn, K.M.; Bastow, T.J.; Medhekar, N.; Doherty, C.M.; Hill, A.J.; Liu, J.Z.; et al. Discriminative separation of gases by a “molecular trapdoor” mechanism in chabazite zeolites. J. Am. Chem. Soc. 2012, 134, 19246–19253. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Li, G.; Webley, P.A.; Liu, J.Z. A density functional theory study for the adsorption of various gases on a caesium-exchanged trapdoor chabazite. Comput. Mater. Sci. 2016, 122, 307–313. [Google Scholar] [CrossRef]
- Li, Z.; Liu, Y.; Zhang, C.; Yang, X.; Ren, J.; Jiang, L. Methane recovery from coal bed gas using modified activated carbons: A combined method for assessing the role of functional groups. Energy Fuels 2015, 29, 6858–6865. [Google Scholar] [CrossRef]
- Li, G.K.; Shang, J.; Gu, Q.; Awati, R.V.; Jensen, N.; Grant, A.; Zhang, X.; Sholl, D.S.; Liu, J.Z.; Webley, P.A.; et al. Temperature-regulated guest admission and release in microporous materials. Nat. Commun. 2017, 8, 15777. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.S.; Vartuli, J.C.; Roth, W.J.; Leonowicz, M.E.; Kresge, C.T.; Schmitt, K.D.; Chu, C.T.W.; Olson, D.H.; Sheppard, E.W.; McCullen, S.B.; et al. A new family of mesoporous molecular sieves prepared with liquid crystal templates. J. Am. Chem. Soc. 1992, 114, 10834–10843. [Google Scholar] [CrossRef]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.S. Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism. Nature 1992, 359, 710. [Google Scholar] [CrossRef]
- Selvam, P.; Bhatia, S.K.; Sonwane, C.G. Recent advances in processing and characterization of periodic mesoporous MCM-41 silicate molecular sieves. Ind. Eng. Chem. Res. 2001, 40, 3237–3261. [Google Scholar] [CrossRef]
- Corma, A. From microporous to mesoporous molecular sieve materials and their use in catalysis. Chem. Rev. 1997, 97, 2373–2420. [Google Scholar] [CrossRef] [PubMed]
- Ciesla, U.; Schüth, F. Ordered mesoporous materials. Microporous Mesoporous Mater. 1999, 27, 131–149. [Google Scholar] [CrossRef]
- Rimola, A.; Costa, D.; Sodupe, M.; Lambert, J.F.; Ugliengo, P. Silica surface features and their role in the adsorption of biomolecules: Computational modeling and experiments. Chem. Rev. 2013, 113, 4216–4313. [Google Scholar] [CrossRef]
- Pajzderska, A.; Gonzalez, M.A.; Mielcarek, J.; Wąsicki, J. Water behavior in MCM-41 as a function of pore filling and temperature studied by NMR and molecular dynamics simulations. J. Phys. Chem. C 2014, 118, 23701–23710. [Google Scholar] [CrossRef]
- Coasne, B.; Galarneau, A.; Di Renzo, F.; Pellenq, R.J. Molecular simulation of nitrogen adsorption in nanoporous silica. Langmuir 2010, 26, 10872–10881. [Google Scholar] [CrossRef]
- Chang, S.C.; Chien, S.Y.; Chen, C.L. Analyzing adsorption characteristics of CO2, N2 and H2O in MCM-41 silica by molecular simulation. Appl. Surf. Sci. 2015, 331, 225–233. [Google Scholar] [CrossRef]
- Coasne, B.; Galarneau, A.; Pellenq, R.J.M.; di Renzo, F. Adsorption, intrusion and freezing in porous silica: The view from the nanoscale. Chem. Soc. Rev. 2013, 42, 4141–4171. [Google Scholar] [CrossRef]
- Coasne, B.; Alba-Simionesco, C.; Audonnet, F.; Dosseh, G.; Gubbins, K.E. Adsorption and structure of benzene on silica surfaces and in nanopores. Langmuir 2009, 25, 10648–10659. [Google Scholar] [CrossRef]
- Ravikovitch, P.I.; Domhnaill, S.C.Ó.; Neimark, A.V.; Schueth, F.; Unger, K.K. Capillary hysteresis in nanopores: Theoretical and experimental studies of nitrogen adsorption on MCM-41. Langmuir 1995, 11, 4765–4772. [Google Scholar] [CrossRef]
- Maddox, M.W.; Gubbins, K.E. Molecular simulation of fluid adsorption in buckytubes and MCM-41. Int. J. Thermophys. 1994, 15, 1115–1123. [Google Scholar] [CrossRef]
- Maddox, M.W.; Olivier, J.P.; Gubbins, K.E. Characterization of MCM-41 using molecular simulation: Heterogeneity effects. Langmuir 1997, 13, 1737–1745. [Google Scholar] [CrossRef]
- Koh, C.A.; Montanari, T.; Nooney, R.I.; Tahir, S.F.; Westacott, R.E. Experimental and computer simulation studies of the removal of carbon dioxide from mixtures with methane using AlPO4-5 and MCM-41. Langmuir 1999, 15, 6043–6049. [Google Scholar] [CrossRef]
- He, Y.; Seaton, N.A. Heats of adsorption and adsorption heterogeneity for methane, ethane, and carbon dioxide in MCM-41. Langmuir 2006, 22, 1150–1155. [Google Scholar] [CrossRef] [PubMed]
- Brodka, A.; Zerda, T.W. Properties of liquid acetone in silica pores: Molecular dynamics simulation. J. Chem. Phys. 1996, 104, 6319–6326. [Google Scholar] [CrossRef]
- Pellenq RJ, M.; Levitz, P.E. Capillary condensation in a disordered mesoporous medium: A grand canonical Monte Carlo study. Mol. Phys. 2002, 100, 2059–2077. [Google Scholar] [CrossRef]
- Gelb, L.D.; Gubbins, K.E. Characterization of porous glasses: Simulation models, adsorption isotherms, and the Brunauer–Emmett–Teller analysis method. Langmuir 1998, 14, 2097–2111. [Google Scholar] [CrossRef]
- Pérez-Sánchez, G.; Gomes, J.R.B.; Jorge, M. Modeling self-assembly of silica/surfactant mesostructures in the templated synthesis of nanoporous solids. Langmuir 2013, 29, 2387–2396. [Google Scholar] [CrossRef]
- Jing, Y.; Wei, L.; Wang, Y.; Yu, Y. Molecular simulation of MCM-41: Structural properties and adsorption of CO2, N2 and flue gas. Chem. Eng. J. 2013, 220, 264–275. [Google Scholar] [CrossRef]
- Ugliengo, P.; Sodupe, M.; Musso, F.; Bush, I.J.; Orlando, R.; Dovesi, R. Realistic models of hydroxylated amorphous silica surfaces and MCM-41 mesoporous material simulated by large-scale periodic B3LYP calculations. Adv. Mater. 2008, 20, 4579–4583. [Google Scholar] [CrossRef]
- Coasne, B.; Pellenq, R.J.M. Grand canonical Monte Carlo simulation of argon adsorption at the surface of silica nanopores: Effect of pore size, pore morphology, and surface roughness. J. Chem. Phys. 2004, 120, 2913–2922. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, S.; Huang, Y.; Hu, J.; Liu, H.; Hu, Y.; Jiang, J. Computer simulation for adsorption of CO2, N2 and flue gas in a mimetic MCM-41. J. Phys. Chem. C 2008, 112, 11295–11300. [Google Scholar] [CrossRef]
- Zhang, X.; Shao, X.; Wang, W.; Cao, D. Molecular modeling of selectivity of single-walled carbon nanotube and MCM-41 for separation of methane and carbon dioxide. Sep. Purif. Technol. 2010, 74, 280–287. [Google Scholar] [CrossRef]
- Bonnaud, P.A.; Coasne, B.; Pellenq, R.J.M. Molecular simulation of water confined in nanoporous silica. J. Phys. Condens. Matter 2010, 22, 284110. [Google Scholar] [CrossRef] [PubMed]
- Brás, A.R.; Fonseca, I.M.; Dionísio, M.; Schönhals, A.; Affouard, F.; Correia, N.T. Influence of nanoscale confinement on the molecular mobility of ibuprofen. J. Phys. Chem. C 2014, 118, 13857–13868. [Google Scholar] [CrossRef]
- Ghoufi, A.; Hureau, I.; Lefort, R.; Morineau, D. Hydrogen-bond-induced supermolecular assemblies in a nanoconfined tertiary alcohol. J. Phys. Chem. C 2011, 115, 17761–17767. [Google Scholar] [CrossRef]
- Herdes, C.; Ferreiro-Rangel, C.A.; Duren, T. Predicting neopentane isosteric enthalpy of adsorption at zero coverage in MCM-41. Langmuir 2011, 27, 6738–6743. [Google Scholar] [CrossRef]
- Lerbret, A.; Lelong, G.; Mason, P.E.; Saboungi, M.L.; Brady, J.W. Molecular dynamics and neutron scattering study of glucose solutions confined in MCM-41. J. Phys. Chem. B 2011, 115, 910–918. [Google Scholar] [CrossRef]
- Huang, J.; Meagher, M.M. Pervaporative recovery of n-butanol from aqueous solutions and ABE fermentation broth using thin-film silicalite-filled silicone composite membranes. J. Membr. Sci. 2001, 192, 231–242. [Google Scholar] [CrossRef]
- Coasne, B.; Fourkas, J.T. Structure and dynamics of benzene confined in silica nanopores. J. Phys. Chem. C 2011, 115, 15471–15479. [Google Scholar] [CrossRef]
- Li, Z.; Liu, Y.; Yang, X.; Xing, Y.; Yang, Q.; Yang, R.T. Adsorption thermodynamics and desorption properties of gaseous polycyclic aromatic hydrocarbons on mesoporous adsorbents. Adsorption 2017, 23, 361–371. [Google Scholar] [CrossRef]
- Li, Z.; Liu, Y.; Yang, X.; Xing, Y.; Tsai, C.J.; Meng, M.; Yang, R.T. Performance of mesoporous silicas and carbon in adsorptive removal of phenanthrene as a typical gaseous polycyclic aromatic hydrocarbon. Microporous Mesoporous Mater. 2017, 239, 9–18. [Google Scholar] [CrossRef]
- Yang, R.T. Gas Separation by Adsorption Processes; Butterworth-Heinemann: Oxford, UK, 2013. [Google Scholar]
- Moreno-Castilla, C. Adsorption of organic molecules from aqueous solutions on carbon materials. Carbon 2004, 42, 83–94. [Google Scholar] [CrossRef]
- Siperstein, F.R.; Gubbins, K.E. Synthesis and characterization of templated mesoporous materials using molecular simulation. Mol. Simul. 2001, 27, 339–352. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Z.; Yang, X.; Xing, Y.; Tsai, C.; Yang, Q.; Wang, Z.; Yang, R.T. Performance of mesoporous silicas (MCM-41 and SBA-15) and carbon (CMK-3) in the removal of gas-phase naphthalene: Adsorption capacity, rate and regenerability. RSC Adv. 2016, 6, 21193–21203. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adsorbate | S | S Values Corresponding States | |
|---|---|---|---|
| Nap | −0.1843 | 0 | disordered |
| Phe | −0.3568 | 1 | vertical |
| Pyr | −0.3629 | −0.5 | parallel |
| Type | Distance (Å) | ||
|---|---|---|---|
| Nap | Phe | Pyr | |
| OMCM41–HPAHs | 3.43 | 3.63 | 3.53 |
| HMCM41–CorePAHs | 4.48 | 4.43 | 4.38 |
| OMCM41–CorePAHs | 4.92 | 4.83 | 4.83 |
| OMCM41–HMCM41 | 0.94 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Zhang, C.; Jiang, L.; Li, Z.; Liu, Y.; Wang, H.; Xing, Y.; Yang, R.T. Molecular Simulation of Naphthalene, Phenanthrene, and Pyrene Adsorption on MCM-41. Int. J. Mol. Sci. 2019, 20, 665. https://doi.org/10.3390/ijms20030665
Yang X, Zhang C, Jiang L, Li Z, Liu Y, Wang H, Xing Y, Yang RT. Molecular Simulation of Naphthalene, Phenanthrene, and Pyrene Adsorption on MCM-41. International Journal of Molecular Sciences. 2019; 20(3):665. https://doi.org/10.3390/ijms20030665
Chicago/Turabian StyleYang, Xiong, Chuanzhao Zhang, Lijun Jiang, Ziyi Li, Yingshu Liu, Haoyu Wang, Yi Xing, and Ralph T. Yang. 2019. "Molecular Simulation of Naphthalene, Phenanthrene, and Pyrene Adsorption on MCM-41" International Journal of Molecular Sciences 20, no. 3: 665. https://doi.org/10.3390/ijms20030665
APA StyleYang, X., Zhang, C., Jiang, L., Li, Z., Liu, Y., Wang, H., Xing, Y., & Yang, R. T. (2019). Molecular Simulation of Naphthalene, Phenanthrene, and Pyrene Adsorption on MCM-41. International Journal of Molecular Sciences, 20(3), 665. https://doi.org/10.3390/ijms20030665