Vasoprotective Functions of High-Density Lipoproteins Relevant to Alzheimer’s Disease Are Partially Conserved in Apolipoprotein B-Depleted Plasma

 ,
,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
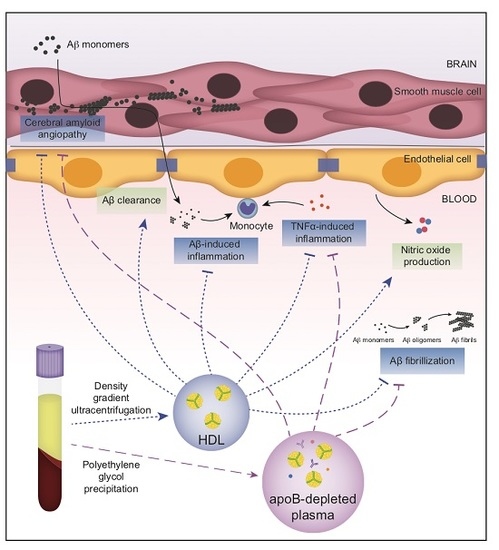
1. Introduction
2. Results
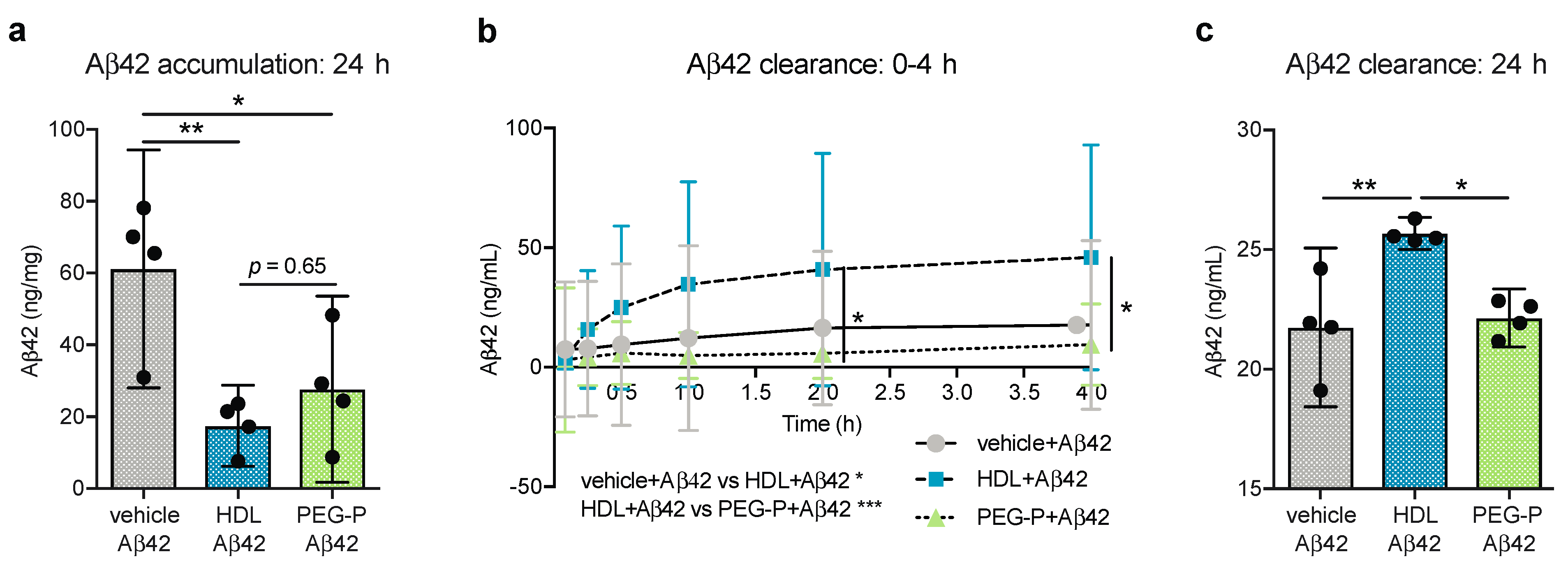
2.1. ApoB-Depleted Plasma Reduces Aβ Accumulation in Bioengineered Vessels Similar to High-Density Lipoprotein (HDL) Isolated by Ultracentrifugation But Does Not Effectively Increase Aβ Transport across the Vessel Wall
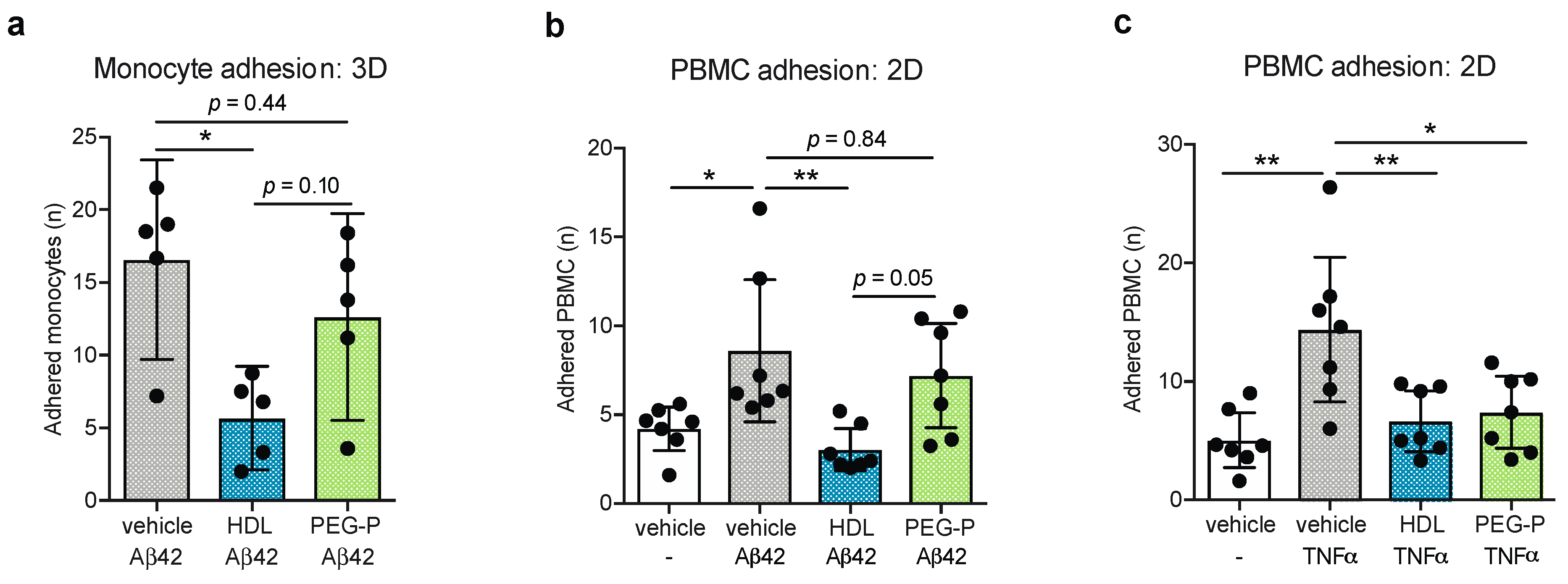
2.2. ApoB-Depleted Plasma Does Not Retain the Ability of HDL to Reduce Aβ-Induced Monocyte Binding to the Endothelium
2.3. ApoB-Depleted Plasma Is Functionally Equivalent to HDL with Respect to Delaying Aβ42 Fibrillization
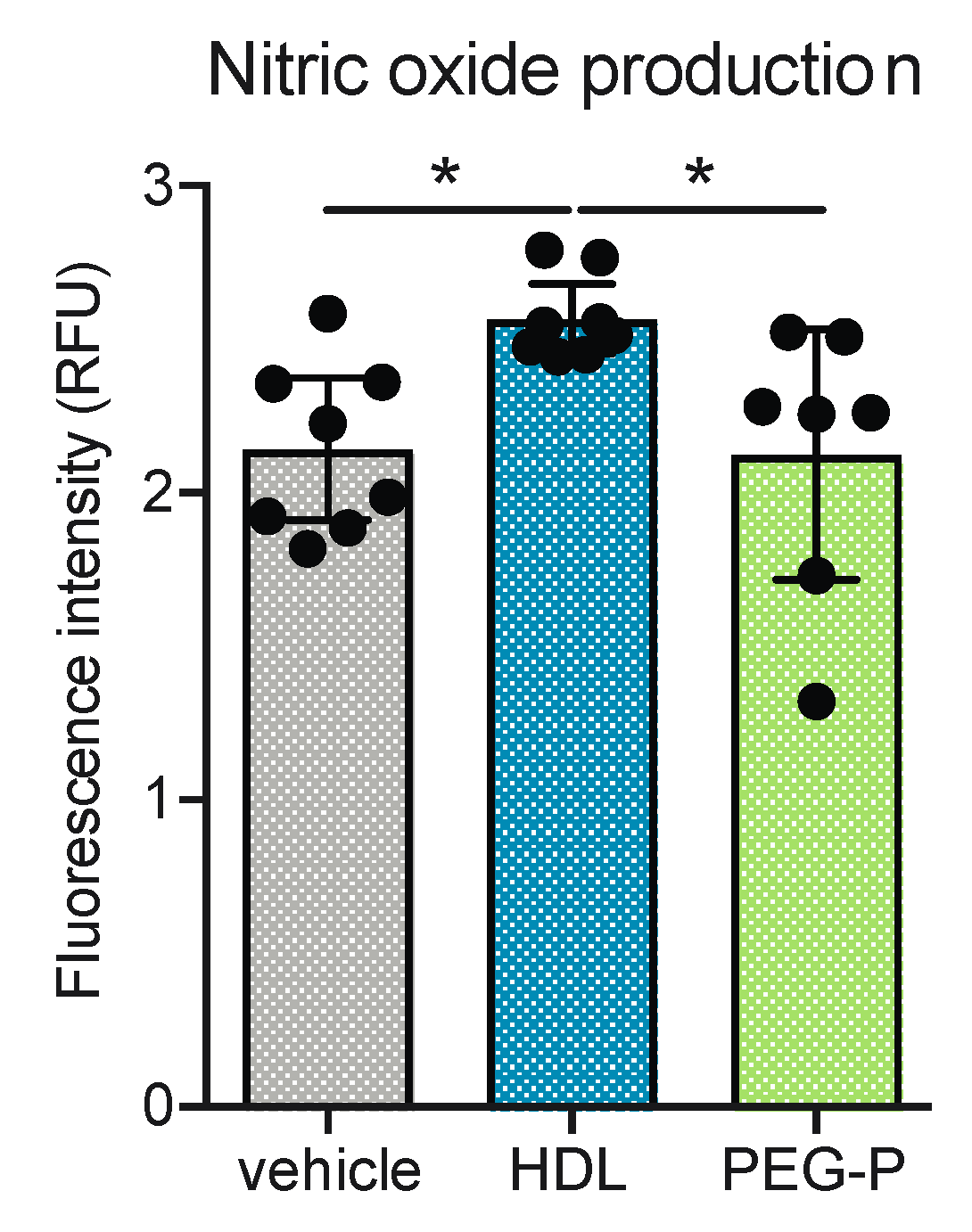
2.4. ApoB-Depleted Plasma Does Not Induce Nitric Oxide (NO) in Human Brain-Derived Endothelial Cells
2.5. Non-HDL Plasma Components in ApoB-Depleted Plasma Interfere with Some Anti-Inflammatory Activities of Purified HDL
3. Discussion
4. Materials and Methods
4.1. Blood Collection
4.2. HDL Isolation
4.3. Enzyme-Linked Immunosorbent Assay (ELISA)
4.4. Cell Culture
4.5. Fabrication of 3D Bioengineered Vessels
4.6. Nitric Oxide Production
4.7. Preparation of Aβ Monomers
4.8. Anti-Inflammatory Function
4.9. Cell-Free Assay of Aβ Fibrillization
4.10. Amyloid Beta Clearance and Accumulation Assays Using Engineered Vessels
4.11. Amyloid Beta Induced Monocyte Adhesion Using Engineered Vessels
4.12. Characterization of HDL Preparations
4.13. Gel Electrophoresis
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Patterson, C. World Alzheimer Report 2018. The State of the Art of Dementia Research: New Frontiers; Alzheimer’s Disease International: London, UK, 2018. [Google Scholar]
- Attems, J.; Jellinger, K.A. The overlap between vascular disease and Alzheimer’s disease—Lessons from pathology. BMC Med. 2014, 12, 206. [Google Scholar] [CrossRef] [PubMed]
- Cupino, T.L.; Zabel, M.K. Alzheimer’s Silent Partner: Cerebral Amyloid Angiopathy. Transl. Stoke Res. 2014, 5, 330–337. [Google Scholar] [CrossRef]
- Corriveau, R.A.; Gladman, J.T.; Jeon, S.; Babcock, D.; Bennett, D.A.; Carmichael, S.T.; Dickinson, S.L.; Emr, M.; Fillit, H.; Greenberg, S.M.; et al. Alzheimer’s Disease—Related Dementias Summit 2016: National research priorities. Am. Acad. Neurol. 2017, 89, 2381–2391. [Google Scholar] [CrossRef]
- Crichton, G.E.; Elias, M.F.; Davey, A.; Sullivan, K.J.; Robbins, M.A. Higher HDL cholesterol is associated with better cognitive function: The Maine-Syracuse study. J. Int. Neuropsychol. Soc. 2014, 20, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Bates, K.A.; Sohrabi, H.R.; Rainey-Smith, S.R.; Weinborn, M.; Bucks, R.S.; Rodrigues, M.; Beilby, J.; Howard, M.; Taddei, K.; Martins, G.; et al. Serum high-density lipoprotein is associated with better cognitive function in a cross-sectional study of aging women. Int. J. Neurosci. 2017, 127, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Tortelli, R.; Lozupone, M.; Guerra, V.; Rosaria, M.; Imbimbo, B.P.; Capozzo, R.; Grasso, A.; Tursi, M.; di Dio, C.; Sardone, R.; et al. Midlife Metabolic Profile and the Risk of Late-Life Cognitive Decline. J. Alzheimer’s Dis. 2017, 59, 121–130. [Google Scholar] [CrossRef]
- Ma, C.; Yin, Z.; Zhu, P.; Luo, J.; Shi, X.; Gao, X. Blood cholesterol in late-life and cognitive decline: A longitudinal study of the Chinese elderly. Mol. Neurobiol. Mol. Neurodegener. 2017, 12, 24. [Google Scholar] [CrossRef]
- Kim, W.; He, Y.; Phan, K.; Ahmed, R.; Rye, K.; Piguet, O.; Hodges, J.R.; Halliday, G.M. Altered High Density Lipoprotein Composition in Behavioral Variant Frontotemporal Dementia. Front. Neurosci. 2018, 12, 847. [Google Scholar] [CrossRef]
- Lefterov, I.; Fitz, N.F.; Cronican, A.A.; Fogg, A.; Lefterov, P.; Kodali, R.; Wetzel, R.; Koldamova, R. Apolipoprotein A-I Deficiency Increases Cerebral Amyloid Angiopathy and Cognitive Deficits in APP/PS1DeltaE9 Mice. J. Biol. Chem. 2010, 285, 36945–36957. [Google Scholar] [CrossRef]
- Lewis, T.L.; Cao, D.; Lu, H.; Mans, R.A.; Su, Y.R.; Jungbauer, L.; Linton, M.F.; Fazio, S.; LaDu, M.J.; Li, L. Overexpression of Human Apolipoprotein A-I Preserves Cognitive Function and Attenuates Neuroinflammation and Cerebral Amyloid Angiopathy in a Mouse Model of Alzheimer. J. Biol. Chem. 2010, 285, 36958–36968. [Google Scholar] [CrossRef]
- Handattu, S.P.; Garber, D.W.; Monroe, C.E.; van Groen, T.; Kadish, I.; Nayyar, G.; Cao, D. Oral apolipoprotein A-I mimetic peptide improves cognitive function and reduces amyloid burden in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2009, 34, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Robert, J.; Stukas, S.; Button, E.; Hang, W.; Lee, M.; Fan, J.; Wilkinson, A.; Kulic, I.; Wright, S.D. Wellington CL. Reconstituted high-density lipoproteins acutely reduce soluble brain A β levels in symptomatic APP/PS1 mice. Biochim. Biophys. Acta 2015, 1862, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Fernández-de-Retana, S.; Cano-Sarabia, M.; Marazuela, P.; Sánchez-Quesada, J.L.; Garcia-Leon, A.; Montañola, A.; Montaner, J.; Maspoch, D.; Hernández-Guillamon, M. Characterization of ApoJ-reconstituted high-density lipoprotein (rHDL) nanodisc for the potential treatment of cerebral β-amyloidosis. Sci. Rep. 2017, 7, 14637. [Google Scholar] [CrossRef] [PubMed]
- Robert, J.; Button, E.B.; Yuen, B.; Gilmour, M.; Kang, K.; Bahrabadi, A.; Stukas, S.; Zhao, W.; Kulic, I.; Wellington, C.L. Clearance of beta-amyloid is facilitated by apolipoprotein E and circulating high- density lipoproteins in bioengineered human vessels. eLife 2017, 6, e29595. [Google Scholar] [CrossRef]
- Robert, J.; Button, E.B.; Stukas, S.; Boyce, G.K.; Gibbs, E.; Cowan, C.M.; Gilmour, M.; Cheng, W.H.; Soo, S.K.; Yuen, B.; et al. High-density lipoproteins suppress Aβ-induced PBMC adhesion to human endothelial cells in bioengineered vessels and in monoculture. Mol. Neurodegener. 2017, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, A.; Khera, A.V.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuganna, I.S.; Rader, D.R.; de Lemos, J.A.; et al. HDL Cholesterol Efflux Capacity and Incident Cardiovascular Events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef] [PubMed]
- Saleheen, D.; Scott, R.; Javad, S.; Zhao, W.; Rodrigues, A.; Picataggi, A.; Lukmanova, D.; Muchsavage, M.L.; Luben, R.; Billheimer, J.; et al. Association of HDL cholesterol efflux capacity with incident coronary heart disease events: A prospective case-control study. Lancet Diabetes Endocrinol. 2015, 3, 507–513. [Google Scholar] [CrossRef]
- Khera, A.V.; Cuchel, M.; de la Llera Moya, M.; Rodrigues, A.; Burke, M.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L.; et al. Cholesterol Efflux Capacity, High-Density Lipoprotein Function, and Atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef]
- Genest, J.; McPherson, R.; Frohlich, J.; Anderson, T.; Campbell, N.; Carpentier, A.; Couture, P.; Dufour, R.; Fodor, G.; Francis, G.A.; et al. 2009 Canadian Cardiovascular Society/Canadian guidelines for the diagnosis and treatment of dyslipidemia and prevention of cardiovascular disease in the adult—2009 recommendations. Can. J. Cardiol. 2009, 25, 567–579. [Google Scholar] [CrossRef]
- Giri, R.; Selvaraj, S.; Miller, C.A.; Hofman, F.; Yan, S.D.; Stern, D.; Zlokovic, B.V.; Kalra, V.K. Effect of endothelial cell polarity on β-amyloid-induced migration of monocytes across normal and AD endothelium. Am. J. Physiol. Physiol. 2002, 283, C895–904. [Google Scholar] [CrossRef]
- Truran, S.; Weissig, V.; Madine, J.; Davies, H.A.; Guzman-Villanueva, D.; Franco, D.A.; Karamanova, N.; Burciu, C.; Serrano, G.; Beach, T.G.; et al. Nanoliposomes protect against human arteriole endothelial dysfunction induced by β-amyloid peptide. J. Cereb. Blood Flow Metab. 2016, 36, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Lamoke, F.; Mazzone, V.; Persichini, T.; Maraschi, A.; Harris, M.B.; Venema, R.C.; Colasanti, M.; Gliozzi, M.; Muscoli, C.; Bartoli, M.; et al. Amyloid β peptide-induced inhibition of endothelial nitric oxide production involves oxidative stress-mediated constitutive eNOS/HSP90 interaction and disruption of agonist-mediated Akt activation. J. Neuroinflamm. 2015, 12, 84. [Google Scholar] [CrossRef] [PubMed]
- Besler, C.; Heinrich, K.; Rohrer, L.; Doerries, C.; Riwanto, M.; Shih, D.M.; Chroni, A.; Yonekawa, K.; Stein, S.; Schaefer, N.; et al. Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J. Clin. Investig. 2011, 121, 2693–2708. [Google Scholar] [CrossRef] [PubMed]
- Davidson, W.S.; Heink, A.; Sexmith, H.; Melchior, J.T.; Gordon, S.M.; Kuklenyik, Z.; Woollett, L.; Barr, J.R.; Jones, J.I.; Toth, C.A.; et al. The effects of apolipoprotein B depletion on HDL subspecies composition and function. J. Lipid Res. 2016, 57, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Kunitake, S.T.; Kane, J.P. Factors affecting the integrity of high density lipoproteins in the ultracentrifuge. J. Lipid Res. 1982, 23, 936–940. [Google Scholar] [PubMed]
- Van’t Hooft, F.; Havel, R. Metabolism of Apolipoprotein E in Plasma High Density Lipoproteins from Normal and Cholesterol-fed Rats. J. Biol. Chem. 1982, 257, 10996–11001. [Google Scholar] [PubMed]
- Holmes, M.V.; Asselbergs, F.W.; Palmer, T.M.; Drenos, F.; Lanktree, M.B.; Nelson, C.P.; Dale, C.E.; Padmanabhan, S.; Finan, C.; Swerdlow, D.I.; et al. Mendelian randomization of blood lipids for coronary heart disease. Eur. Heart J. 2015, 36, 539–550. [Google Scholar] [CrossRef]
- Boyce, G.; Button, E.; Soo, S.; Wellington, C. The pleiotropic vasoprotective functions of high density lipoproteins (HDL). J. Biomed. Res. 2018, 32, 164–182. [Google Scholar]
- Khalil, A.; Berrougui, H.; Pawelec, G.; Fulop, T. Impairment of the ABCA1 and SR-BI-mediated cholesterol efflux pathways and HDL anti-inflammatory activity in Alzheimer’s disease. Mech. Ageing Dev. 2012, 133, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Hutchinson, L.; Kirk, R. High drug attrition rates—Where are we going wrong? Nat. Rev. Clin. Oncol. 2011, 8, 189–190. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, S.A.; Besler, C.; Rohrer, L.; Meyer, M.; Heinrich, K.; Bahlmann, F.H.; Mueller, M.; Horváth, T.; Doerries, C.; Heinemann, M.; et al. Endothelial-Vasoprotective Effects of High-Density Lipoprotein Are Impaired in Patients with Type 2 Diabetes Mellitus but Are Improved after Extended-Release Niacin Therapy. Circulation 2010, 121, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Stahlman, M.; Davidsson, P.; Kanmert, I.; Rosengren, B.; Boren, J.; Fagerberg, B.; Camejo, G. Proteomics and lipids of lipoproteins isolated at low salt concentrations in D2O/sucrose or in KBr. J. Lipid Res. 2008, 49, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Hafiane, A.; Genest, J. HDL-Mediated Cellular Cholesterol Efflux Assay Method. Ann. Clin. Lab. Sci. 2015, 45, 659–668. [Google Scholar] [PubMed]
- Gordon, S.M.; Deng, J.; Lu, L.J.; Davidson, W.S. Proteomic characterization of human plasma high density lipoprotein fractionated by gel filtration chromatography. J. Proteome Res. 2010, 9, 5239–5249. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.S.P.; Liu, C.S.; Rau, A.; Lanctôt, K.L.; Köhler, C.A.; Pakosh, M.; Carvalho, A.F.; Herrmann, N. Peripheral inflammatory markers in Alzheimer’s disease: A systematic review and meta-analysis of 175 studies. J. Neurol. Neurosurg. Psychiatry 2017, 88, 876–882. [Google Scholar] [CrossRef]
- O’Neill, F.; Riwanto, M.; Charakida, M.; Colin, S.; Manz, J.; McLoughlin, E.; Khan, T.; Klein, N.; Kay, C.W.; Patel, K.; et al. Structural and functional changes in HDL with low grade and chronic inflammation. Int. J. Cardiol. 2015, 188, 111–116. [Google Scholar] [CrossRef]
- Ortiz-munoz, G.; Couret, D.; Lapergue, B.; Bruckert, E.; Meseguer, E.; Amarenco, P.; Meilhac, O. Dysfunctional HDL in acute stroke. Atherosclerosis 2016, 253, 75–80. [Google Scholar] [CrossRef]
- Charakida, M.; Besler, C.; Batuca, J.R.; Wang, G. Vascular Abnormalities, Paraoxonase in Primary Antiphospholipid Syndrome. JAMA 2009, 302, 1210–1217. [Google Scholar] [CrossRef]
- Fan, J.; Zareyan, S.; Zhao, W.; Shimizu, Y.; Pfeifer, T.A.; Tak, J.-H.; Isman, M.B.; van den Hoven, B.; Duggan, M.E.; Wood, W.; et al. Identification of a Chrysanthemic Ester as an Apolipoprotein E Inducer in Astrocytes. PLoS ONE 2016, 11, e0162384. [Google Scholar] [CrossRef] [PubMed]
- Robert, J.; Weber, B.; Frese, L.; Emmert, M.Y.; Schmidt, D.; Von Eckardstein, A.; Rohrer, L.; Hoerstrug, S.P. A three-dimensional engineered artery model for in vitro atherosclerosis research. PLoS ONE 2013, 8, e79821. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Button, E.B.; Gilmour, M.; Cheema, H.K.; Martin, E.M.; Agbay, A.; Robert, J.; Wellington, C.L. Vasoprotective Functions of High-Density Lipoproteins Relevant to Alzheimer’s Disease Are Partially Conserved in Apolipoprotein B-Depleted Plasma. Int. J. Mol. Sci. 2019, 20, 462. https://doi.org/10.3390/ijms20030462
Button EB, Gilmour M, Cheema HK, Martin EM, Agbay A, Robert J, Wellington CL. Vasoprotective Functions of High-Density Lipoproteins Relevant to Alzheimer’s Disease Are Partially Conserved in Apolipoprotein B-Depleted Plasma. International Journal of Molecular Sciences. 2019; 20(3):462. https://doi.org/10.3390/ijms20030462
Chicago/Turabian StyleButton, Emily B., Megan Gilmour, Harleen K. Cheema, Emma M. Martin, Andrew Agbay, Jérôme Robert, and Cheryl L. Wellington. 2019. "Vasoprotective Functions of High-Density Lipoproteins Relevant to Alzheimer’s Disease Are Partially Conserved in Apolipoprotein B-Depleted Plasma" International Journal of Molecular Sciences 20, no. 3: 462. https://doi.org/10.3390/ijms20030462
APA StyleButton, E. B., Gilmour, M., Cheema, H. K., Martin, E. M., Agbay, A., Robert, J., & Wellington, C. L. (2019). Vasoprotective Functions of High-Density Lipoproteins Relevant to Alzheimer’s Disease Are Partially Conserved in Apolipoprotein B-Depleted Plasma. International Journal of Molecular Sciences, 20(3), 462. https://doi.org/10.3390/ijms20030462