Cancer Cell-Derived Granulocyte-Macrophage Colony-Stimulating Factor Is Dispensable for the Progression of 4T1 Murine Breast Cancer


 and
and 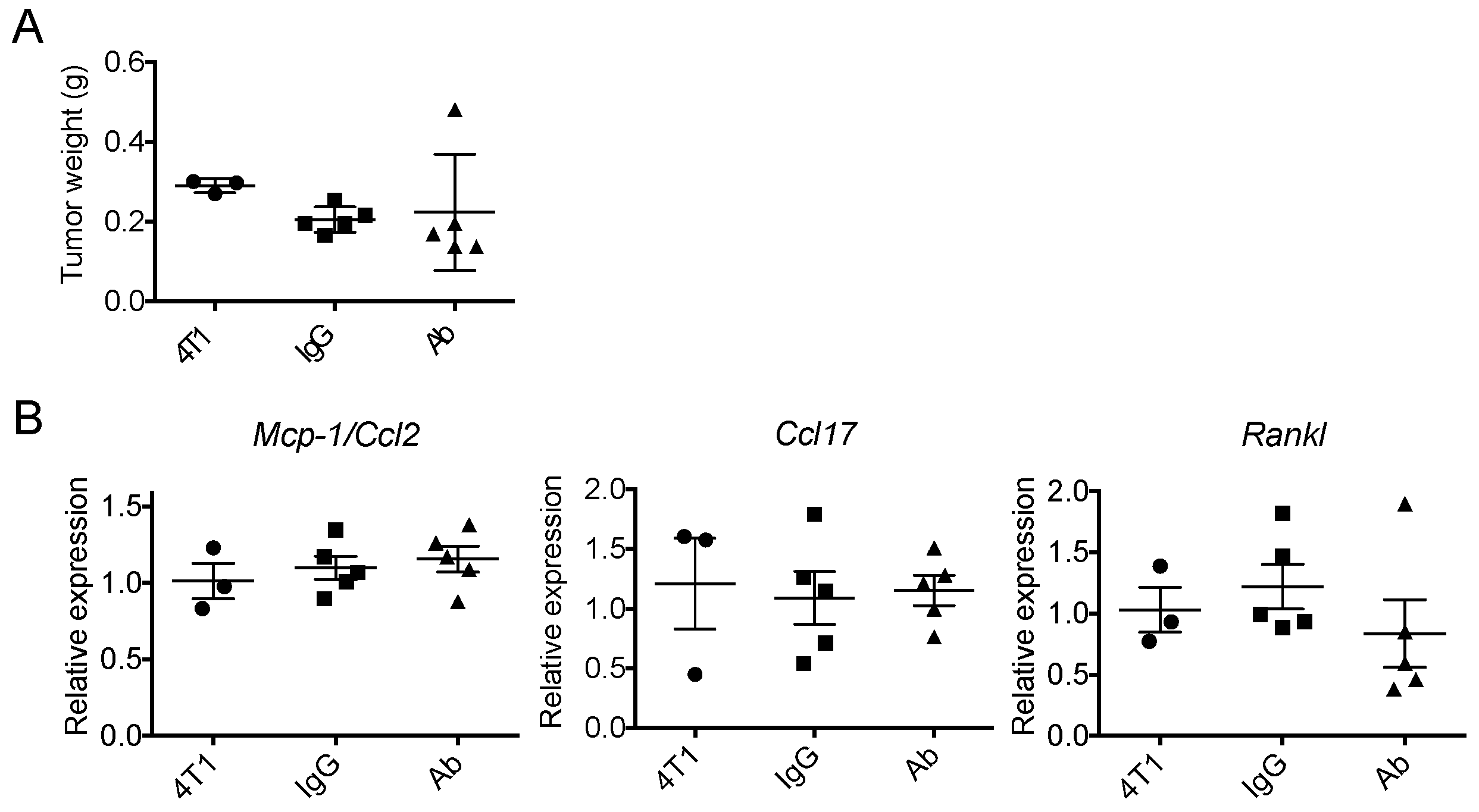{kind=link}
{kind=link}
{kind=link}
{kind=link}
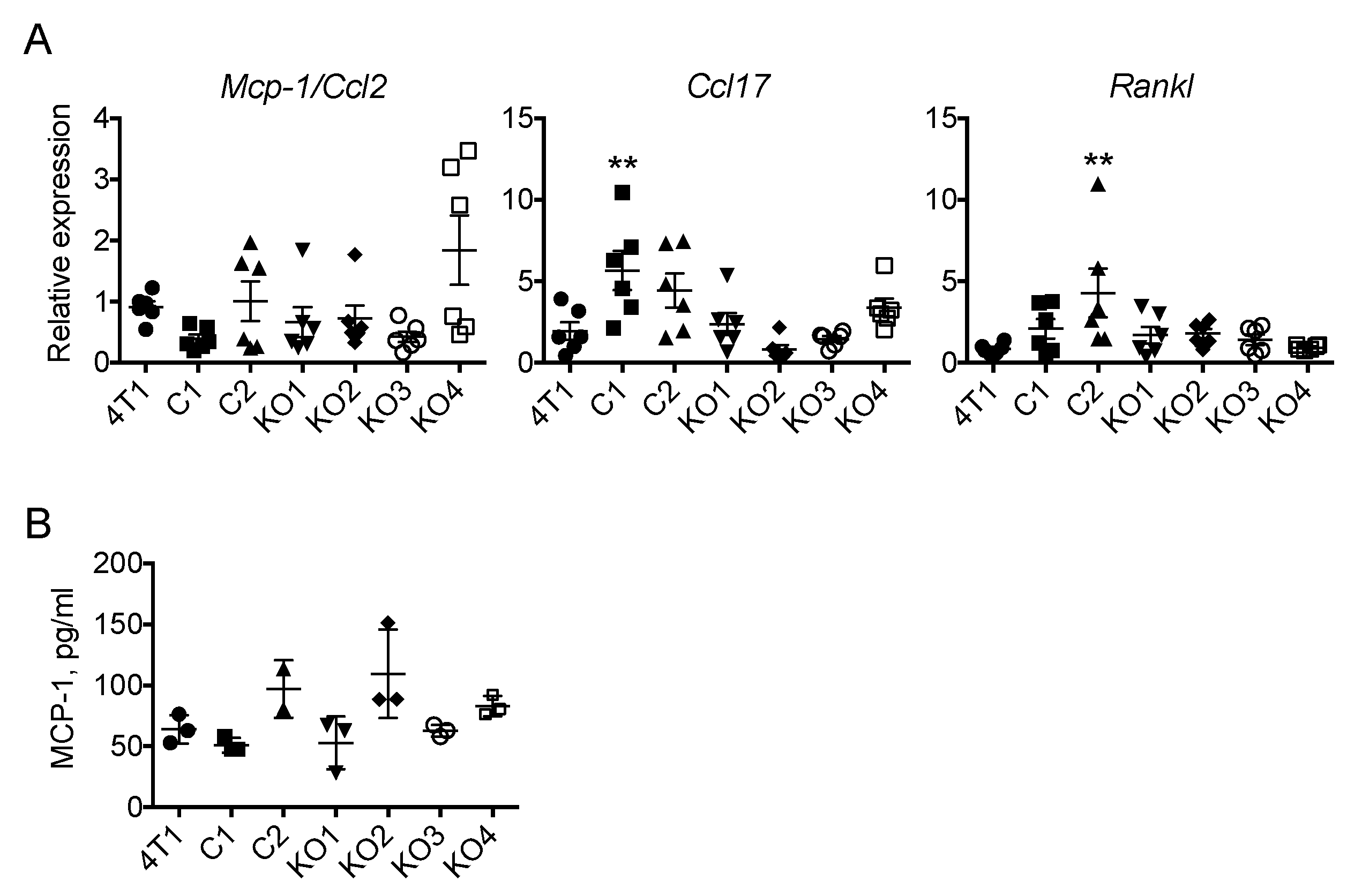{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Neutralization of GM-CSF Does not Affect the Expression of Mcp-1, Ccl17 or Rankl mRNA by 4T1 tumors
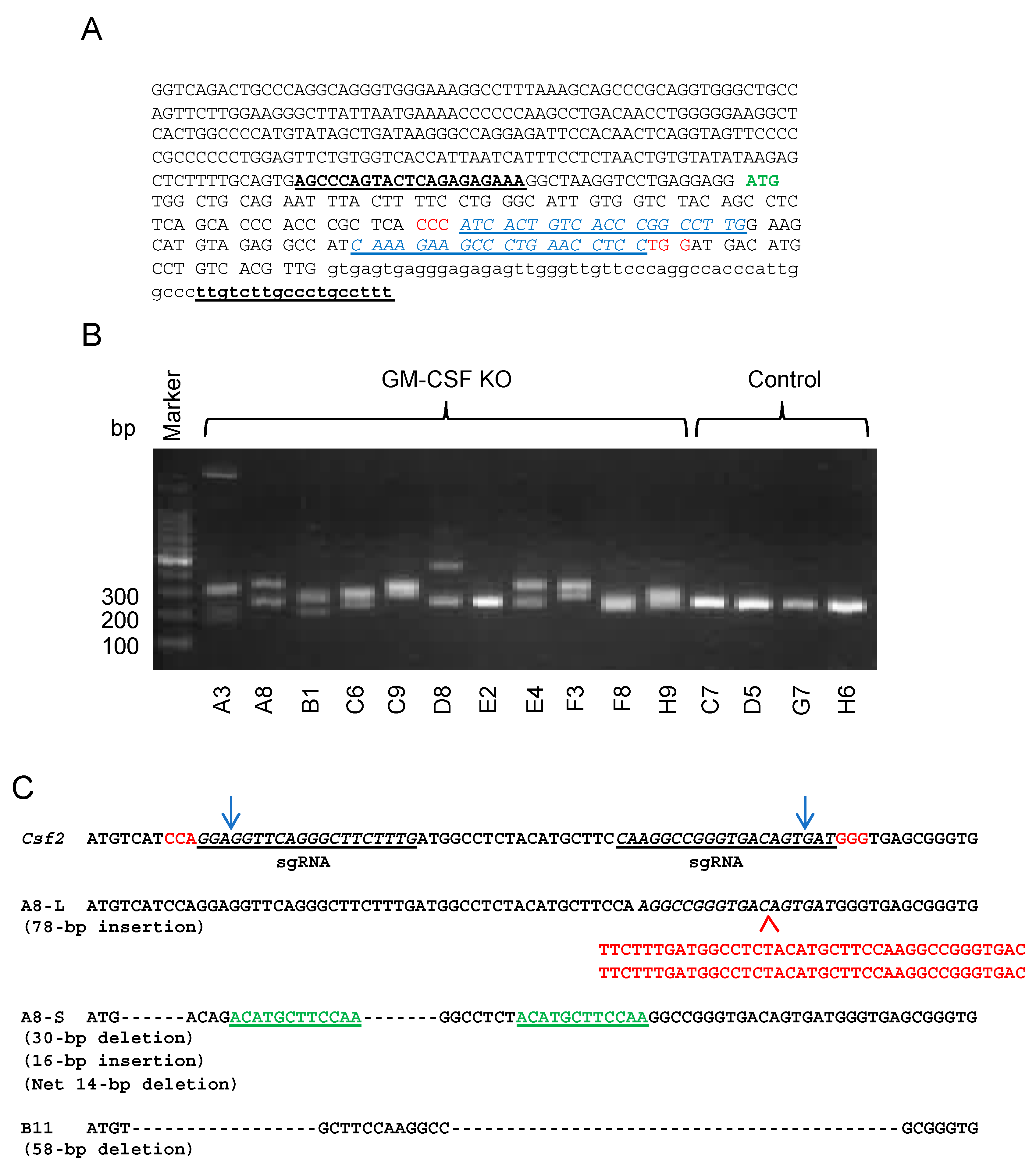
2.2. Generation of GM-CSF-Deficient 4T1 Cells Using the CRISPR-Cas9 System
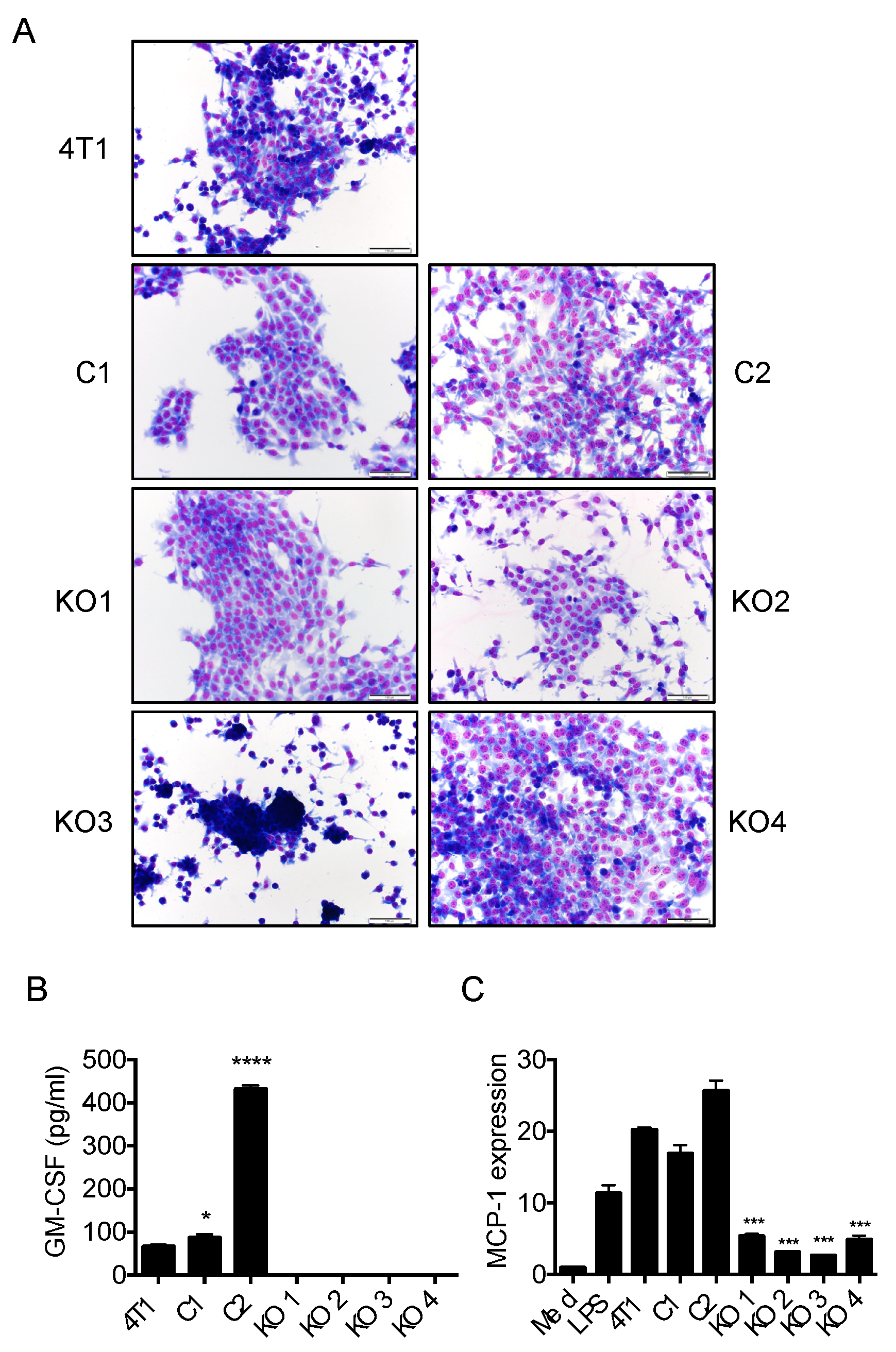
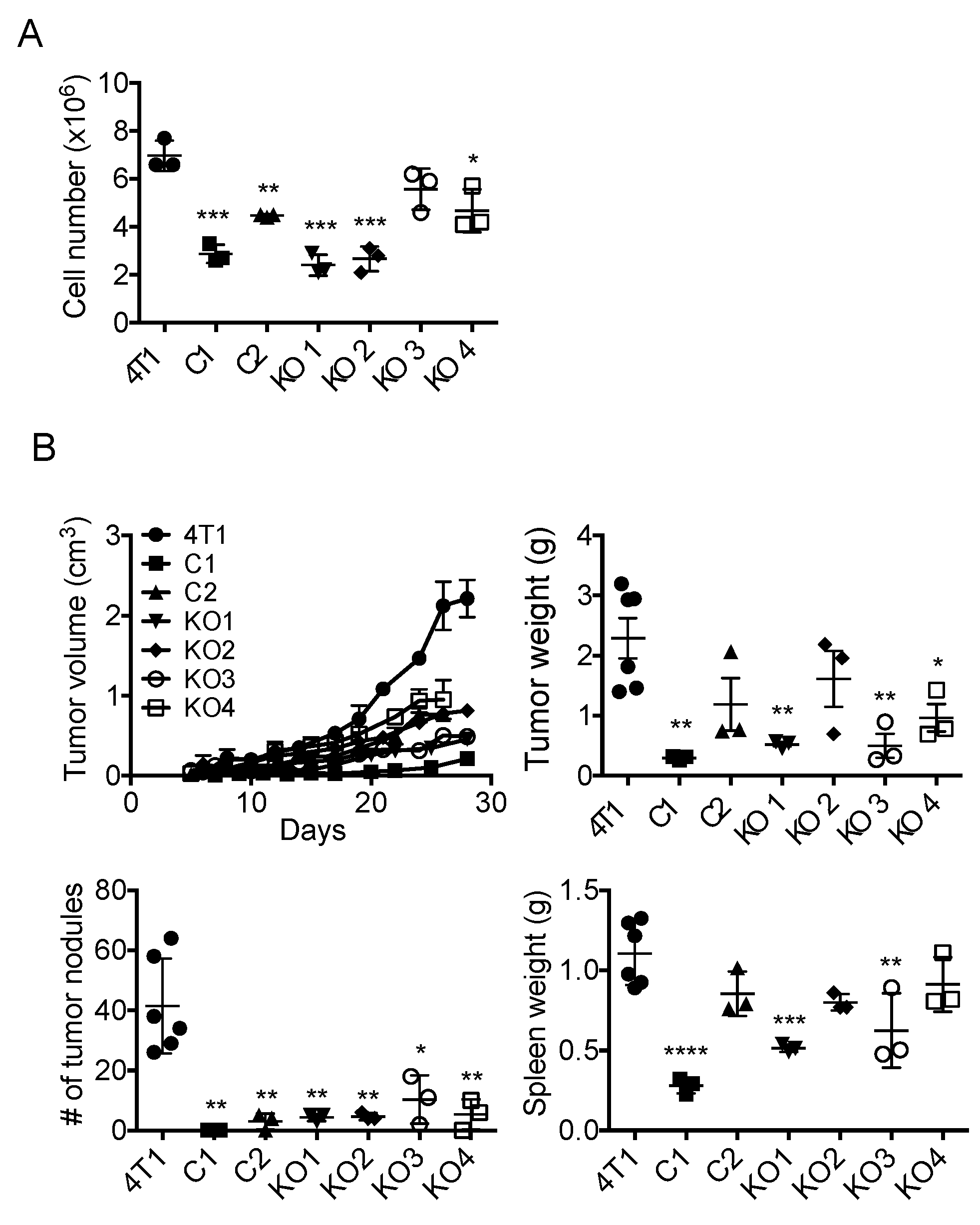
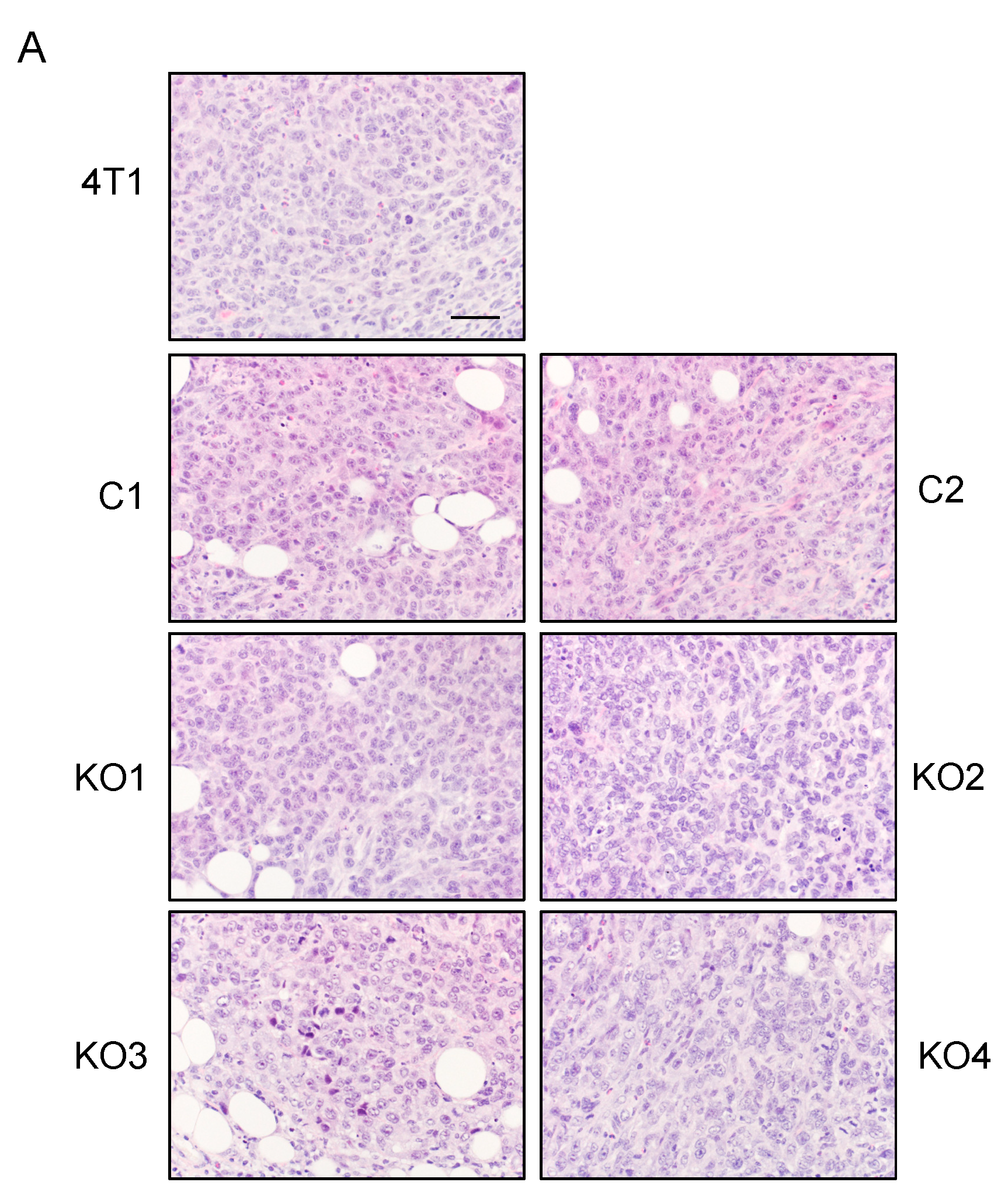
2.3. Single Cell Cloning of 4T1 Cells Reduced the Progression of 4T1 Breast Cancer, Independent of GM-CSF Production
2.4. The Expression of Mcp-1/Ccl2, Ccl17 or Rankl by 4T1 Tumors Is Independent of Tumor Cell-Derived GM-CSF
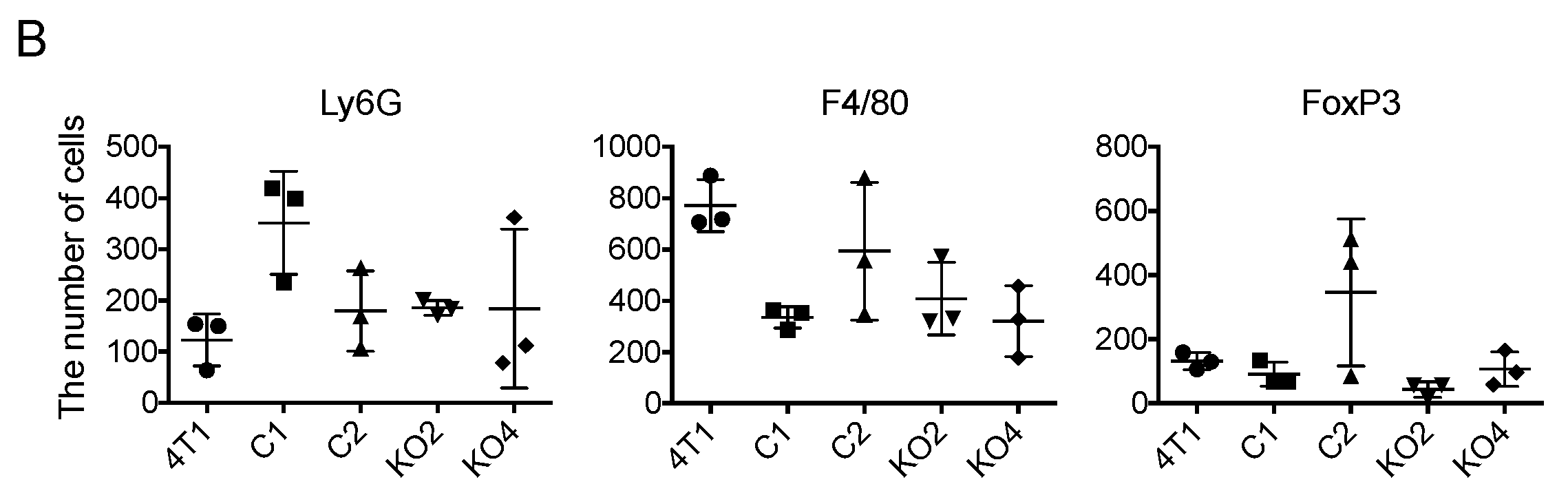
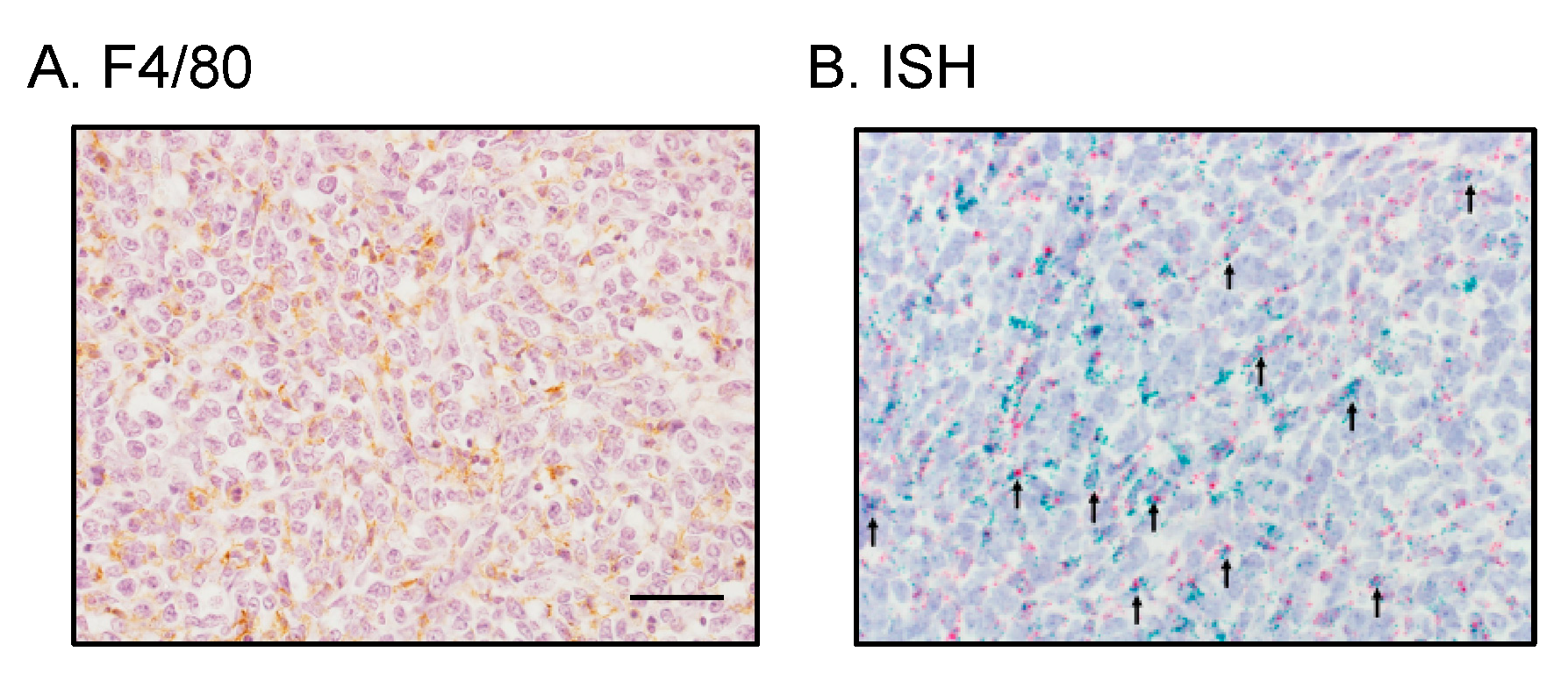
2.5. Tumor-Infiltrating Macrophages Express MCP-1 mRNA Independent of Cancer Cell-Derived GM-CSF
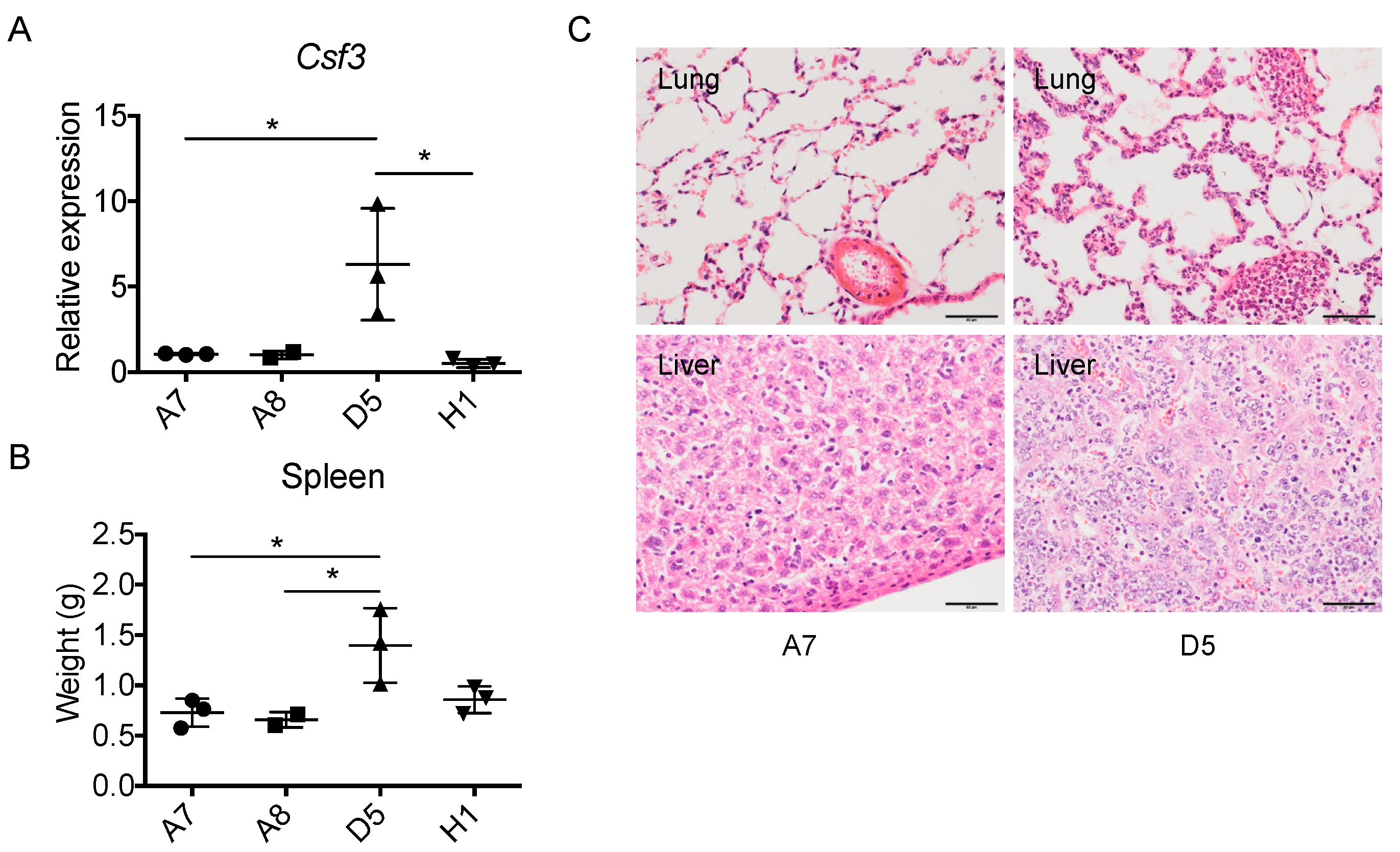
2.6. The Level of Cancer Cell-Derived G-CSF Correlates with Splenomegaly and Tissue Congestion
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Transplantation of 4T1 Cells and Neutralization of GM-CSF
4.3. Generation and Transplantation of GM-CSF-Deficient 4T1 Cell Lines
4.4. Activation of Macrophages in Vitro
4.5. Quantification of Cytokine/Chemokine Concentration
4.6. qRT-PCR
4.7. Immunohistochemistry (IHC)
4.8. In Situ Hybridization (ISH)
4.9. Statistical Analysis
4.10. Ethics Statement
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| G-CSF | Granulocyte colony-stimulating factor |
| RANKL | Receptor activator nuclear factor-κB ligand |
References
- Sund, M.; Kalluri, R. Tumor stroma derived biomarkers in cancer. Cancer Metastasis Rev. 2009, 28, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Sporn, M.B. The tumour microenvironment as a target for chemoprevention. Nat. Rev. 2007, 7, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Weiseman, B.S.; Werb, Z. Stromal effects on mammary gland development and breast cancer. Science 2002, 296, 1046–1049. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, T.; Howard, O.M.Z.; Ito, T.; Kuwabara, M.; Matsukawa, A.; Chen, K.; Liu, Y.; Liu, M.; Oppenheim, J.J.; Wang, J.M. Monocyte chemoattractant protein-1/CCL2 produced by stromal cells promotes lung metastasis of 4T1 murine breast cancer cells. PLoS ONE 2013, 8, e58791. [Google Scholar] [CrossRef]
- Fujimoto, H.; Sangai, T.; Ishii, G.; Ikehara, A.; Nagashima, T.; Miyazaki, M.; Ochiai, A. Stromal MCP-1 in mammary tumors induces tumo-associated macrophage infiltration and contributes to tumor progression. Int. J. Cancer 2009, 125, 1276–1284. [Google Scholar] [CrossRef]
- Yoshimura, T.; Imamichi, T.; Weiss, J.M.; Sato, M.; Li, L.; Matsukawa, A.; Wang, J.M. Induction of monocyte chemoattractant proteins in macrophages via the production of granulocyte/macrophage colony-stimulating factor by breast cancer cells. Front. Immunol. 2016, 7, 2. [Google Scholar] [CrossRef]
- Yoshie, O.; Matsushima, K. CCR4 and its ligands: From bench to bedside. Int. Immunol. 2015, 27, 11–20. [Google Scholar] [CrossRef]
- Nolan, E.; Vaillant, F.; Branstetter, D.; Pal, B.; Giner, G.; Whitehead, L.; Lok, S.W.; Mann, G.B.; Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer (kConFab); Rohrbach, K.; et al. RANK ligand as a potential target for breast cancer prevention in BRCA1-mutation carriers. Nat. Med. 2016, 22, 933–939. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef]
- Hamilton, J.A. Colony-stimulating factors in inflammation and autoimmunity. Nat. Rev. Immunol. 2008, 8, 533–544. [Google Scholar] [CrossRef]
- Dranoff, G. GM-CSF-based cancer vaccines. Immunol. Rev. 2002, 188, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Hatakeyama, K.; Tsuchiya, Y.; Rikiishi, H.; Kumagai, K. A correlation between GM-CSF gene expression and metastases in murine tumors. Int. J. Cancer 1996, 47, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Park, B.K.; Zhang, H.; Zeng, Q.; Dai, J.; Keller, E.T.; Giordano, T.; Gu, K.; Shah, V.; Pei, L.; Zarbo, R.J.; et al. NF-kB in breast cancer cells promotes osteolytic bone metastasis by inducing osteoclastogenesis via GM-CSF. Nat. Med. 2006, 13, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Liu, Q.; Chen, J.; Chen, J.; Chen, F.; He, C.; Huang, D.; Wu, W.; Lin, L.; Huang, W.; et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 2014, 25, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, A.; Murata, Y.; Wang, K.-Y.; Tsutsui, M.; Kohno, K.; Sasaguri, Y. Monocyte chemoattractant protein-1 expression Is enhanced by granulocyte-macrophage colony-stimulating factor via Jak2-Stat5 signaling and inhibited by atorvastatin in human monocytic U937 cells. J. Biol. Chem. 2008, 283, 4643–4651. [Google Scholar] [CrossRef]
- Colotta, F.; Borrë, A.; Wang, J.M.; Tattanelli, M.; Maddalena, F.; Polentarutti, N.; Peri, G.; Mantovani, A. Expression of a monocyte chemotactic cytokine by human mononuclear phagocytes. J. Immunol. 1992, 148, 760–765. [Google Scholar]
- Popivanova, B.K.; Kitamura, K.; Wu, Y.; Kondo, T.; Kagaya, T.; Kaneko, S.; Oshima, M.; Fujii, C.; Mukaida, N. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Investig. 2008, 118, 560–570. [Google Scholar]
- Yoshimura, T.; Liu, M.; Chen, X.; Li, L.; Wang, J.M. Crosstalk between tumor cells and macrophages in stroma renders tumor cells as the primary source of MCP-1/CCL2 in Lewis lung carcinoma. Front. Immunol. 2015, 6, 332. [Google Scholar] [CrossRef]
- DuPré, S.A.; Redelman, D.; Hunter, K.W., Jr. The mouse mammary carcinoma 4T1: Characterization of the cellular landscape of primary tumours and metastatic tumour foci. Int. J. Exp. Pathol. 2007, 88, 351–360. [Google Scholar] [CrossRef]
- Petráčková, M.; Staněk, L.; Mandys, V.; Dundr, P.; Vonka, V. Properties of bcr-abl-transformed mouse 12B1 cells secreting interleukin-2 and granulocyte-macrophage colony stimulating factor (GM-CSF): II. Adverse effects of GM-CSF. Int. J. Oncol. 2012, 40, 1915–1922. [Google Scholar] [PubMed]
- Ravindranathan, S.; Nguyen, K.G.; Kurtz, S.L.; Frazier, H.N.; Smith, S.G.; prasanth Koppolu, B.; Rajaram, N.; Zaharoff, D.A. Tumor-derived granulocyte colonystimulating factor diminishes efficacy of breast tumor cell vaccines. Breast Cancer Res. 2018, 20, 126. [Google Scholar] [CrossRef] [PubMed]
- Wagenblast, E.; Soto, M.; Gutiérrez-Ángel, S.; Hartl, C.A.; Gable, A.L.; Maceli, A.R.; Erard, N.; Williams, A.M.; Kim, S.Y.; Dickopf, S.; et al. A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis. Nature 2015, 520, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.L.; Kowalski, H.M.; Blazar, B.A.; Fligiel, Z.; Vogel, R.; Heppner, G.H. Heterogeneity of tumor cells from a single mouse mammary tumor. Cancer Res. 1978, 38, 3174–3181. [Google Scholar] [PubMed]
- Miller, B.E.; Miller, F.R.; Heppner, G.H. Interactions between tumor subpopulations affecting their sensitivity to the antineoplastic agents cyclophosphamide and methotrexate. Cancer Res. 1981, 41, 4378–4381. [Google Scholar]
- Yamashina, K.; Miller, B.E.; Heppner, G.H. Macrophage-mediated induction of drug-resistant variants in a mouse mammary tumor cell line. Cancer Res. 1986, 46, 2396–2401. [Google Scholar]
- Aslakson, C.J.; Miller, F.R. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992, 52, 1399–1405. [Google Scholar]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshimura, T.; Nakamura, K.; Li, C.; Fujisawa, M.; Shiina, T.; Imamura, M.; Li, T.; Mukaida, N.; Matsukawa, A. Cancer Cell-Derived Granulocyte-Macrophage Colony-Stimulating Factor Is Dispensable for the Progression of 4T1 Murine Breast Cancer. Int. J. Mol. Sci. 2019, 20, 6342. https://doi.org/10.3390/ijms20246342
Yoshimura T, Nakamura K, Li C, Fujisawa M, Shiina T, Imamura M, Li T, Mukaida N, Matsukawa A. Cancer Cell-Derived Granulocyte-Macrophage Colony-Stimulating Factor Is Dispensable for the Progression of 4T1 Murine Breast Cancer. International Journal of Molecular Sciences. 2019; 20(24):6342. https://doi.org/10.3390/ijms20246342
Chicago/Turabian StyleYoshimura, Teizo, Kaoru Nakamura, Chunning Li, Masayoshi Fujisawa, Tsuyoshi Shiina, Mayu Imamura, Tiantian Li, Naofumi Mukaida, and Akihiro Matsukawa. 2019. "Cancer Cell-Derived Granulocyte-Macrophage Colony-Stimulating Factor Is Dispensable for the Progression of 4T1 Murine Breast Cancer" International Journal of Molecular Sciences 20, no. 24: 6342. https://doi.org/10.3390/ijms20246342
APA StyleYoshimura, T., Nakamura, K., Li, C., Fujisawa, M., Shiina, T., Imamura, M., Li, T., Mukaida, N., & Matsukawa, A. (2019). Cancer Cell-Derived Granulocyte-Macrophage Colony-Stimulating Factor Is Dispensable for the Progression of 4T1 Murine Breast Cancer. International Journal of Molecular Sciences, 20(24), 6342. https://doi.org/10.3390/ijms20246342