Molecular Insights into the Classification of Luminal Breast Cancers: The Genomic Heterogeneity of Progesterone-Negative Tumors


Abstract
1. Introduction
2. Results
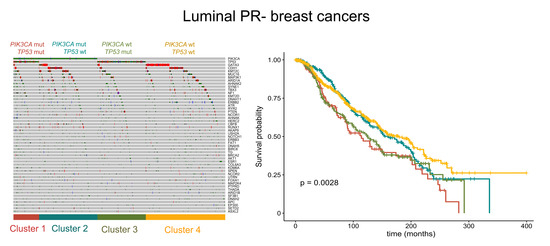
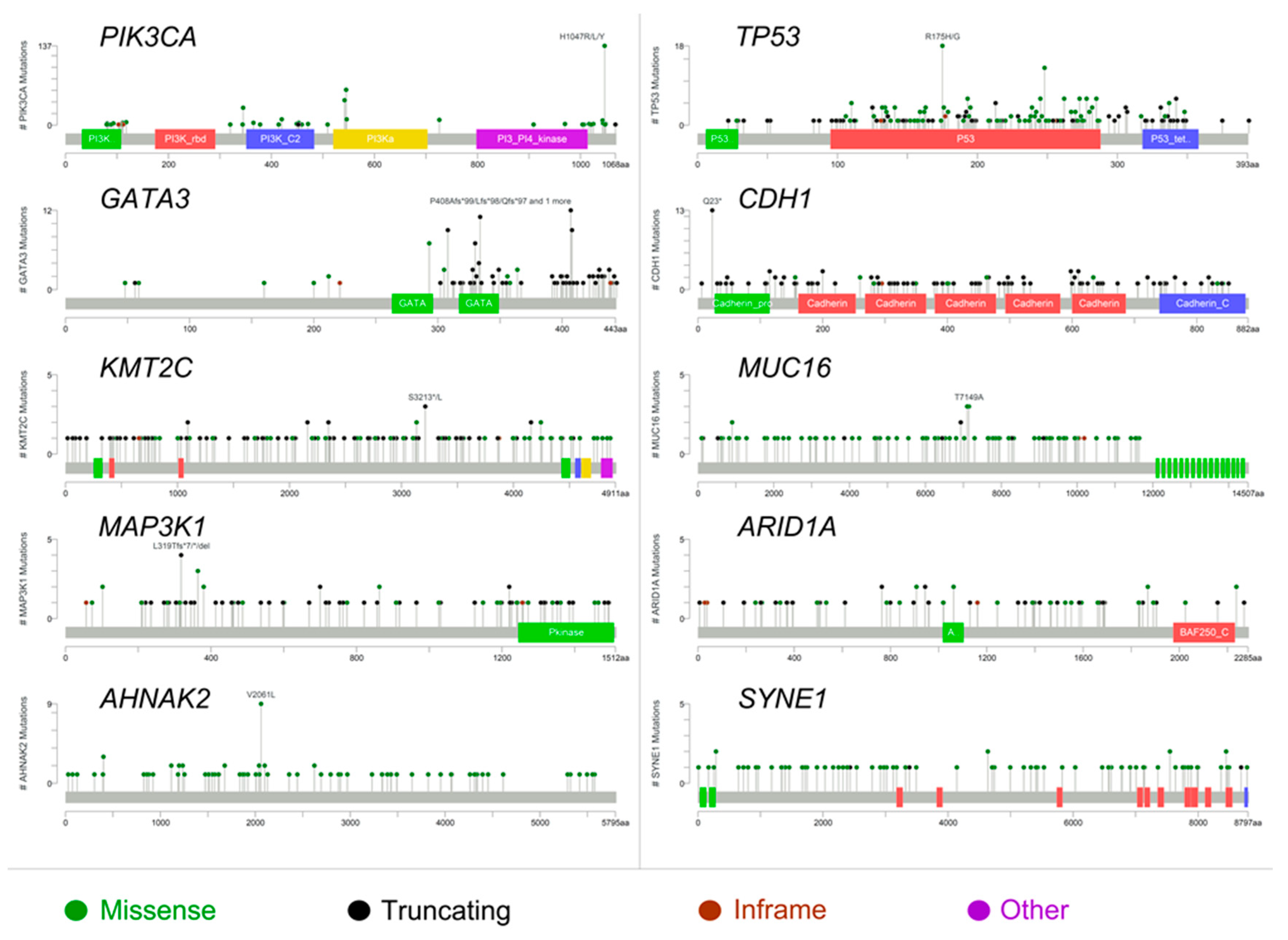
2.1. The Molecular Landscape of ER+/PR− Breast Cancers
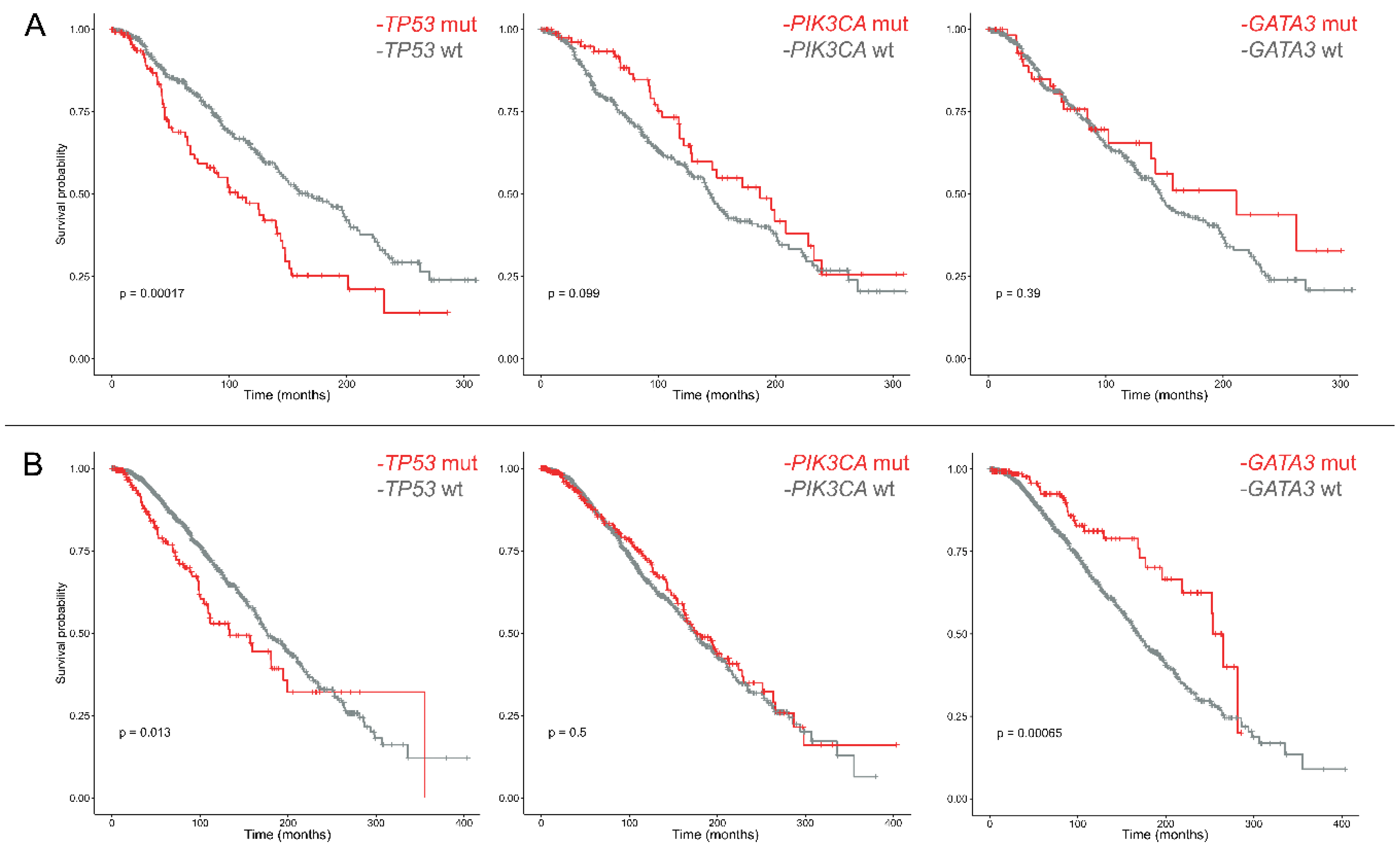
2.2. The Prognostic Role of PIK3CA and TP53 in ER+/PR− Breast Cancers
3. Discussion
4. Materials and Methods
4.1. Case Selection and Definitions
4.2. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thakkar, J.P.; Mehta, D.G. A review of an unfavorable subset of breast cancer: Estrogen receptor positive progesterone receptor negative. Oncologist 2011, 16, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Arpino, G.; Weiss, H.; Lee, A.V.; Schiff, R.; De Placido, S.; Osborne, C.K.; Elledge, R.M. Estrogen receptor-positive, progesterone receptor-negative breast cancer: Association with growth factor receptor expression and tamoxifen resistance. J. Natl. Cancer Inst. 2005, 97, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- WHO Classification of Tumours of the Breast; International Agency for Research on Cancer: Lyon, France, 2012.
- Purdie, C.A.; Quinlan, P.; Jordan, L.B.; Ashfield, A.; Ogston, S.; Dewar, J.A.; Thompson, A.M. Progesterone receptor expression is an independent prognostic variable in early breast cancer: A population-based study. Br. J. Cancer 2014, 110, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Van Mackelenbergh, M.T.; Denkert, C.; Nekljudova, V.; Karn, T.; Schem, C.; Marme, F.; Stickeler, E.; Jackisch, C.; Hanusch, C.; Huober, J.; et al. Outcome after neoadjuvant chemotherapy in estrogen receptor-positive and progesterone receptor-negative breast cancer patients: A pooled analysis of individual patient data from ten prospectively randomized controlled neoadjuvant trials. Breast Cancer Res. Treat. 2018, 167, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.D.; Liu, G.Y.; Di, G.H.; Wu, J.; Lu, J.S.; Shen, K.W.; Shen, Z.Z.; Shao, Z.M. Progesterone receptor status provides predictive value for adjuvant endocrine therapy in older estrogen receptor-positive breast cancer patients. Breast 2007, 16, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Neven, P.; Pochet, N.; Drijkoningen, M.; Amant, F.; De Smet, F.; Paridaens, R.; Christiaens, M.R.; Vergote, I. Progesterone receptor in estrogen receptor-positive breast cancer: The association between her-2 and lymph node involvement is age related. J. Clin. Oncol. 2006, 24, 2595–2597. [Google Scholar] [CrossRef] [PubMed]
- Viale, G.; Regan, M.M.; Maiorano, E.; Mastropasqua, M.G.; Dell’Orto, P.; Rasmussen, B.B.; Raffoul, J.; Neven, P.; Orosz, Z.; Braye, S.; et al. Prognostic and predictive value of centrally reviewed expression of estrogen and progesterone receptors in a randomized trial comparing letrozole and tamoxifen adjuvant therapy for postmenopausal early breast cancer: Big 1-98. J. Clin. Oncol. 2007, 25, 3846–3852. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, I.A.; Truong, P.T.; Speers, C.H.; Bernstein, V.; Allan, S.J.; Kelly, S.J.; Lesperance, M.L. Time to stop progesterone receptor testing in breast cancer management. J. Clin. Oncol. 2004, 22, 1769–1770. [Google Scholar] [CrossRef] [PubMed]
- Burstein, H.J.; Prestrud, A.A.; Seidenfeld, J.; Anderson, H.; Buchholz, T.A.; Davidson, N.E.; Gelmon, K.E.; Giordano, S.H.; Hudis, C.A.; Malin, J.; et al. American society of clinical oncology clinical practice guideline: Update on adjuvant endocrine therapy for women with hormone receptor-positive breast cancer. J. Clin. Oncol. 2010, 28, 3784–3796. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Shou, J.; Massarweh, S.; Schiff, R. Crosstalk between estrogen receptor and growth factor receptor pathways as a cause for endocrine therapy resistance in breast cancer. Clin. Cancer Res. 2005, 11, 865s–870s. [Google Scholar]
- Cui, X.; Schiff, R.; Arpino, G.; Osborne, C.K.; Lee, A.V. Biology of progesterone receptor loss in breast cancer and its implications for endocrine therapy. J. Clin. Oncol. 2005, 23, 7721–7735. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Kent Osborne, C.; van de Vijver, M.J.; Foekens, J.A.; Klijn, J.G.; Horlings, H.M.; Nuyten, D.; Wang, Y.; Zhang, Y.; Chamness, G.C.; et al. Molecular profiles of progesterone receptor loss in human breast tumors. Breast Cancer Res. Treat. 2009, 114, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Maggi, A. Liganded and unliganded activation of estrogen receptor and hormone replacement therapies. Biochim. Biophys. Acta 2011, 1812, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jia, M.; Qin, X.; Hu, J.; Zhang, X.; Zhou, G. Harmful effect of erbeta on bcrp-mediated drug resistance and cell proliferation in eralpha/PR−negative breast cancer. FEBS J. 2013, 280, 6128–6140. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Tyson, J.J.; Dixon, J.M. Endocrine resistance in breast cancer--an overview and update. Mol. Cell Endocrinol. 2015, 418 Pt 3, 220–234. [Google Scholar] [CrossRef]
- Johnston, S.R.; Saccani-Jotti, G.; Smith, I.E.; Salter, J.; Newby, J.; Coppen, M.; Ebbs, S.R.; Dowsett, M. Changes in estrogen receptor, progesterone receptor, and ps2 expression in tamoxifen-resistant human breast cancer. Cancer Res. 1995, 55, 3331–3338. [Google Scholar] [PubMed]
- Thomas, C.; Gustafsson, J.A. Progesterone receptor-estrogen receptor crosstalk: A novel insight. Trends Endocrinol. Metab. 2015, 26, 453–454. [Google Scholar] [CrossRef] [PubMed]
- Piscuoglio, S.; Ng, C.K.; Murray, M.P.; Guerini-Rocco, E.; Martelotto, L.G.; Geyer, F.C.; Bidard, F.C.; Berman, S.; Fusco, N.; Sakr, R.A.; et al. The genomic landscape of male breast cancers. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Fusco, N.; Geyer, F.C.; De Filippo, M.R.; Martelotto, L.G.; Ng, C.K.; Piscuoglio, S.; Guerini-Rocco, E.; Schultheis, A.M.; Fuhrmann, L.; Wang, L.; et al. Genetic events in the progression of adenoid cystic carcinoma of the breast to high-grade triple-negative breast cancer. Mod. Pathol. 2016, 29, 1292–1305. [Google Scholar] [CrossRef]
- Kim, J.; Geyer, F.C.; Martelotto, L.G.; Ng, C.K.Y.; Lim, R.S.; Selenica, P.; Li, A.; Pareja, F.; Fusco, N.; Edelweiss, M.; et al. Mybl1 rearrangements and myb amplification in breast adenoid cystic carcinomas lacking the myb-nfib fusion gene. J. Pathol. 2018, 244, 143–150. [Google Scholar] [CrossRef]
- Fusco, N.; Colombo, P.E.; Martelotto, L.G.; De Filippo, M.R.; Piscuoglio, S.; Ng, C.K.; Lim, R.S.; Jacot, W.; Vincent-Salomon, A.; Reis-Filho, J.S.; et al. Resolving quandaries: Basaloid adenoid cystic carcinoma or breast cylindroma? The role of massively parallel sequencing. Histopathology 2016, 68, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Marchiò, C.; De Filippo, M.R.; Ng, C.K.; Piscuoglio, S.; Soslow, R.A.; Reis-Filho, J.S.; Weigelt, B. Piking the type and pattern of pi3k pathway mutations in endometrioid endometrial carcinomas. Gynecol. Oncol. 2015, 137, 321–328. [Google Scholar] [CrossRef] [PubMed]
- De Mattos-Arruda, L.; Bidard, F.C.; Won, H.H.; Cortes, J.; Ng, C.K.; Peg, V.; Nuciforo, P.; Jungbluth, A.A.; Weigelt, B.; Berger, M.F.; et al. Establishing the origin of metastatic deposits in the setting of multiple primary malignancies: The role of massively parallel sequencing. Mol. Oncol. 2014, 8, 150–158. [Google Scholar] [CrossRef]
- Ng, C.K.; Pemberton, H.N.; Reis-Filho, J.S. Breast cancer intratumor genetic heterogeneity: Causes and implications. Expert. Rev. Anticancer Ther. 2012, 12, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Fusco, N.; Lopez, G.; Corti, C.; Pesenti, C.; Colapietro, P.; Ercoli, G.; Gaudioso, G.; Faversani, A.; Gambini, D.; Michelotti, A.; et al. Mismatch repair protein loss as a prognostic and predictive biomarker in breast cancers regardless of microsatellite instability. JNCI Cancer Spectrum 2018, 2, pky056. [Google Scholar] [CrossRef]
- Ercoli, G.; Lopez, G.; Ciapponi, C.; Corti, C.; Despini, L.; Gambini, D.; Runza, L.; Blundo, C.; Sciarra, A.; Fusco, N. Building up a high-throughput screening platform to assess the heterogeneity of HER2 gene amplification in breast cancers. J. Vis. Exp. 2017, 13, 233–236. [Google Scholar] [CrossRef]
- Fusco, N.; Bosari, S. HER2 aberrations and heterogeneity in cancers of the digestive system: Implications for pathologists and gastroenterologists. World J. Gastroenterol. 2016, 22, 7926–7937. [Google Scholar] [CrossRef]
- Vuong, D.; Simpson, P.T.; Green, B.; Cummings, M.C.; Lakhani, S.R. Molecular classification of breast cancer. Virchows Arch. 2014, 465, 1–14. [Google Scholar] [CrossRef]
- Russnes, H.G.; Lingjaerde, O.C.; Borresen-Dale, A.L.; Caldas, C. Breast cancer molecular stratification: From intrinsic subtypes to integrative clusters. Am. J. Pathol. 2017, 187, 2152–2162. [Google Scholar] [CrossRef]
- Chakrabarty, A.; Bhola, N.E.; Sutton, C.; Ghosh, R.; Kuba, M.G.; Dave, B.; Chang, J.C.; Arteaga, C.L. Trastuzumab-resistant cells rely on a her2-pi3k-foxo-survivin axis and are sensitive to pi3k inhibitors. Cancer Res. 2013, 73, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Garbe, C.; Abusaif, S.; Eigentler, T.K. Vemurafenib. Recent Results Cancer Res. 2014, 201, 215–225. [Google Scholar] [PubMed]
- Fumagalli, C.; Bianchi, F.; Raviele, P.R.; Vacirca, D.; Bertalot, G.; Rampinelli, C.; Lazzeroni, M.; Bonanni, B.; Veronesi, G.; Fusco, N.; et al. Circulating and tissue biomarkers in early-stage non-small cell lung cancer. Ecancermedicalscience 2017, 11, 717. [Google Scholar] [CrossRef] [PubMed]
- Fda Approved Drug: Neratinib. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208051s000lbl.pdf (accessed on 15 December 2018).
- Miettinen, M.; McCue, P.A.; Sarlomo-Rikala, M.; Rys, J.; Czapiewski, P.; Wazny, K.; Langfort, R.; Waloszczyk, P.; Biernat, W.; Lasota, J.; et al. Gata3: A multispecific but potentially useful marker in surgical pathology: A systematic analysis of 2500 epithelial and nonepithelial tumors. Am. J. Surg. Pathol. 2014, 38, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.K.; Schultheis, A.M.; Bidard, F.C.; Weigelt, B.; Reis-Filho, J.S. Breast cancer genomics from microarrays to massively parallel sequencing: Paradigms and new insights. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Silwal-Pandit, L.; Vollan, H.K.; Chin, S.F.; Rueda, O.M.; McKinney, S.; Osako, T.; Quigley, D.A.; Kristensen, V.N.; Aparicio, S.; Borresen-Dale, A.L.; et al. Tp53 mutation spectrum in breast cancer is subtype specific and has distinct prognostic relevance. Clin. Cancer Res. 2014, 20, 3569–3580. [Google Scholar] [CrossRef] [PubMed]
- Cran—Package Cgdsr. Available online: https://cran.r-project.org/web/packages/cgdsr/index.html (accessed on 15 December 2018).
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 15 December 2018).
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell 2018, 34, 427–438. [Google Scholar] [CrossRef]
- Cran—Package Gplots. Available online: https://cran.r-project.org/web/packages/gplots/index.html (accessed on 15 December 2018).
- Wickham, H. Ggplot2—Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2018. [Google Scholar]
- Cran—Package Survival. Available online: https://cran.r-project.org/web/packages/survival/index.html (accessed on 15 December 2018).
- Kaplan, E.L.; Meier, P. Nonparametric estimation from incomplete observations. J. Am. Stat. Assoc. 1958, 53, 457–481. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TCGA (%) | MSK (%) | METABRIC (%) | |
|---|---|---|---|
| PR− (n = 959) | 110 (12) | 396 (41) | 453 (47) |
| PR+ (n = 2611) | 608 (23) | 1031 (40) | 972 (37) |
| Total (n = 3570) | 718 (20) | 1427 (40) | 1425 (40) |
| Mutation | PR+ (%) | PR− (%) | p Value |
|---|---|---|---|
| ARID1A_Q766SfsX67 | 0 | 2 (0.20) | 0.019 |
| ATR_A14S | 0 | 2 (0.20) | 0.019 |
| BCL6_K474EfsX26 | 0 | 2 (0.20) | 0.019 |
| BRAF_V600E | 0 | 2 (0.20) | 0.019 |
| CARD11_D200E | 0 | 2 (0.20) | 0.019 |
| CDH1_R598X | 0 | 2 (0.20) | 0.019 |
| CDH1_E138X | 0 | 2 (0.20) | 0.019 |
| CDH1_E497RfsX25 | 0 | 2 (0.20) | 0.019 |
| AXIN2_S493L | 0 | 3 (0.31) | 0.004 |
| GATA3_R364T | 0 | 3 (0.31) | 0.005 |
| CDH1_V202CfsX7 | 0 | 3 (0.31) | 0.006 |
| MUC16_T7149A | 0 | 3 (0.31) | 0.007 |
| CCDC82_E175del | 0 | 3 (0.31) | 0.008 |
| RUNX1_D123GfsX15 | 0 | 4 (0.41) | <0.001 |
| TBX3_W113X | 0 | 4 (0.41) | <0.001 |
| CDH1_T115NfsX53 | 1 (0.04) | 3 (0.31) | 0.029 |
| FOXA1_D226N | 1 (0.04) | 3 (0.31) | 0.029 |
| FOXA1_I176M | 1 (0.04) | 3 (0.31) | 0.029 |
| GATA3_X444LfsX63 | 1 (0.04) | 3 (0.31) | 0.029 |
| TERT_Promoter | 1 (0.04) | 3 (0.31) | 0.029 |
| TP53_P278S | 1 (0.04) | 3 (0.31) | 0.029 |
| SMAD4_Q245X | 1 (0.04) | 3 (0.31) | 0.029 |
| TP53_I195T | 1 (0.04) | 5 (0.52) | 0.002 |
| ERBB2_E770_A771insGIRD | 1 (0.04) | 8 (0.83) | 0.003 |
| ERBB2_S310F | 2 (0.08) | 4 (0.41) | 0.027 |
| MAP3K1_R364W | 2 (0.08) | 4 (0.41) | 0.027 |
| TP53_H179R | 2 (0.08) | 4 (0.41) | 0.027 |
| TP53_R342X | 5 (0.19) | 7 (0.72) | 0.013 |
| GATA3_D335GfsX17 | 16 (0.61) | 13 (1.35) | 0.028 |
| TP53_R175H | 21 (0.80) | 18 (1.87) | 0.006 |
| ESR1_Y537S | 29 (1.11) | 3 (0.31) | 0.024 |
| ESR1_D538G | 47 (1.80) | 7 (0.72) | 0.020 |
| SF3B1_K700E | 60 (2.29) | 10 (1.04) | 0.016 |
| GATA3_X308_splice | 70 (2.68) | 9 (0.94) | 0.002 |
| AKT1_E17K | 106 (4.05) | 25 (2.60) | 0.04 |
| PIK3CA_E545K | 251 (9.61) | 68 (7.09) | 0.019 |
| PIK3CA_H1047R | 482 (18.46) | 134 (13.97) | 0.002 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopez, G.; Costanza, J.; Colleoni, M.; Fontana, L.; Ferrero, S.; Miozzo, M.; Fusco, N. Molecular Insights into the Classification of Luminal Breast Cancers: The Genomic Heterogeneity of Progesterone-Negative Tumors. Int. J. Mol. Sci. 2019, 20, 510. https://doi.org/10.3390/ijms20030510
Lopez G, Costanza J, Colleoni M, Fontana L, Ferrero S, Miozzo M, Fusco N. Molecular Insights into the Classification of Luminal Breast Cancers: The Genomic Heterogeneity of Progesterone-Negative Tumors. International Journal of Molecular Sciences. 2019; 20(3):510. https://doi.org/10.3390/ijms20030510
Chicago/Turabian StyleLopez, Gianluca, Jole Costanza, Matteo Colleoni, Laura Fontana, Stefano Ferrero, Monica Miozzo, and Nicola Fusco. 2019. "Molecular Insights into the Classification of Luminal Breast Cancers: The Genomic Heterogeneity of Progesterone-Negative Tumors" International Journal of Molecular Sciences 20, no. 3: 510. https://doi.org/10.3390/ijms20030510
APA StyleLopez, G., Costanza, J., Colleoni, M., Fontana, L., Ferrero, S., Miozzo, M., & Fusco, N. (2019). Molecular Insights into the Classification of Luminal Breast Cancers: The Genomic Heterogeneity of Progesterone-Negative Tumors. International Journal of Molecular Sciences, 20(3), 510. https://doi.org/10.3390/ijms20030510