TUDCA-Treated Mesenchymal Stem Cells Protect against ER Stress in the Hippocampus of a Murine Chronic Kidney Disease Model


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
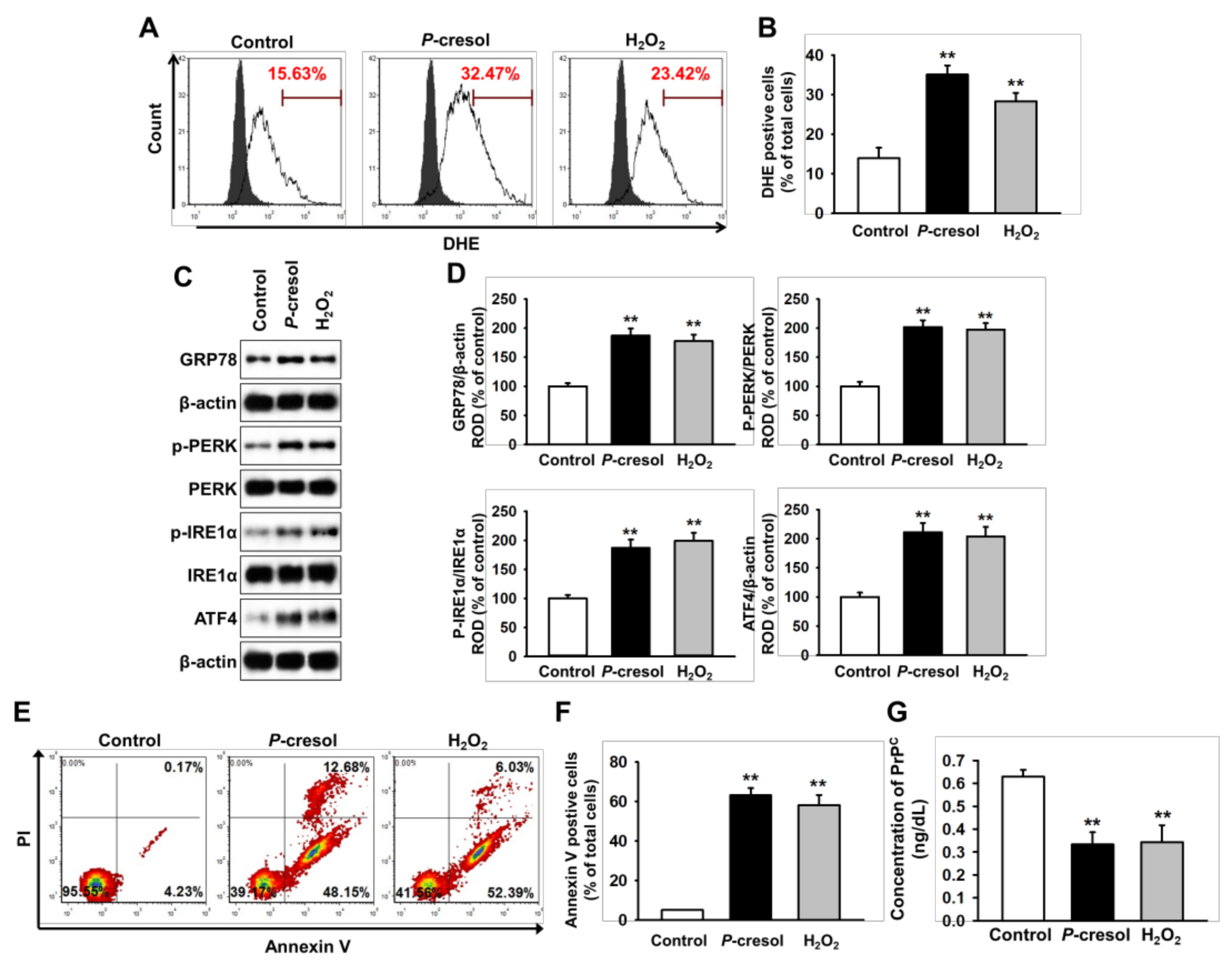
2.1. CKD Increases Neuronal Cell Death via Induction of ER Stress
2.2. Tudca-Stimulated CKD-hMSCS Protect SH-SY5Y Cells against Uremic Toxin-Induced Oxidative Stress
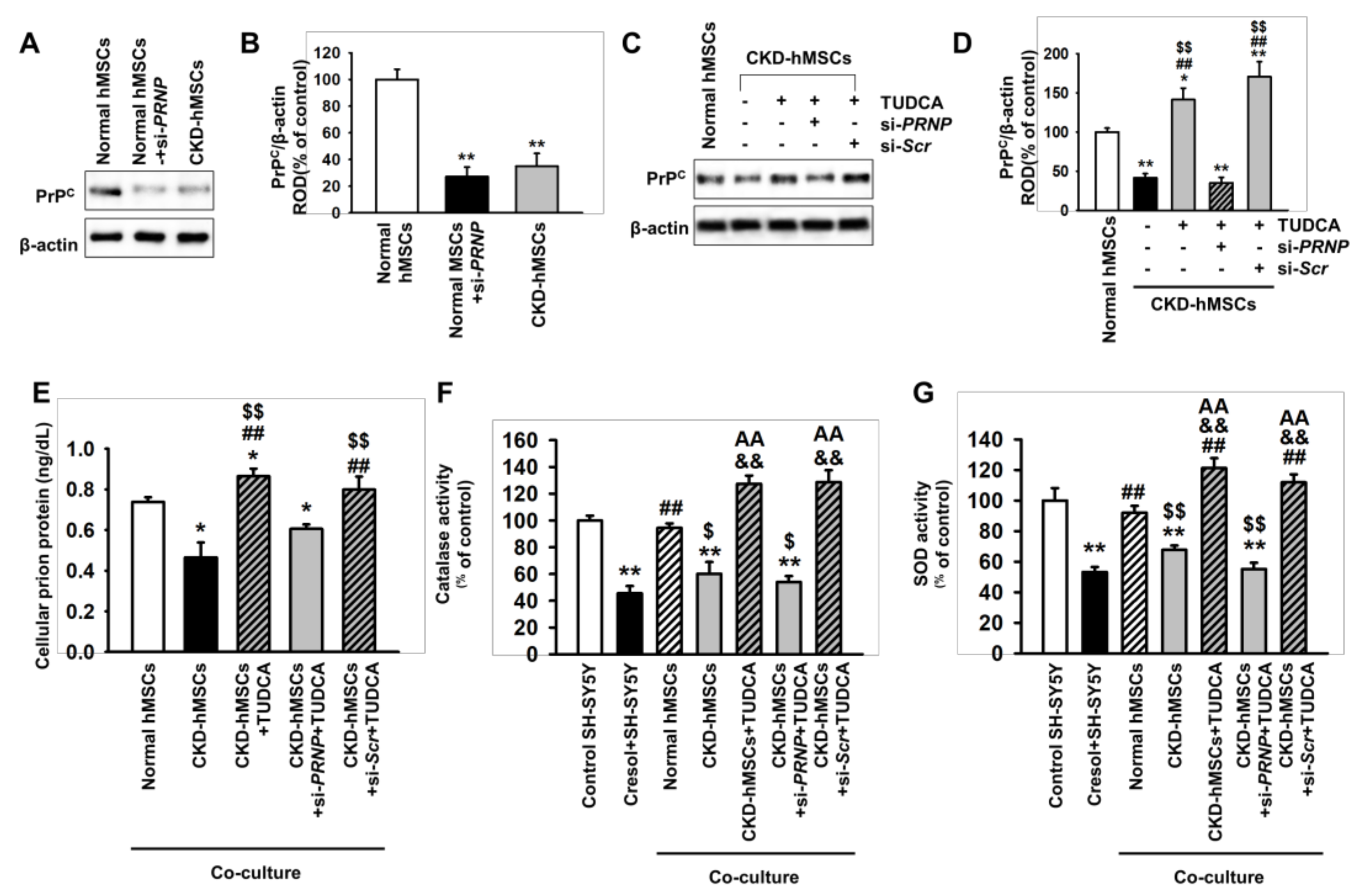
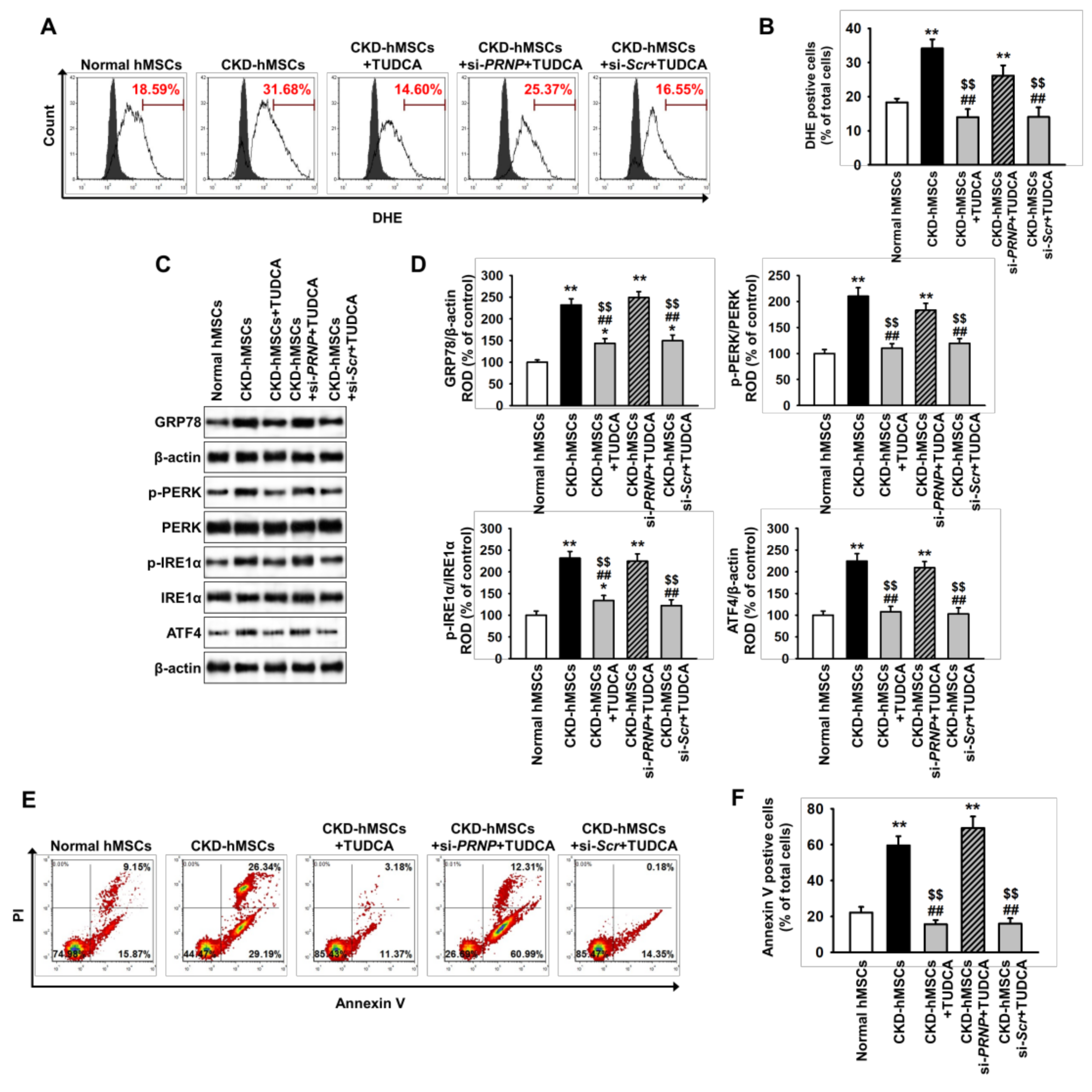
2.3. TUDCA-Treated CKD-hMSCs Suppress Uremic Toxin-Induced ER Stress in SH-SY5Y Cells via Upregulation of PrPC
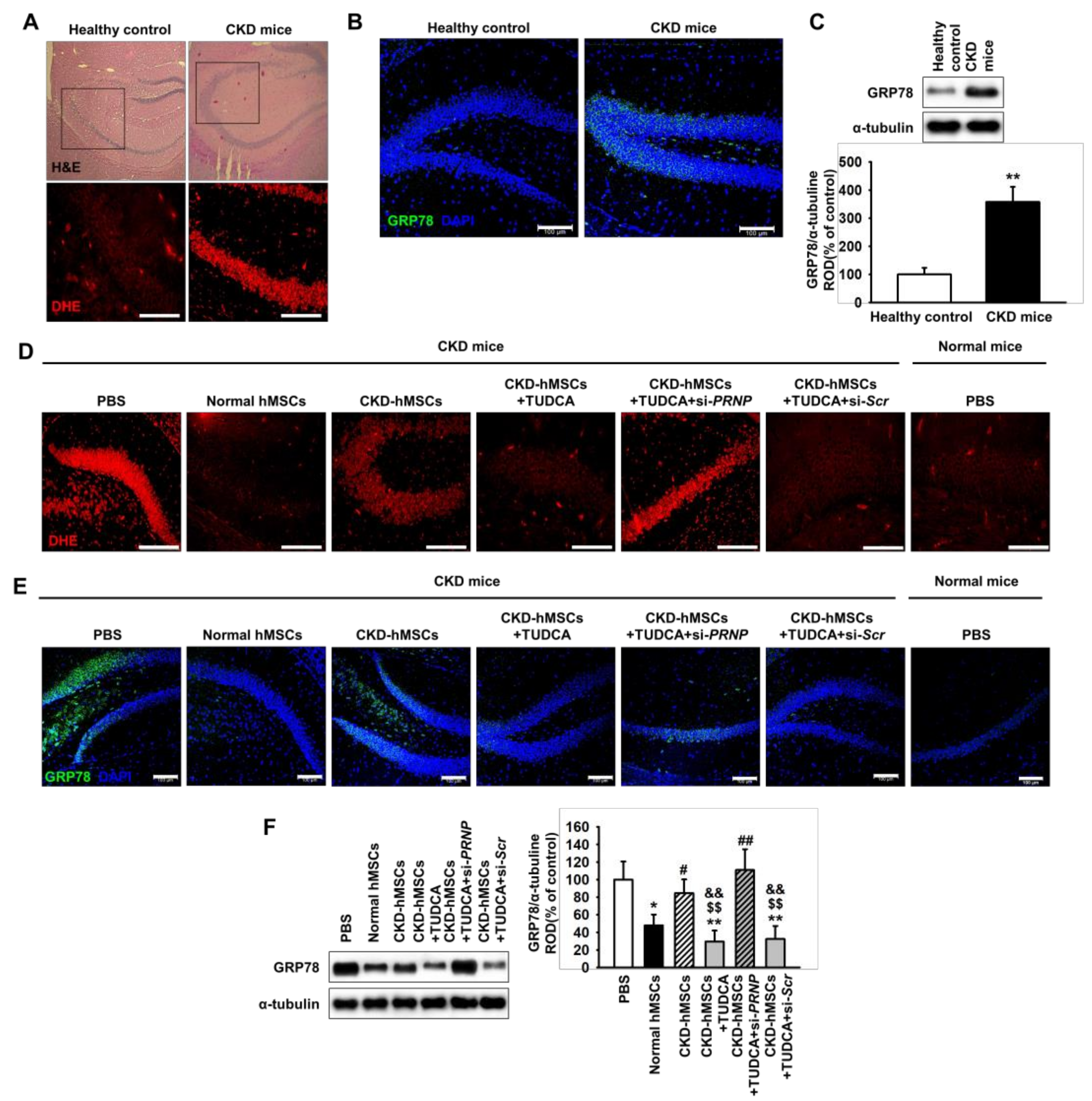
2.4. TUDCA-Treated CKD-hMSCs Prevent ROS-Mediated ER Stress in The Hippocampus of CKD Mice through Prpc Expression
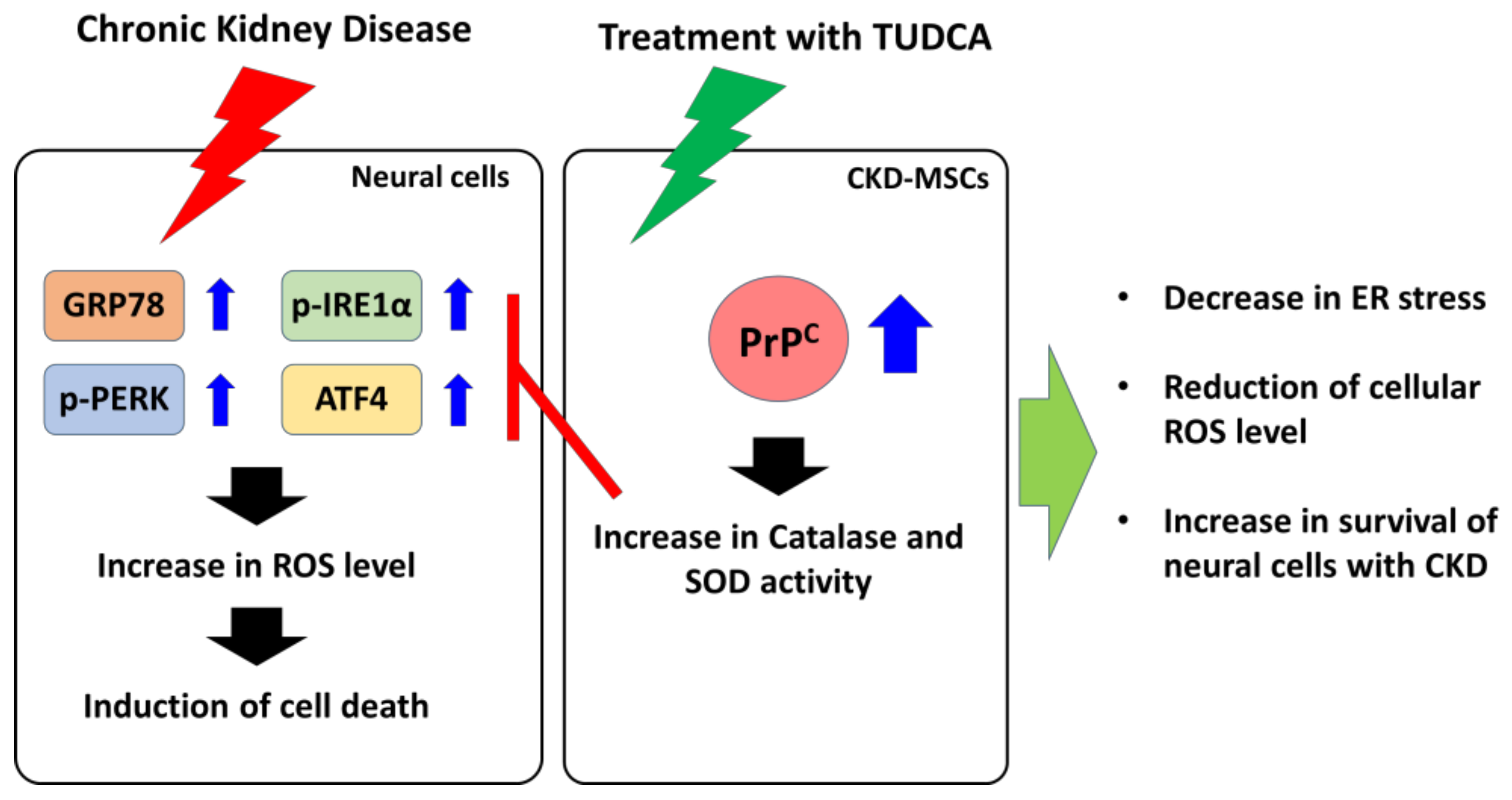
3. Discussion
4. Materials and Methods
4.1. Cell Culture of Human MSCs
4.2. Culture of SH-SY5Y Cells
4.3. Co-Culture of SH-SY5Y Cells with hMSCs
4.4. Western Blot Assay
4.5. Silencing of PrPC Expression by RNA Interference
4.6. Flow Cytometric Analysis
4.7. Immunohistochemistry
4.8. DHE Staining
4.9. Catalase activity
4.10. Superoxide Dismutase Activity
4.11. Ethics Statement
4.12. The CKD Model
4.13. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Peired, A.J.; Sisti, A.; Romagnani, P. Mesenchymal Stem Cell-Based Therapy for Kidney Disease: A Review of Clinical Evidence. Stem Cells Int. 2016, 2016, 4798639. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argiles, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Vaziri, N.D.; Jabbari, B.; Ni, Z.; Yan, X.X. Increased tyrosine nitration of the brain in chronic renal insufficiency: Reversal by antioxidant therapy and angiotensin-converting enzyme inhibition. J. Am. Soc. Nephrol. 2001, 12, 1892–1899. [Google Scholar] [PubMed]
- Fujisaki, K.; Tsuruya, K.; Yamato, M.; Toyonaga, J.; Noguchi, H.; Nakano, T.; Taniguchi, M.; Tokumoto, M.; Hirakata, H.; Kitazono, T. Cerebral oxidative stress induces spatial working memory dysfunction in uremic mice: Neuroprotective effect of tempol. Nephrol. Dial. Transpl. 2014, 29, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, M.L.; Bonan, N.B.; Dias, G.; Brehm, F.; Steiner, T.M.; Souza, W.M.; Stinghen, A.E.; Barreto, F.C.; Elifio-Esposito, S.; Pecoits-Filho, R.; et al. p-Cresyl sulfate affects the oxidative burst, phagocytosis process, and antigen presentation of monocyte-derived macrophages. Toxicol. Lett. 2016, 263, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yun, C.W.; Hur, J.; Lee, S.H. Fucoidan Rescues p-Cresol-Induced Cellular Senescence in Mesenchymal Stem Cells via FAK-Akt-TWIST Axis. Mar. Drugs 2018, 16, 121. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Yoon, Y.M.; Lee, J.H.; Kook, M.; Han, Y.S.; Jung, S.K.; Lee, S.H. Tauroursodeoxycholic Acid Protects against the Effects of P-Cresol-Induced Reactive Oxygen Species via the Expression of Cellular Prion Protein. Int. J. Mol. Sci. 2018, 19, 352. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.M.; Han, Y.S.; Yun, C.W.; Lee, J.H.; Kim, R.; Lee, S.H. Pioglitazone Protects Mesenchymal Stem Cells against P-Cresol-Induced Mitochondrial Dysfunction via Up-Regulation of PINK-1. Int. J. Mol. Sci. 2018, 19, 2898. [Google Scholar] [CrossRef]
- Lv, F.J.; Tuan, R.S.; Cheung, K.M.; Leung, V.Y. Concise review: The surface markers and identity of human mesenchymal stem cells. Stem Cells 2014, 32, 1408–1419. [Google Scholar] [CrossRef]
- Liang, X.; Ding, Y.; Zhang, Y.; Tse, H.F.; Lian, Q. Paracrine mechanisms of mesenchymal stem cell-based therapy: Current status and perspectives. Cell Transpl. 2014, 23, 1045–1059. [Google Scholar] [CrossRef]
- Westergard, L.; Christensen, H.M.; Harris, D.A. The cellular prion protein (PrP(C)): Its physiological function and role in disease. Biochim. Biophys. Acta 2007, 1772, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Han, Y.S.; Lee, S.H. Potentiation of biological effects of mesenchymal stem cells in ischemic conditions by melatonin via upregulation of cellular prion protein expression. J. Pineal Res. 2017, 62, e12385. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Han, Y.S.; Yoon, Y.M.; Yun, C.W.; Yun, S.P.; Kim, S.M.; Kwon, H.Y.; Jeong, D.; Baek, M.J.; Lee, H.J.; et al. Role of HSPA1L as a cellular prion protein stabilizer in tumor progression via HIF-1alpha/GP78 axis. Oncogene 2017, 36, 6555–6567. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yun, C.W.; Han, Y.S.; Kim, S.; Jeong, D.; Kwon, H.Y.; Kim, H.; Baek, M.J.; Lee, S.H. Melatonin and 5-fluorouracil co-suppress colon cancer stem cells by regulating cellular prion protein-Oct4 axis. J. Pineal Res. 2018, 65, e12519. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.S.; Yun, S.P.; Lee, J.H.; Kwon, S.H.; Kim, S.; Hur, J.; Lee, S.H. C-Met-Activated Mesenchymal Stem Cells Rescue Ischemic Damage via Interaction with Cellular Prion Protein. Cell. Physiol. Biochem. 2018, 46, 1835–1848. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, F.; Carpi, A.; Menabo, R.; Giorgio, M.; Schulz, R.; Valen, G.; Baysa, A.; Massimino, M.L.; Sorgato, M.C.; Bertoli, A.; et al. The cellular prion protein counteracts cardiac oxidative stress. Cardiovasc. Res. 2014, 104, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.M.; Lee, J.H.; Yun, S.P.; Han, Y.S.; Yun, C.W.; Lee, H.J.; Noh, H.; Lee, S.J.; Han, H.J.; Lee, S.H. Tauroursodeoxycholic acid reduces ER stress by regulating of Akt-dependent cellular prion protein. Sci. Rep. 2016, 6, 39838. [Google Scholar] [CrossRef]
- Cho, J.G.; Lee, J.H.; Hong, S.H.; Lee, H.N.; Kim, C.M.; Kim, S.Y.; Yoon, K.J.; Oh, B.J.; Kim, J.H.; Jung, S.Y.; et al. Tauroursodeoxycholic acid, a bile acid, promotes blood vessel repair by recruiting vasculogenic progenitor cells. Stem Cells 2015, 33, 792–805. [Google Scholar] [CrossRef]
- Impellizzeri, D.; Esposito, E.; Attley, J.; Cuzzocrea, S. Targeting inflammation: New therapeutic approaches in chronic kidney disease (CKD). Pharmacol. Res. 2014, 81, 91–102. [Google Scholar] [CrossRef]
- Inagi, R. Endoplasmic reticulum stress in the kidney as a novel mediator of kidney injury. Nephron Exp. Nephrol. 2009, 112, e1–e9. [Google Scholar] [CrossRef]
- Kosuge, Y.; Osada, N.; Shimomura, A.; Miyagishi, H.; Wada, T.; Ishige, K.; Shimba, S.; Ito, Y. Relevance of the hippocampal endoplasmic reticulum stress response in a mouse model of chronic kidney disease. Neurosci. Lett. 2018, 677, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Dokka, S.; Shi, X.; Leonard, S.; Wang, L.; Castranova, V.; Rojanasakul, Y. Interleukin-10-mediated inhibition of free radical generation in macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L1196–L1202. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, A.; Filesi, I.; Vetrugno, V.; Pocchiari, M.; Sy, M.S.; Biocca, S. Trapping prion protein in the endoplasmic reticulum impairs PrPC maturation and prevents PrPSc accumulation. J. Boil. Chem. 2005, 280, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.S.; Harris, D.A. A transmembrane form of the prion protein is localized in the Golgi apparatus of neurons. J. Boil. Chem. 2005, 280, 15855–15864. [Google Scholar] [CrossRef] [PubMed]
- Vang, S.; Longley, K.; Steer, C.J.; Low, W.C. The Unexpected Uses of Urso- and Tauroursodeoxycholic Acid in the Treatment of Non-liver Diseases. Glob. Adv. Health Med. 2014, 3, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Gronbeck, K.R.; Rodrigues, C.M.; Mahmoudi, J.; Bershad, E.M.; Ling, G.; Bachour, S.P.; Divani, A.A. Application of Tauroursodeoxycholic Acid for Treatment of Neurological and Non-neurological Diseases: Is There a Potential for Treating Traumatic Brain Injury? Neurocrit. Care 2016, 25, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.; Booth, S.A.; Beniac, D.R.; Coulthart, M.B.; Booth, T.F.; McNicol, A. Cellular prion protein is released on exosomes from activated platelets. Blood 2006, 107, 3907–3911. [Google Scholar] [CrossRef] [PubMed]
- Stahl, N.; Borchelt, D.R.; Hsiao, K.; Prusiner, S.B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987, 51, 229–240. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Groth, D.F.; Bolton, D.C.; Kent, S.B.; Hood, L.E. Purification and structural studies of a major scrapie prion protein. Cell 1984, 38, 127–134. [Google Scholar] [CrossRef]
- Black, S.A.; Stys, P.K.; Zamponi, G.W.; Tsutsui, S. Cellular prion protein and NMDA receptor modulation: Protecting against excitotoxicity. Front. Cell Dev. Biol 2014, 2, 45. [Google Scholar] [CrossRef]
- Shi, F.; Yang, Y.; Wang, T.; Kouadir, M.; Zhao, D.; Hu, S. Cellular Prion Protein Promotes Neuronal Differentiation of Adipose-Derived Stem Cells by Upregulating miRNA-124. J. Mol. Neurosci. 2016, 59, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.H.; Santos, T.G. Prion potency in stem cells biology. Prion 2012, 6, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Dounousi, E.; Papavasiliou, E.; Makedou, A.; Ioannou, K.; Katopodis, K.P.; Tselepis, A.; Siamopoulos, K.C.; Tsakiris, D. Oxidative stress is progressively enhanced with advancing stages of CKD. Am. J. Kidney Dis. 2006, 48, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Shyu, W.C.; Lin, S.Z.; Chiang, M.F.; Ding, D.C.; Li, K.W.; Chen, S.F.; Yang, H.I.; Li, H. Overexpression of PrPC by adenovirus-mediated gene targeting reduces ischemic injury in a stroke rat model. J. Neurosci. 2005, 25, 8967–8977. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Nicholas, R.S.; Canevari, L. Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J. Neurosci. Res. 2002, 67, 211–224. [Google Scholar] [CrossRef]
- Weise, J.; Sandau, R.; Schwarting, S.; Crome, O.; Wrede, A.; Schulz-Schaeffer, W.; Zerr, I.; Bahr, M. Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase-3 activation, and exacerbation of ischemic brain injury. Stroke 2006, 37, 1296–1300. [Google Scholar] [CrossRef]
- Yang, H.C.; Zuo, Y.; Fogo, A.B. Models of chronic kidney disease. Drug Discov. Today Dis. Models 2010, 7, 13–19. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.H.; Yoon, Y.M.; Lee, S.H. TUDCA-Treated Mesenchymal Stem Cells Protect against ER Stress in the Hippocampus of a Murine Chronic Kidney Disease Model. Int. J. Mol. Sci. 2019, 20, 613. https://doi.org/10.3390/ijms20030613
Lee JH, Yoon YM, Lee SH. TUDCA-Treated Mesenchymal Stem Cells Protect against ER Stress in the Hippocampus of a Murine Chronic Kidney Disease Model. International Journal of Molecular Sciences. 2019; 20(3):613. https://doi.org/10.3390/ijms20030613
Chicago/Turabian StyleLee, Jun Hee, Yeo Min Yoon, and Sang Hun Lee. 2019. "TUDCA-Treated Mesenchymal Stem Cells Protect against ER Stress in the Hippocampus of a Murine Chronic Kidney Disease Model" International Journal of Molecular Sciences 20, no. 3: 613. https://doi.org/10.3390/ijms20030613
APA StyleLee, J. H., Yoon, Y. M., & Lee, S. H. (2019). TUDCA-Treated Mesenchymal Stem Cells Protect against ER Stress in the Hippocampus of a Murine Chronic Kidney Disease Model. International Journal of Molecular Sciences, 20(3), 613. https://doi.org/10.3390/ijms20030613