The Major Factors Causing the Microspore Abortion of Genic Male Sterile Mutant NWMS1 in Wheat (Triticum aestivum L.)

Abstract

1. Introduction
2. Results
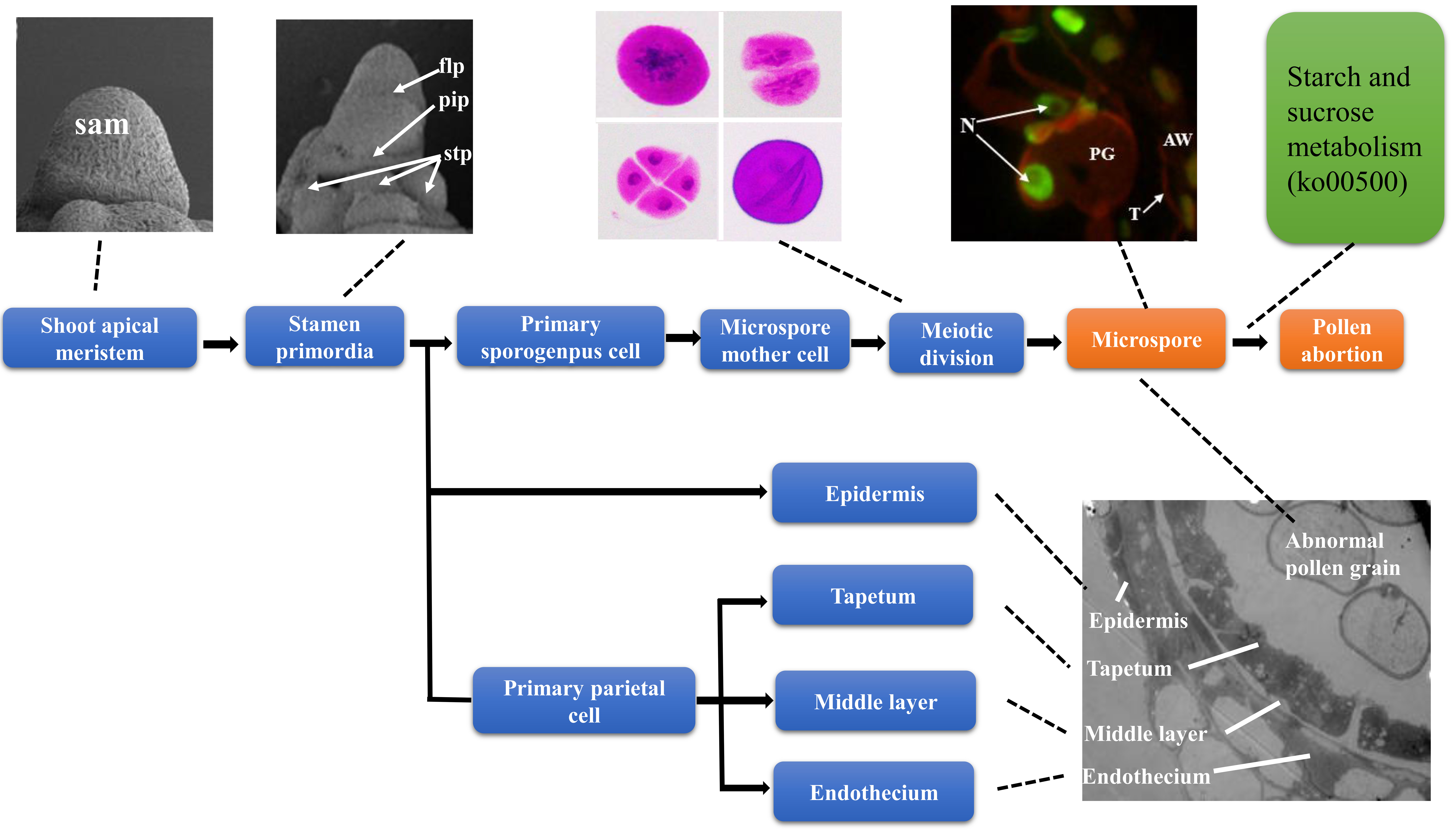
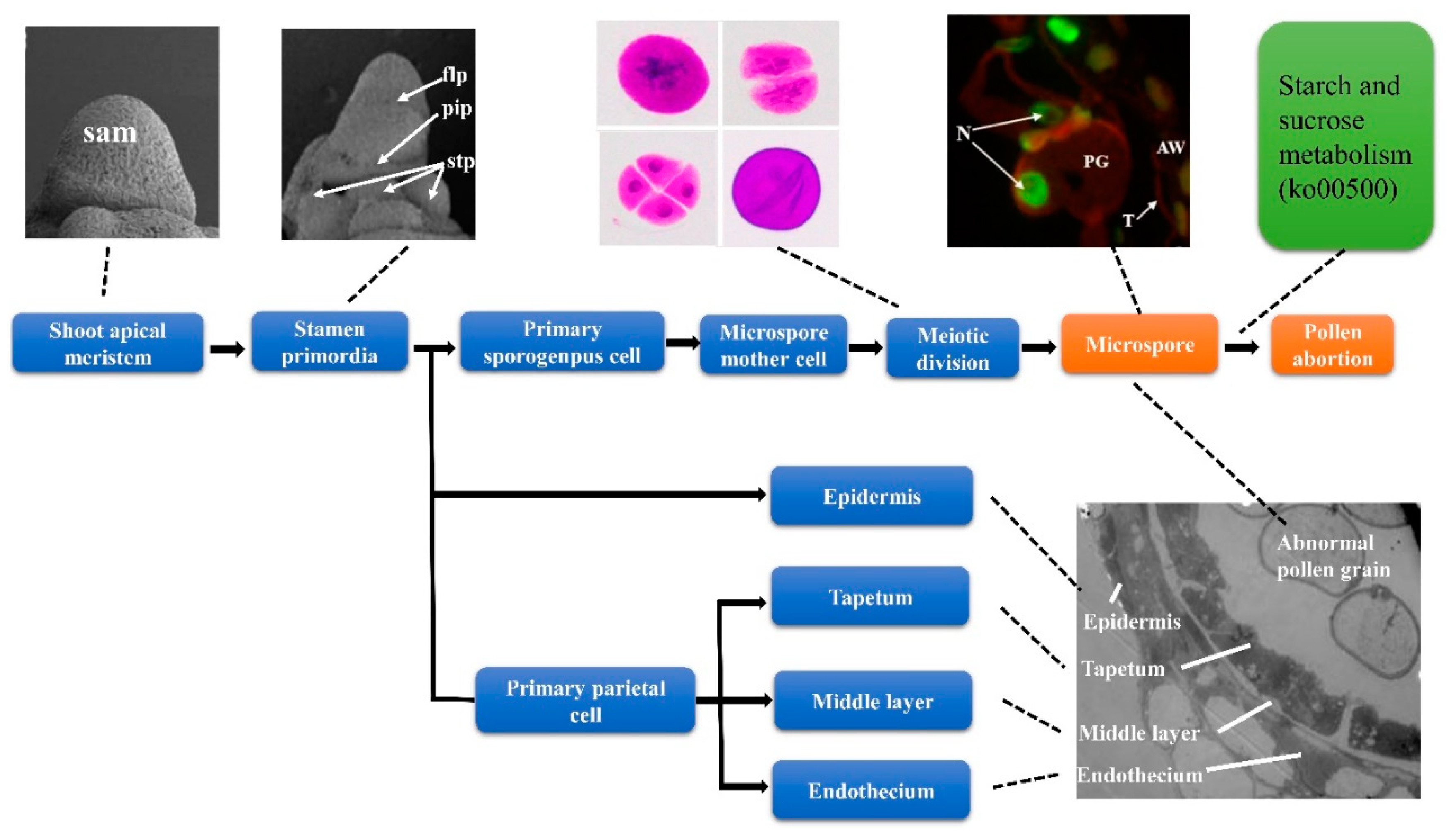
2.1. Morphology of the Mutant NWMS1 and Shengnong 1
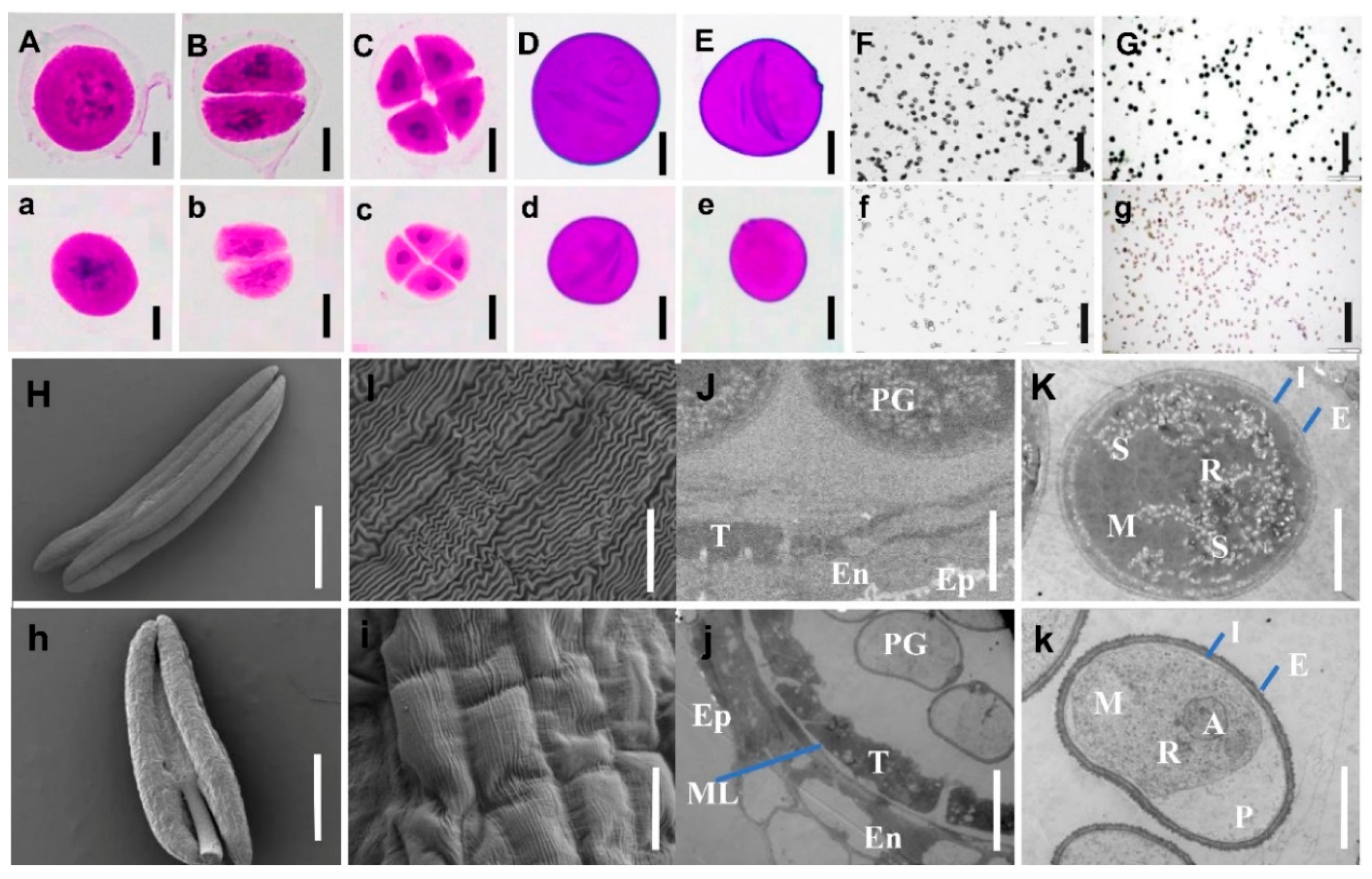
2.2. Anther and Microspore Structures of Mutant NWMS1 and Shengnong 1
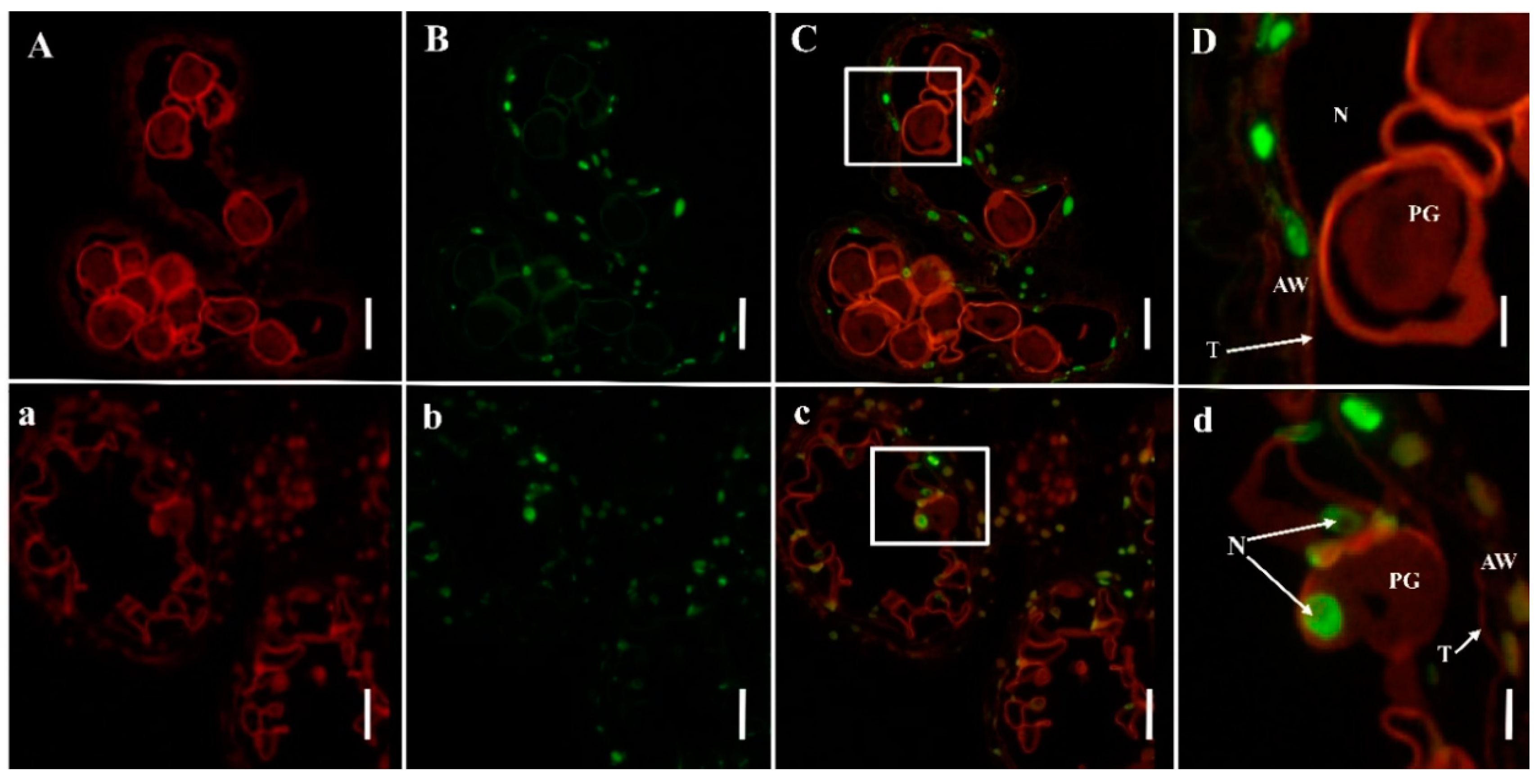
2.3. PCD Detection in Anthers
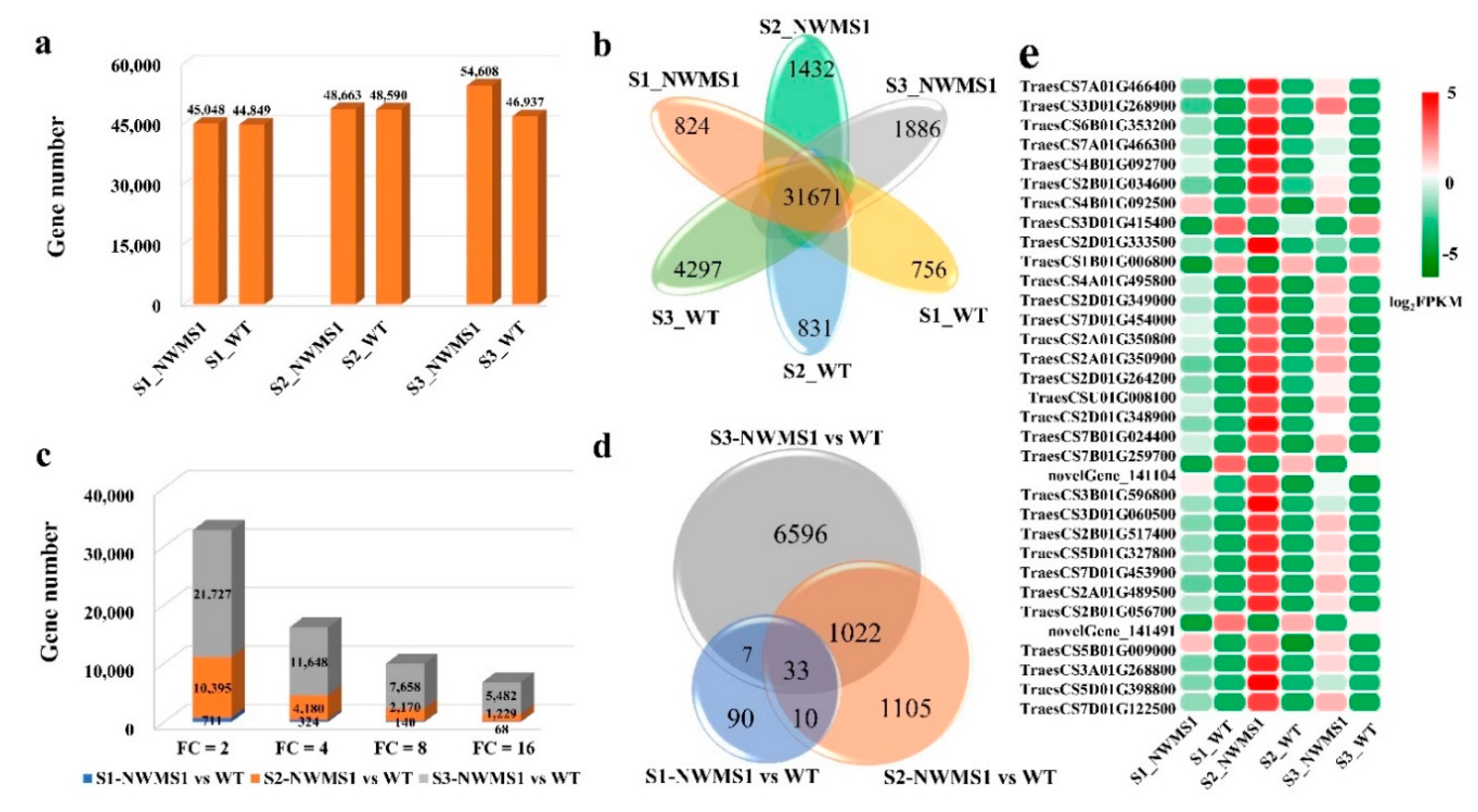
2.4. Overview of the Transcriptome Data
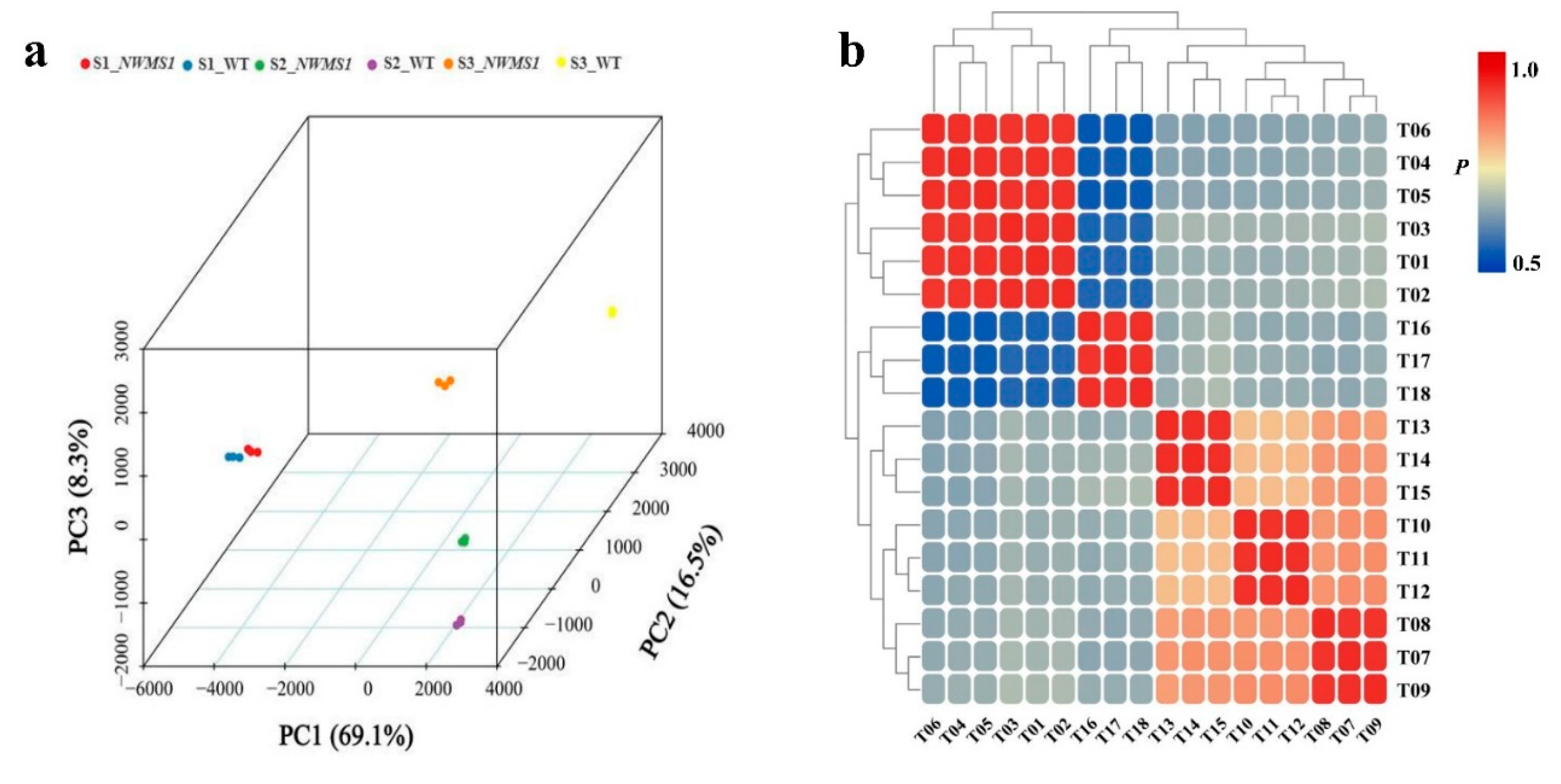
2.5. DEGs among the Six Samples
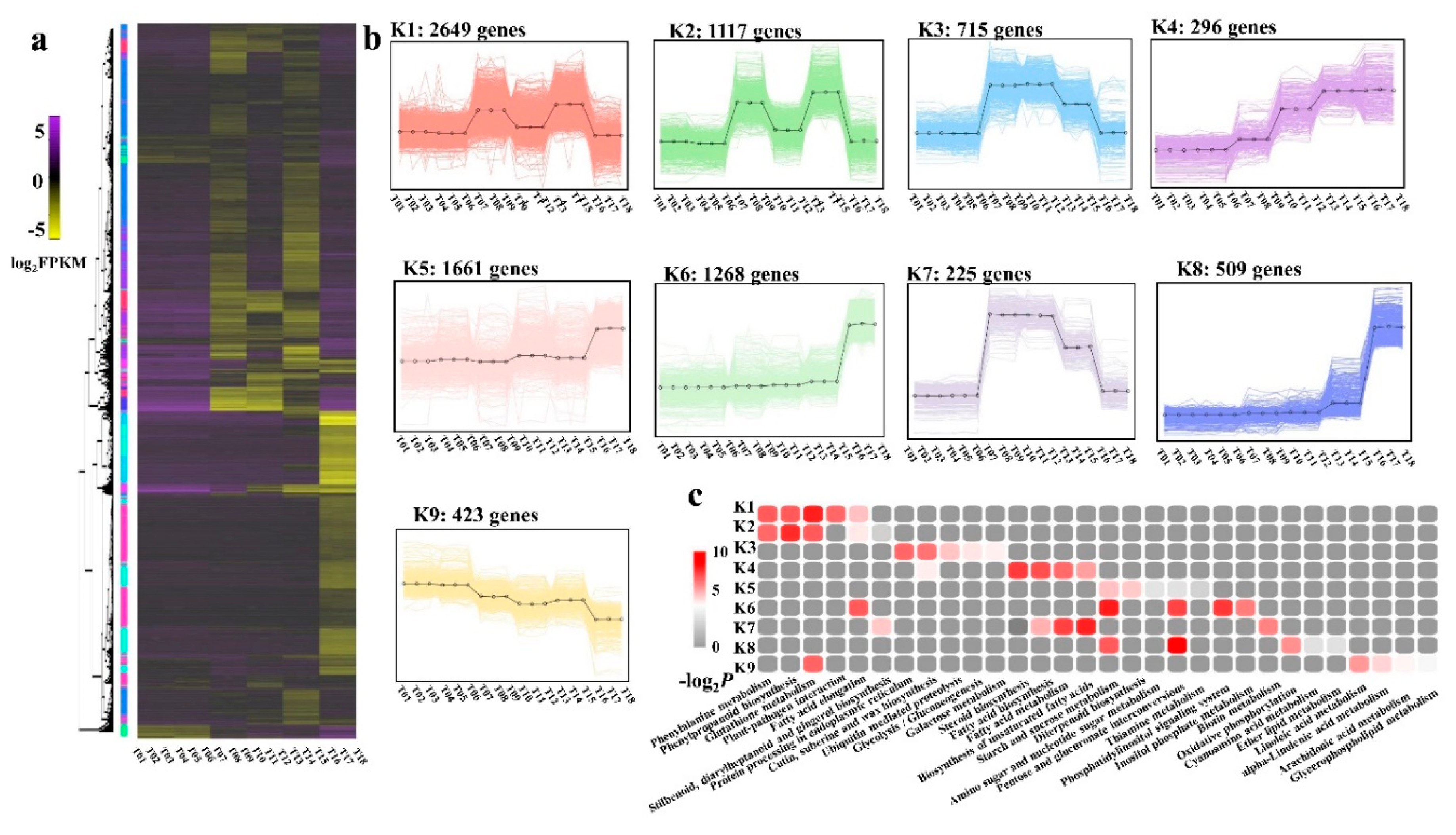
2.6. DEG Co-Expression Clusters
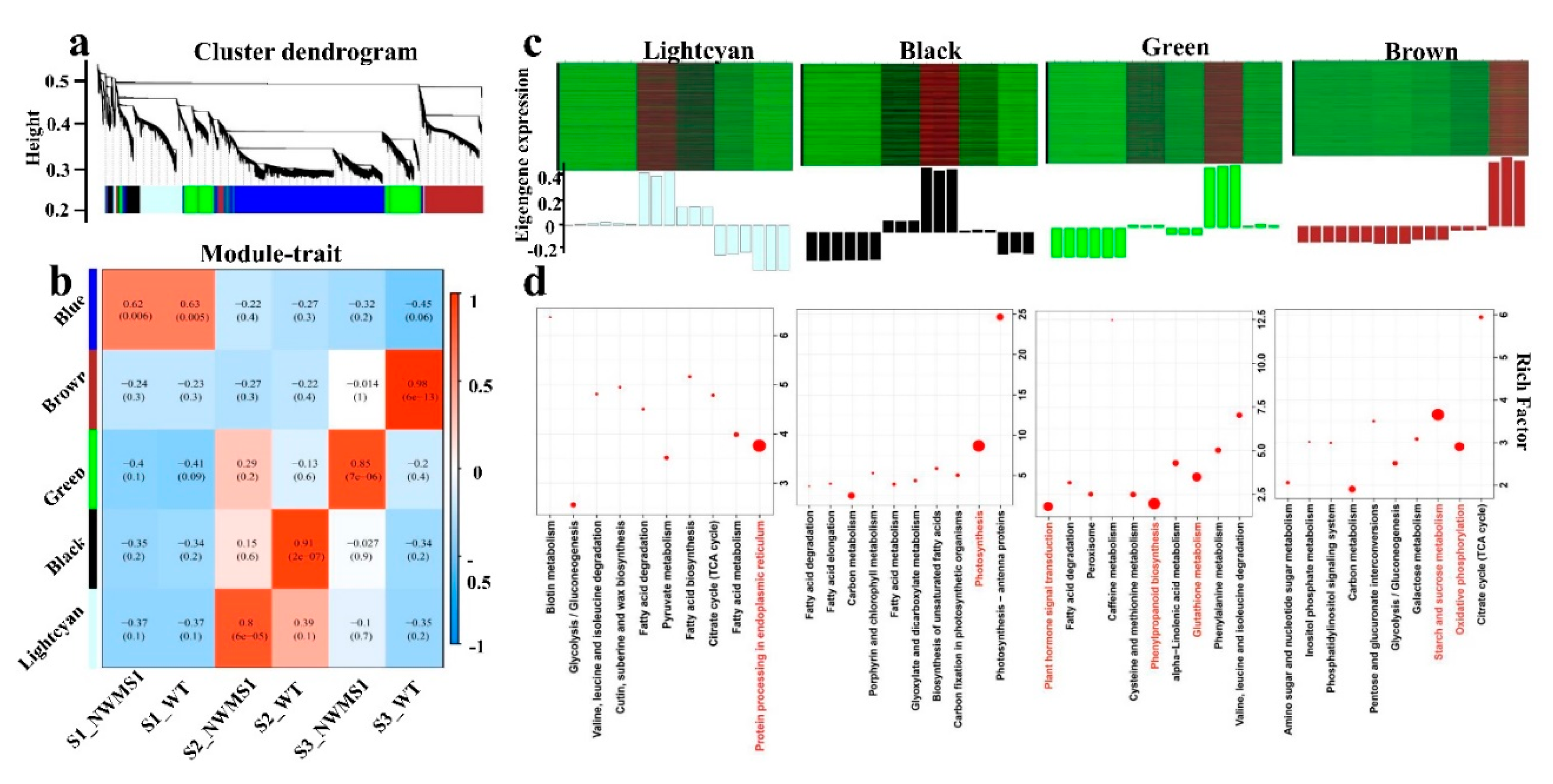
2.7. Valid Gene Co-Expression Network Analysis with WGCNA
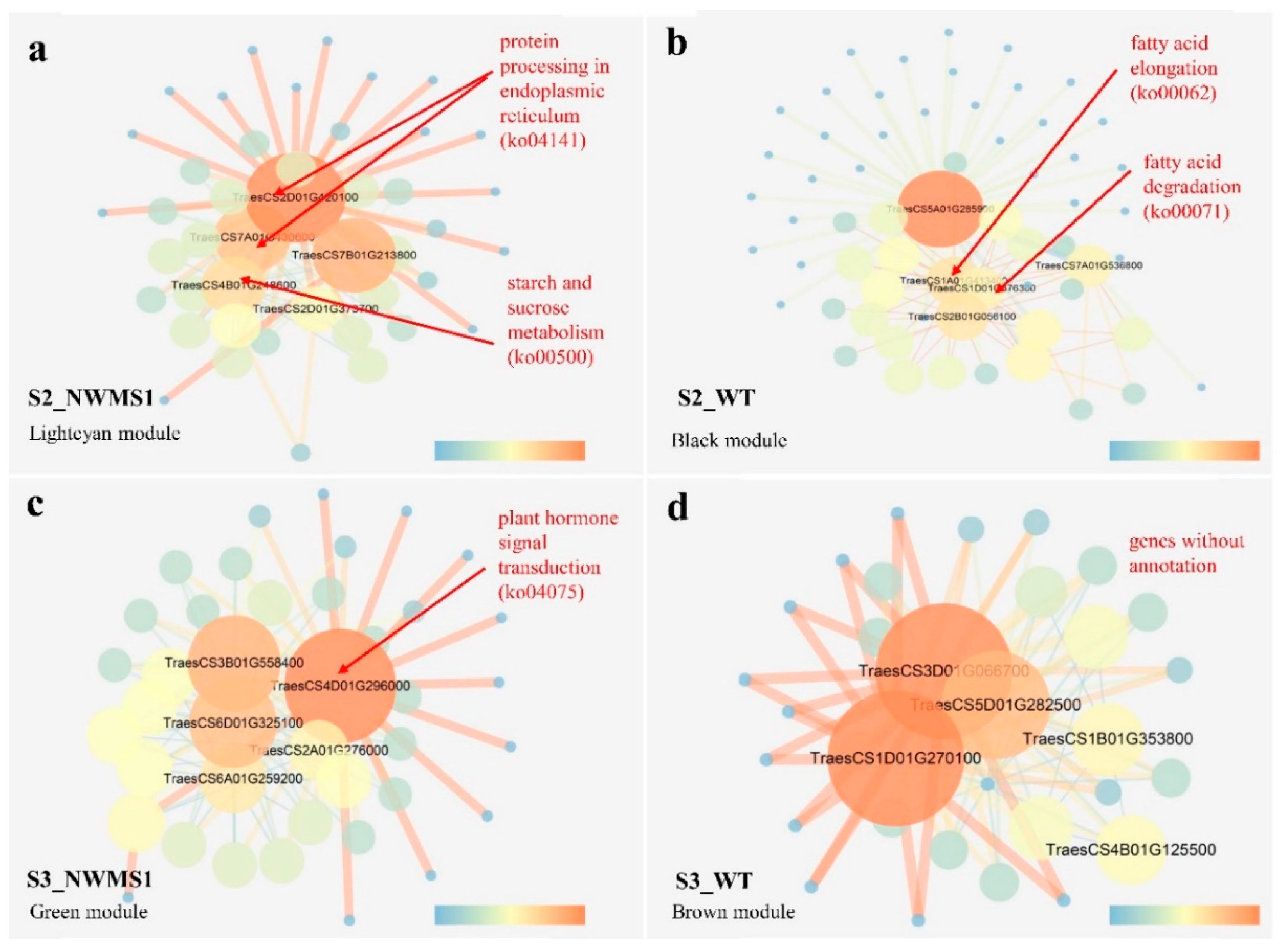
2.8. Key Genes Involved in Abnormal Anther Development of NWMS1 Screened via WGCNA
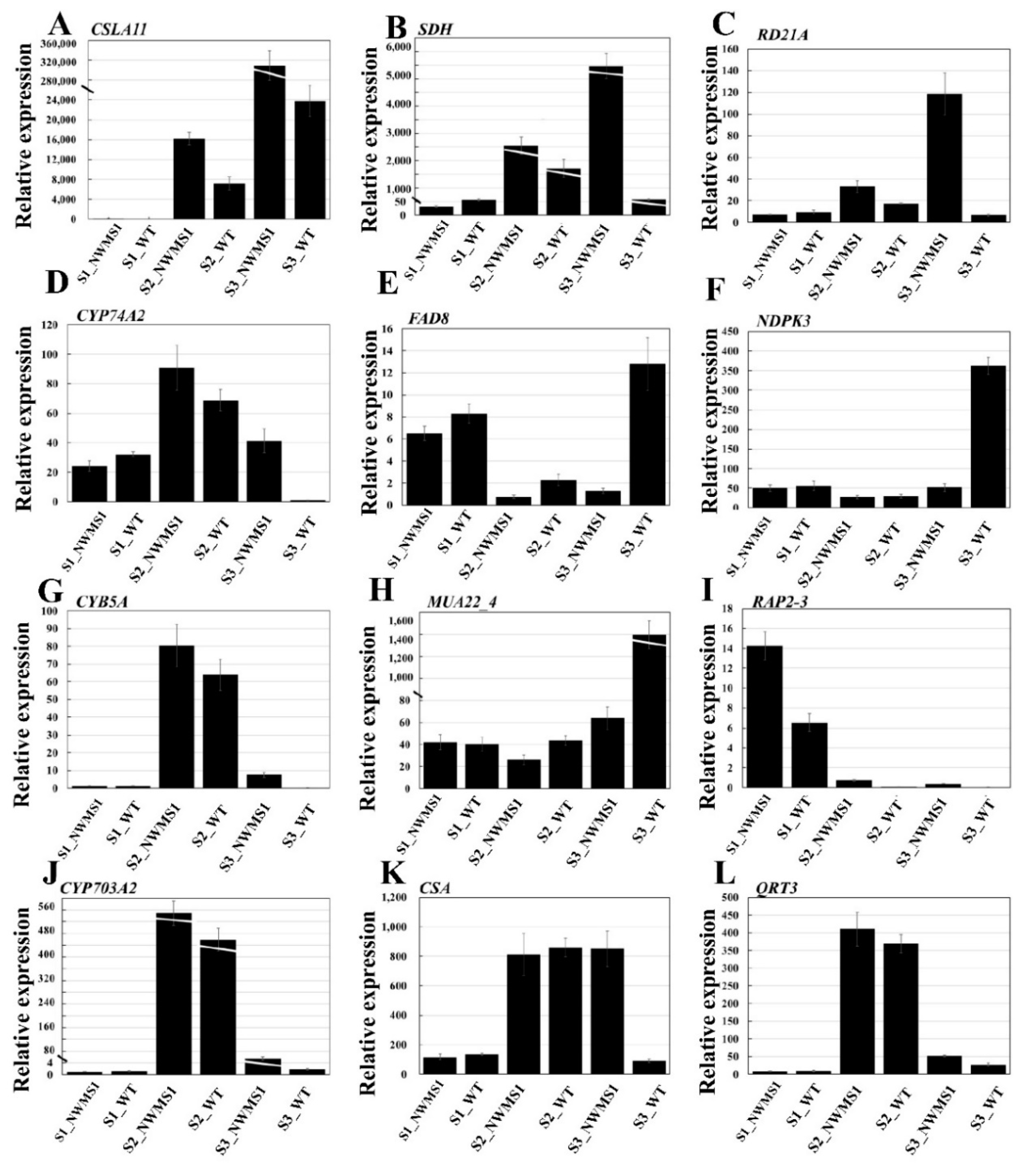
2.9. The Expression Profiles of Key Genes Involved in Anther Development
3. Discussion
3.1. NWMS1 Belongs to Binucleate Microspore Abortion
3.2. Global Gene Transcription Changes in NWMS1 and Shengnong 1
3.3. Complex Network Regulating Anther Development of NWMS1
3.4. Starch and Sucrose Metabolism is a Major Factor Causing Male Sterility in NWMS1
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Morphology Observation
4.3. Micrstructure and Ultrastructure Observation
4.4. TUNEL Assay
4.5. Sample Preparation and RNA Extraction for RNA-seq
4.6. Transcriptome Sequencing and Data Analysis
4.7. Weighted Gene Co-Expression Network Analysis (WGCNA)
4.8. qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pingali, P.L. Green Revolution: Impacts, limits, and the path ahead. Proc. Natl. Acad. Sci. USA 2012, 109, 12302–12308. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, M.; Foulkes, J.; Furbank, R.; Griffiths, S.; King, J.; Murchie, E.; Parry, M.; Slafer, G. Achieving yield gains in wheat. Plant Cell Environ. 2012, 35, 1799–1823. [Google Scholar] [CrossRef] [PubMed]
- Voss-Fels, K.P.; Stahl, A.; Hickey, L.T. Q&A: Modern crop breeding for future food security. BMC Biol. 2019, 17, 18. [Google Scholar] [CrossRef]
- Juliana, P.; Poland, J.; Huerta-Espino, J.; Shrestha, S.; Crossa, J.; Crespo-Herrera, L.; Toledo, F.H.; Govindan, V.; Mondal, S.; Kumar, U.; et al. Improving grain yield, stress resilience and quality of bread wheat using large-scale genomics. Nat. Genet. 2019, 51, 1530–1539. [Google Scholar] [CrossRef]
- Zheng, S.; Li, J.; Ma, L.; Wang, H.; Zhou, H.; Ni, E.; Jiang, D.; Liu, Z.; Zhuang, C. OsAGO2 controls ROS production and the initiation of tapetal PCD by epigenetically regulating OsHXK1 expression in rice anthers. Proc. Natl. Acad. Sci. USA 2019, 116, 7549–7558. [Google Scholar] [CrossRef]
- Bruns, R.; Peterson, C.J. Yield and stability factors associated with hybrid wheat. Euphytica 1998, 100, 1–5. [Google Scholar] [CrossRef]
- Vanderkley, F.K. Male sterility and its importance in breeding heterosis varieties. Euphytica 1954, 3, 117–124. [Google Scholar] [CrossRef]
- Mayr, E. Joseph Gottlieb Kolreuter’s Contributions to Biology. Osiris 1986, 2, 135–176. [Google Scholar] [CrossRef]
- Vedel, F.; Pla, M.; Vitart, V.; Gutierres, S.; Chetrit, P.; Paepe, R.D. Molecular basis of nuclear and cytoplasmic male sterility in higher plants. Plant Physiol. Biochem. 1994, 32, 601–618. [Google Scholar] [CrossRef]
- Horn, R.; Gupta, K.J.; Colombo, N. Mitochondrion role in molecular basis of cytoplasmic male sterility. Mitochondrion 2014, 19, 198–205. [Google Scholar] [CrossRef]
- Kim, Y.J.; Zhang, D. Molecular control of male fertility for crop hybrid breeding. Trends Plant Sci. 2018, 23, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Kaul, M.L.R. Male Sterility in Higher Plants. Springer-Verl. Berl. Heidelb. 1988, 10, 775–797. [Google Scholar] [CrossRef]
- Schnable, P.S.; Wise, R.P. The molecular basis of cytoplasmic male sterility and fertility restoration. Trends Plant Sci. 1998, 3, 175–180. [Google Scholar] [CrossRef]
- Murai, K.; Tsunewaki, K. Photoperiod-sensitive cytoplasmic male sterility in wheat with Aegilops crassa cytoplasm. Euphytica 1993, 67, 41–48. [Google Scholar] [CrossRef]
- Li, Y.F.; Zhao, C.P.; Zhang, F.T.; Sun, H.; Sun, D.F. Fertility alteration in the photo-thermo-sensitive male sterile line BS20 of wheat (Triticum aestivum L.). Euphytica 2006, 151, 207–213. [Google Scholar] [CrossRef]
- Gómez, J.F.; Talle, B.; Wilson, Z.A. Anther and pollen development: A conserved developmental pathway. J. Integr. Plant Biol. 2015, 57, 876–891. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.K.; Devi, K.U.; Arundhati, A. Applications of Genie Male Sterility in Plant Breeding. Plant Breed. 1990, 105, 1–25. [Google Scholar] [CrossRef]
- Endo, T.R.; Mukai, Y.; Yamamoto, M.; Gill, B. Physical mapping of a male-fertility gene of common wheat. Jpn. J. Genet. 1991, 66, 291–295. [Google Scholar] [CrossRef]
- Liu, B.H.; Deng, J.Y. A Dominant Gene for Male Sterility in Wheat. Plant Breed. 1986, 97, 204–209. [Google Scholar] [CrossRef]
- Maan, S.S.; Carlson, K.M.; Williams, N.D.; Yang, T. Chromosomal Arm Location and Gene-Centromere Distance of a Dominant Gene for Male Sterility in Wheat1. Crop Sci. 1987, 27, 494–500. [Google Scholar] [CrossRef]
- Qi, L.L.; Gill, B.S. High-density physical maps reveal that the dominant male-sterile gene Ms3 is located in a genomic region of low recombination in wheat and is not amenable to map-based cloning. Theor. Appl. Genet. 2001, 103, 998–1006. [Google Scholar] [CrossRef]
- Maan, S.S.; Kianian, S.F. Third dominant male sterility gene in common wheat. Wheat Inf. Serv. 2001, 93, 27–31. [Google Scholar] [CrossRef]
- Klindworth, D.L.; Williams, N.D.; Maan, S.S. Chromosomal location of genetic male sterility genes in four mutants of hexaploid wheat. Crop Sci. 2002, 42, 1447–1450. [Google Scholar] [CrossRef]
- Whitford, R.; Fleury, D.; Reif, J.C.; Garcia, M.; Okada, T.; Korzun, V.; Langridge, P. Hybrid breeding in wheat: Technologies to improve hybrid wheat seed production. J. Exp. Bot. 2013, 64, 5411–5428. [Google Scholar] [CrossRef]
- Tucker, E.J.; Baumann, U.; Kouidri, A.; Suchecki, R.; Baes, M.; Garcia, M.; Okada, T.; Dong, C.; Wu, Y.; Sandhu, A.; et al. Molecular identification of the wheat male fertility gene Ms1 and its prospects for hybrid breeding. Nat. Commun. 2017, 8, 869. [Google Scholar] [CrossRef]
- Wang, Z.; Li, J.; Chen, S.; Heng, Y.; Chen, Z.; Yang, J.; Zhou, K.; Pei, J.; He, H.; Deng, X.W.; et al. Poaceae-specific MS1 encodes a phospholipid-binding protein for male fertility in bread wheat. Proc. Natl. Acad. Sci. USA 2017, 114, 12614–12619. [Google Scholar] [CrossRef]
- Ni, F.; Qi, J.; Hao, Q.; Lyu, B.; Luo, M.C.; Wang, Y.; Chen, F.; Wang, S.; Zhang, C.; Epstein, L.; et al. Wheat Ms2 encodes for an orphan protein that confers male sterility in grass species. Nat. Commun. 2017, 8, 15121. [Google Scholar] [CrossRef]
- Pallotta, M.A.; Warner, P.; Kouidri, A.; Tucker, E.J.; Baes, M.; Suchecki, R.; Watson-Haigh, N.; Okada, T.; Garcia, M.; Sandhu, A.; et al. Wheat ms5 male-sterility is induced by recessive homoeologous A and D genome non-specificlipid transfer proteins. Plant J. 2019, 99, 673–685. [Google Scholar] [CrossRef]
- Okada, A.; Arndell, T.; Borisjuk, N.; Sharma, N.; Watson-Haigh, N.S.; Tucker, E.J.; Baumann, U.; Langridge, P.; Whitford, R. CRISPR/Cas9-mediated knockout of Ms1 enables the rapid generation of male-sterile hexaploid wheat lines for use in hybrid seed production. Plant Biotechnol. J. 2019, 17, 1905–1913. [Google Scholar] [CrossRef]
- Wang, A.; Xia, Q.; Xie, W.; Dumonceaux, T.; Zou, J.; Datla, R.; Selvaraj, G. Male gametophyte development in bread wheat (Triticum aestivum L.): Molecular, cellular, and biochemical analyses of a sporophytic contribution to pollen wall ontogeny. Plant J. 2002, 30, 613–623. [Google Scholar] [CrossRef]
- Wang, A.M.; Xia, Q.; Xie, W.S.; Datla, R.; Selvaraj, G. The classical Ubisch bodies carry a sporophytically produced structural protein (RAFTIN) that is essential for pollen development. Proc. Natl. Acad. Sci. USA 2003, 100, 14487–14492. [Google Scholar] [CrossRef] [PubMed]
- Cigan, A.M.; Singh, M.; Benn, G.; Feigenbutz, L.; Kumar, M.; Cho, M.J.; Svitashev, S.; Young, J. Targeted mutagenesis of a conserved anther-expressed P450 gene confers male sterility in monocots. Plant Biotechol. J. 2017, 15, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Ni, F.; Wang, X.; Sun, M.; Cui, Y.; Wu, J.; Caplan, A.; Fu, D. The anther-specific CYP704B is potentially responsible for MSG26 male sterility in barley. Theor. Appl. Genet. 2019, 132, 2413–2423. [Google Scholar] [CrossRef]
- Singh, M.; Kumar, M.; Thilges, K.; Cho, M.; Cigan, A.M. MS26/CYP704B is required for anther and pollen wall development in bread wheat (Triticum aestivum L.) and combining mutations in all three homeologs causes male sterility. PLoS ONE 2017, 12, e0177632. [Google Scholar] [CrossRef]
- Singh, M.; Kumar, M.; Albertsen, M.C.; Young, J.K.; Cigan, A.M. Concurrent modifications in the three homeologs of Ms45 gene with CRISPR-Cas9 lead to rapid generation of male sterile bread wheat (Triticum aestivum L.). Plant Mol. Biol. 2018, 97, 371–383. [Google Scholar] [CrossRef]
- Tang, Z.; Zhang, L.; Yang, D.; Zhao, C.; Zheng, Y. Cold stress contributes to aberrant cytokinesis during male meiosis I in a wheat thermosensitive genic male sterile line. Plant Cell Environ. 2011, 34, 389–405. [Google Scholar] [CrossRef]
- Tang, Z.; Zhang, L.; Xu, C.; Yuan, S.; Zhang, F.; Zheng, Y.; Zhao, C. Uncovering Small RNA-Mediated Responses to Cold Stress in a Wheat Thermosensitive Genic Male-Sterile Line by Deep Sequencing. Plant Physiol. 2012, 159, 721–738. [Google Scholar] [CrossRef]
- Su, Y.; Liu, J.; Liang, W.; Dou, Y.; Fu, R.; Li, W.; Feng, C.; Gao, C.; Zhang, D.; Kang, Z.; et al. Wheat AGAMOUS LIKE 6 transcription factors function in stamen development by regulating the expression of TaAPETALA3. Development 2019, 146, dev177527. [Google Scholar] [CrossRef]
- Sun, Y.; Zhu, X.; He, R.; Li, J.; An, J.; Jiao, Z.; Jiang, Y.; Li, Q.; Niu, J. Selection and Identification of the EMS-Induced Wheat Mutants from Shengnong 1. J. Triticeae Crop. 2018, 38, 782–790. [Google Scholar] [CrossRef]
- Laser, K.D.; Lersten, N.R. Anatomy and Cytology of Microsporogenesis in Cytoplasmic Male Sterile Angiosperms. Bot. Rev. 1972, 38, 425–454. [Google Scholar] [CrossRef]
- Rao, Y.S. Cytohistology of cytoplasmic male sterile lines in hybrid rice. In Proceedings of the Hybrid Rice: Proceedings of the International Symposium on Hybrid Rice, Manila, Philippines, 6–10 October 1986; pp. 115–128. [Google Scholar]
- Sui, X.; Sun, L. Studies and utilizations of taigu-male-sterile wheat. J. Shandong Agric. Univ. 2001, 32, 239–244. [Google Scholar] [CrossRef]
- Liu, Z.; Shi, X.; Li, S.; Hu, G.; Zhang, L.; Song, X. Tapetal-Delayed programmed cell death (PCD) and oxidative stress-induced male sterility of aegilops uniaristata cytoplasm in wheat. Int. J. Mol. Sci. 2018, 19, 1708. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Sun, G.; Shi, C.; Sun, D. Transcriptome analysis reveals new microRNAs-mediated pathway involved in anther development in male sterile wheat. BMC Genom. 2018, 19. [Google Scholar] [CrossRef]
- Guo, J.X.; Liu, Y.G. Molecular control of male reproductive development and pollen fertility in rice. J. Integr. Plant Biol. 2012, 54, 967–978. [Google Scholar] [CrossRef]
- Nonomura, K.I.; Nakano, M.; Fukuda, T.; Eiguchi, M.; Miyao, A.; Hirochika, H.; Kurata, N. The Novel Gene HOMOLOGOUS PAIRING ABERRATION IN RICE MEIOSIS1 of Rice Encodes a Putative Coiled-Coil Protein Required for Homologous Chromosome Pairing in Meiosis. Plant Cell 2004, 16, 1008–1020. [Google Scholar] [CrossRef]
- Nonomura, K.I.; Nakano, M.; Murata, K.; Miyoshi, K.; Eiguchi, M.; Miyao, A.; Hirochika, H.; Kurata, N. An insertional mutation in the rice PAIR2 gene, the ortholog of Arabidopsis ASY1, results in a defect in homologous chromosome pairing during meiosis. Mol. Genet. Genom. 2004, 271, 121–129. [Google Scholar] [CrossRef]
- Nonomura, K.I.; Nakano, M.; Eiguchi, M.; Suzuki, T.; Kurata, N. PAIR2 is essential for homologous chromosome synapsis in rice meiosis I. J. Cell Sci. 2006, 119, 217–225. [Google Scholar] [CrossRef]
- Kitajima, T.S.; Kawashima, S.A.; Watanabe, Y. The conserved kinetochore protein shugoshin protects centromeric cohesion during meiosis. Nature 2004, 427, 510–517. [Google Scholar] [CrossRef]
- Wang, M.; Tang, D.; Luo, Q.; Jin, Y.; Shen, Y.; Wang, K.; Cheng, Z. BRK1, a bub1-related kinase, is essential for generating proper tension between homologous kinetochores at metaphase i of rice meiosis. Plant Cell 2012, 24, 4961–4973. [Google Scholar] [CrossRef]
- Werck-Reichhart, D.; Bak, S.; Paquette, S. Cytochromes P450. Arab. Book 2002, e0028. [Google Scholar] [CrossRef]
- Morant, M.; Bak, S.; Moller, B.L.; Werck-Reichhart, D. Plant cytochromes P450: Tools for pharmacology, plant protection and phytoremediation. Curr. Opin. Biotechnol. 2003, 14, 151–162. [Google Scholar] [CrossRef]
- Morant, M.; Jørgensen, K.; Schaller, H.; Pinot, F.; Møller, B.L.; Werck-Reichhart, D.; Bak, S. CYP703 Is an Ancient Cytochrome P450 in Land Plants Catalyzing in-Chain Hydroxylation of Lauric Acid to Provide Building Blocks for Sporopollenin Synthesis in Pollen. Plant Cell 2007, 19, 1473–1487. [Google Scholar] [CrossRef] [PubMed]
- Phan, H.A.; Li, S.F.; Parish, R.W. MYB80, a regulator of tapetal and pollen development, is functionally conserved in crops. Plant Mol. Biol. 2012, 78, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Higginson, T.; Li, S.F.; Parish, R.W. AtMYB103 regulates tapetum and trichome development in Arabidopsis thaliana. Plant J. 2003, 35, 177–192. [Google Scholar] [CrossRef]
- Li, S.F.; Iacuone, S.; Parish, R.W. Suppression and restoration of male fertility using a transcription factor. Plant Biotechnol. J. 2007, 5, 297–312. [Google Scholar] [CrossRef]
- Zhang, Z.B.; Zhu, J.; Gao, J.F.; Wang, C.; Li, H.; Li, H.; Zhang, H.Q.; Zhang, S.; Wang, D.M.; Wang, Q.X.; et al. Transcription factor AtMYB103 is required for anther development by regulating tapetum development, callose dissolution and exine formation in Arabidopsis. Plant J. 2007, 52, 528–538. [Google Scholar] [CrossRef]
- Li, N.; Zhang, D.S.; Liu, H.S.; Yin, C.S.; Li, X.; Liang, W.; Yuan, Z.; Xu, B.; Chu, H.W.; Wang, J.; et al. The Rice Tapetum Degeneration Retardation Gene Is Required for Tapetum Degradation and Anther Development. Plant Cell 2006, 18, 2999–3014. [Google Scholar] [CrossRef]
- Ishiguro, S.; Kawai-Oda, A.; Ueda, J.; Nishida, I.; Okada, K. The DEFECTIVE IN ANTHER DEHISCENCE1 Gene Encodes a Novel Phospholipase A1 Catalyzing the Initial Step of Jasmonic Acid Biosynthesis, Which Synchronizes Pollen Maturation, Anther Dehiscence, and Flower Opening in Arabidopsis. Plant Cell 2001, 13, 2191–2209. [Google Scholar] [CrossRef]
- Kaneko, M.; Inukai, Y.; Ueguchi-Tanaka, M.; Itoh, H.; Izawa, T.; Kobayashi, Y.; Hattori, T.; Miyao, A.; Hirochika, H.; Ashikari, M.; et al. Loss-offunction mutations of the rice GAMYB gene impair α-amylase expression in aleurone and flower development. Plant Cell 2004, 16, 33–44. [Google Scholar] [CrossRef]
- Aya, K.; Ueguchi-Tanaka, M.; Kondo, M.; Hamada, K.; Yano, K.; Nishimura, M.; Matsuoka, M. Gibberellin modulates anther development in rice via the transcriptional regulation of GAMYB. Plant Cell 2009, 21, 1453–1472. [Google Scholar] [CrossRef]
- Hord, C.L.H.; Chen, C.; DeYoung, B.J.; Clark, S.E.; Ma, H. The BAM1/BAM2 Receptor-Like Kinases Are Important Regulators of Arabidopsis Early Anther Development. Plant Cell 2006, 18, 1667–1680. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liang, W.; Yang, X.; Luo, X.; Jiang, N.; Ma, H.; Zhang, D. Carbon Starved Anther Encodes a MYB Domain Protein That Regulates Sugar Partitioning Required for Rice Pollen Development. Plant Cell 2010, 22, 672–689. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y.; Osborne, E.; Poindexter, P.D.; Somerville, C.R. Microspore Separation in the quartet 3 Mutants of Arabidopsis Is Impaired by a Defect in a Developmentally Regulated Polygalacturonase Required for Pollen Mother Cell Wall Degradation. Plant Physiol. 2003, 133, 1170–1180. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; De Palma, J.; Oane, R.; Gamuyao, R.; Luo, M.; Chaudhury, A.; Herve, P.; Xue, Q.; Bennett, J. OsTDL1A binds to the LRR domain of rice receptor kinase MSP1, and is required to limit sporocyte numbers. Plant J. 2008, 54, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Meng, Z.; Liang, W.; Behera, S.; Kudla, J.; Tucker, M.R.; Luo, Z.; Chen, M.; Xu, D.; Zhao, G.; et al. A Rice Ca Binding Protein Is Required for Tapetum Function and Pollen Formation. Plant Physiol. 2016, 172, 1772–1786. [Google Scholar] [CrossRef]
- Yang, L.; Diao, X. Progress in Identification of Plant Male Sterility Related Nuclear Genes. Plant Genet. Resour. 2013, 14, 1108–1117. [Google Scholar] [CrossRef]
- Zhu, Q.; Song, Y.; Zhang, G.; Ju, L.; Zhang, J.; Yu, Y.; Niu, N.; Wang, J.; Ma, S. De Novo Assembly and Transcriptome Analysis of Wheat with Male Sterility Induced by the Chemical Hybridizing Agent SQ-1. PLoS ONE 2015, 10, e0123556. [Google Scholar] [CrossRef]
- Goetz, M.; Godt, D.E.; Guivarc, A.; Kahmann, U.; Chriqui, D.; Roitsch, T. Induction of male sterility in plants by metabolic engineering of the carbohydrate supply. Proc. Natl. Acad. Sci. USA 2001, 98, 6522–6527. [Google Scholar] [CrossRef]
- Sanders, P.M.; Lee, P.Y.; Biesgen, C.; Boone, J.D.; Beals, T.P.; Weiler, E.W.; Goldberg, R.B. The Arabidopsis DELAYED DEHISCENCE1 Gene Encodes an Enzyme in the Jasmonic Acid Synthesis Pathway. Plant Cell 2000, 12, 1041–1062. [Google Scholar] [CrossRef]
- Caldelari, D.; Wang, G.; Farmer, E.E.; Dong, X. Arabidopsislox3 lox4double mutants are male sterile and defective in global proliferative arrest. Plant Mol. Biol. 2011, 75, 25–33. [Google Scholar] [CrossRef]
- Stintzi, A.; Browse, J. The Arabidopsis male-sterile mutant, opr3, lacks the 12-oxophytodienoic acid reductase required for jasmonate synthesis. Proc. Natl. Acad. Sci. USA 2000, 97, 10625–10630. [Google Scholar] [CrossRef] [PubMed]
- Clément, C.; Audran, J.C. Anther wall layers control pollen sugar nutrition in Lilium. Protoplasma 1995, 187, 172–181. [Google Scholar] [CrossRef]
- Jiang, S.Y.; Chi, Y.H.; Wang, J.Z.; Zhou, J.X.; Cheng, Y.S.; Zhang, B.L.; Ma, A.; Vanitha, J.; Ramachandran, S. Sucrose metabolism gene families and their biological functions. Sci. Rep. 2015, 5, 17583. [Google Scholar] [CrossRef] [PubMed]
- Min, L.; Zhu, L.; Tu, L.; Deng, F.; Yuan, D.; Zhang, X. Cotton GhCKI disrupts normal male reproduction by delaying tapetum programmed cell death via inactivating starch synthase. Plant J. 2013, 75, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Dorion, S.; Lalonde, S.; Saini, H.S. lnduction of male sterility in wheat by meiotic-stage water deficit is preceded by a decline in invertase activity and changes in carbohydrate metabolism in anthers. Plant Physiol. 1996, 111, 137–145. [Google Scholar] [CrossRef]
- Sun, L.; Sui, X.; Lucas, W.J.; Li, Y.; Feng, S.; Ma, S.; Fan, J.; Gao, L.; Zhang, Z. Down-regulation of the sucrose transporter CsSUT1 causes male sterility by altering carbohydrate supply. Plant Physiol. 2019, 180, 986–997. [Google Scholar] [CrossRef]
- Sivitz, A.B.; Reinders, A.; Ward, J.M. Arabidopsis sucrose transporter AtSUC1 is important for pollen germination and sucrose-induced anthocyanin accumulation. Plant Physiol. 2008, 147, 92–100. [Google Scholar] [CrossRef]
- Hirose, T.; Zhang, Z.; Miyao, A.; Hirochika, H.; Ohsugi, R.; Terao, T. Disruption of a gene for rice sucrose transporter, OsSUT1, impairs pollen function but pollen maturation is unaffected. J. Exp. Bot. 2010, 61, 3639–3646. [Google Scholar] [CrossRef]
- Basnet, R.; Hussain, N.; Shu, Q. OsDGD2β is the sole digalactosyldiacylglycerol synthase gene highly expressed in anther, and its mutation confers male sterility in rice. Rice 2019, 12, 66. [Google Scholar] [CrossRef]
- Lee, S.K.; Eom, J.S.; Hwang, S.K.; Shin, D.; An, G.; Okita, T.W.; Jeon, J.S. Plastidic phosphoglucomutase and ADP-glucose pyrophosphorylase mutants impair starch synthesis in rice pollen grains and cause male sterility. J. Exp. Bot. 2016, 67, 5557–5569. [Google Scholar] [CrossRef]
- Muller, J.; Aeschbacher, R.A.; Wingler, A.; Boller, T.; Wiemken, A. Trehalose and trehalase in Arabidopsis. Plant Physiol. 2001, 125, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.Y.; Qin, Z.; Jiang, Y.M.; Shen, C.C.; Duan, Z.B.; Niu, J.S. Screening wheat genotypes for resistance to black point and the effects of diseased kernels on seed germination. J. Plant Dis. Prot. 2014, 121, 79–88. [Google Scholar] [CrossRef]
- Zadoks, J.C.; Chang, T.T.; Konzak, C.F. A decimal code for the growth stages of cereals. Weed Res. 1974, 14, 415–421. [Google Scholar] [CrossRef]
- Vahamidis, P.; Karamanos, A.; Economou, G.; Fasseas, C. A new scale for the assessment of wheat spike morphogenesis. Ann. Appl. Biol. 2014, 164, 220–231. [Google Scholar] [CrossRef]
- Geng, D.; Chen, P.; Shen, X.; Zhang, Y.; Li, X.; Jiang, L.; Xie, Y.; Niu, C.; Zhang, J.; Huang, X.; et al. MdMYB88 and MdMYB124 enhance drought tolerance by modulating root vessels and cell walls in apple. Plant Physiol. 2018, 178, 1296–1309. [Google Scholar] [CrossRef]
- Phan, H.A.; Iacuone, S.; Li, S.F.; Parish, R.W. The MYB80 transcription factor is required for pollen development and the regulation of tapetal programmed cell death in Arabidopsis thaliana. Plant Cell 2011, 23, 2209–2224. [Google Scholar] [CrossRef]
- He, R.; Ni, Y.; Li, J.; Jiao, Z.; Zhu, X.; Jiang, Y.; Li, Q.; Niu, J. Quantitative Changes in the Transcription of Phytohormone-Related Genes: Some Transcription Factors Are Major Causes of the Wheat Mutant dmc Not Tillering. Int. J. Mol. Sci. 2018, 19, 1324. [Google Scholar] [CrossRef]
- Florea, L.; Song, L.; Salzberg, S.L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Res. 2013, 2, 188. [Google Scholar] [CrossRef]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2009, 26, 136–138. [Google Scholar] [CrossRef]
- Pearson, E.S. The Test of Significance for the Correlation Coefficient: Some Further Results. J. Am. Stat. Assoc. 1931, 26, 128–134. [Google Scholar] [CrossRef]
- Zou, H.; Hastie, T.; Tibshirani, R. Sparse Principal Component Analysis. J. Comput. Graph. Stat. 2010, 15, 265–286. [Google Scholar] [CrossRef]
- Hartiganm, J.A.; Wong, M.A. Algorithm AS 136: A K-Means Clustering Algorithm. J. R. Stat. Soc. Ser. C 1979, 28, 100–108. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 29, 559. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆Ct method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | Shengnong 1 | NWMS1 |
|---|---|---|
| Plant height (cm) | 70.59 ± 1.54 | 68.19 ± 3.45 * |
| Spike length (cm) | 10.6 ± 0.39 | 8.73 ± 0.51 * |
| Flag leaf length (cm) | 20.02 ± 3.37 | 18.03 ± 2.03 * |
| Flag leaf width (cm) | 2.3 ± 0.15 | 1.88 ± 0.19 * |
| Internode length under spike (cm) | 31.83 ± 2.39 | 29.24 ± 5.21 * |
| Effective tiller number | 13.4 ± 2.55 | 10.2 ± 2.53 * |
| Internode number of the main stem | 5.00 ± 0.00 | 5.00 ± 0.00 |
| Spikelet number on the main stem | 20.4 ± 1.35 | 19.4 ± 1.84 |
| Spike type | Rectangle | Rectangle |
| Awn color | White | White |
| Heading stage (Month-Day) | 04-10 | 04-17 ** |
| Anthesis stage (Month-Day) | 04-23 | 05-01 ** |
| Maturity stage (Month-Day) | 05-22 | 05-31 ** |
| Sample Pair | Class | Annotation | GO ID | Corrected p-Value |
|---|---|---|---|---|
| S1-NWMS1 vs. WT | biological process | response to cadmium ion | GO:0046686 | 1.9 × 10−23 |
| cellular component | cytoplasmic membrane-bounded vesicle | GO:0016023 | 1.0 × 10−30 | |
| molecular function | electron carrier activity | GO:0009055 | 3.9 × 10−19 | |
| S2-NWMS1 vs. WT | biological process | pentose-phosphate shunt | GO:0006098 | 1.0 × 10−30 |
| cellular component | cytoplasmic vesicle | GO:0016023 | 1.0 × 10−30 | |
| molecular function | heme binding | GO:0020037 | 2.2 × 10−22 | |
| S3-NWMS1 vs. WT | biological process | oxidation-reduction process | GO:0055114 | 4.8 × 10−25 |
| cellular component | cytoplasmic membrane-bounded vesicle | GO:0016023 | 1.0 × 10−30 | |
| molecular function | heme binding | GO:0020037 | 1.6 × 10−29 |
| Sample Pair | Pathway | Ko ID | Corrected p-Value |
|---|---|---|---|
| S1-NWMS1 vs. WT | amino sugar and nucleotide sugar metabolism | ko00520 | 0.031039284 |
| S2-NWMS1 vs. WT | phenylalanine metabolism | ko00360 | 0 |
| phenylpropanoid biosynthesis | ko00940 | 0 | |
| glutathione metabolism | ko00480 | 1.50 × 10−10 | |
| S3-NWMS1 vs. WT | phenylalanine metabolism | ko00360 | 0 |
| glutathione metabolism | ko00480 | 0 | |
| starch and sucrose metabolism | ko00500 | 0 |
| Gene ID | Modules | Clusters | S1_NWMS1 | S1_WT | S2_NWMS1 | S2_WT | S3_NWMS1 | S3_WT | Gene Name |
|---|---|---|---|---|---|---|---|---|---|
| TraesCS2D01G420100 | light cyan | K3 | 0.00 | 0.00 | 21.36 | 14.53 | 5.23 | 0.50 | SKP1B |
| TraesCS7B01G213800 | light cyan | K3 | 0.00 | 0.02 | 3.35 | 2.62 | 0.81 | 0.02 | Os03g0802700 |
| TraesCS7A01G430600 | light cyan | K3 | 0.01 | 0.00 | 21.98 | 16.60 | 5.20 | 0.09 | BIP5 |
| TraesCS4B01G248600 | light cyan | K3 | 2.07 | 2.56 | 43.93 | 34.65 | 12.54 | 0.68 | BGLU6 |
| TraesCS2D01G373700 | light cyan | K3 | 0.00 | 0.00 | 41.16 | 23.75 | 4.36 | 0.42 | F775_16360 |
| TraesCS5A01G285900 | black | K3 | 0.00 | 0.00 | 2.95 | 5.69 | 2.04 | 0.03 | PAP27 |
| TraesCS1A01G413400 | black | K3 | 0.00 | 0.00 | 9.60 | 18.89 | 5.89 | 0.49 | KCS11 |
| TraesCS2B01G056100 | black | K3 | 0.00 | 0.00 | 23.47 | 43.09 | 11.88 | 0.05 | SWEET6B |
| TraesCS1D01G376300 | black | K3 | 0.09 | 0.10 | 8.40 | 17.96 | 6.75 | 0.25 | ADH3 |
| TraesCS7A01G536800 | black | K3 | 0.01 | 0.00 | 8.33 | 32.95 | 6.53 | 0.12 | At5g03610 |
| TraesCS4D01G296000 | green | K2 | 0.41 | 0.04 | 41.70 | 16.73 | 65.78 | 5.44 | TIFY10B |
| TraesCS3B01G558400 | green | K2 | 0.60 | 0.52 | 8.20 | 5.28 | 15.95 | 1.75 | CSLA11 |
| TraesCS6D01G325100 | green | K2 | 0.54 | 0.89 | 44.31 | 29.72 | 94.55 | 9.71 | SDH |
| TraesCS6A01G259200 | green | K2 | 0.21 | 0.02 | 18.59 | 7.12 | 31.49 | 2.80 | ATL41 |
| TraesCS2A01G276000 | green | K2 | 0.10 | 0.09 | 4.03 | 1.87 | 7.64 | 0.69 | EXO70B1 |
| TraesCS1D01G270100 | brown | K4 | 0.06 | 0.00 | 0.25 | 8.62 | 112.57 | 445.69 | GASA2 |
| TraesCS3D01G066700 | brown | K4 | 0.00 | 0.00 | 1.20 | 10.69 | 126.30 | 507.99 | F775_31707 |
| TraesCS5D01G282500 | brown | K4 | 0.00 | 0.01 | 0.09 | 1.87 | 34.21 | 146.67 | SAG12 |
| TraesCS1B01G353800 | brown | K4 | 0.00 | 0.01 | 0.04 | 0.74 | 21.35 | 94.06 | STP8 |
| TraesCS4B01G125500 | brown | K6 | 0.00 | 0.00 | 0.04 | 0.56 | 4.34 | 23.94 | At1g26850 |
| Gene ID | S1_NWMS1 | S1_WT | S2_NWMS1 | S2_WT | S3_NWMS1 | S3_WT | Gene Name | Reported Function |
|---|---|---|---|---|---|---|---|---|
| novelGene_53829 | 0.00 | 0.00 | 0.22 | 0.03 | 0.01 | 0.02 | PAIR1 | 3 |
| novelGene_56612 | 0.20 | 0.23 | 0.09 | 0.20 | 0.22 | 0.16 | PAIR2 | 3 |
| TraesCS7A01G316600 | 1.40 | 1.26 | 0.79 | 0.56 | 0.96 | 1.11 | PSS1 | 3, 5 |
| TraesCS2A01G226000 | 8.31 | 8.92 | 2.03 | 2.59 | 1.98 | 1.69 | BUB1 | 2, 3 |
| TraesCS6A01G364300 | 8.81 | 9.02 | 1.34 | 1.02 | 1.60 | 1.39 | SGO1 | 2, 3 |
| TraesCS7A01G309300 | 0.48 | 0.47 | 260.82 | 220.0 | 26.93 | 0.83 | CYP703A2 | 4, 5, 6 |
| novelGene_128161 | 0.54 | 0.68 | 3.13 | 1.28 | 0.56 | 0.26 | MYB80 | 4, 6 |
| novelGene_128276 | 0.00 | 0.07 | 0.08 | 0.00 | 0.06 | 0.09 | DEX1 | 4, 5 |
| TraesCS1B01G247200 | 4.19 | 5.16 | 9.20 | 8.53 | 9.42 | 7.83 | DAD1 | 5, 6 |
| TraesCS7A01G305700 | 2.57 | 2.33 | 0.36 | 0.59 | 0.19 | 0.24 | TDR | 4 |
| TraesCS7A01G458700 | 0.00 | 0.00 | 0.00 | 0.03 | 0.17 | 25.50 | GAMYB | 5, 6 |
| TraesCS5A01G481600 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | BAM1 | 1 |
| TraesCS1A01G444500 | 3.78 | 3.90 | 0.23 | 0.11 | 0.18 | 0.12 | BAM2 | 1 |
| TraesCS2D01G407700 | 28.12 | 32.21 | 183.32 | 193.7 | 192.5 | 20.81 | CSA | 6, 7 |
| novelGene_21887 | 0.02 | 0.00 | 0.01 | 0.00 | 0.00 | 0.20 | TDL1B | 1, 3 |
| novelGene_139463 | 2.67 | 2.34 | 3.78 | 2.67 | 2.63 | 2.35 | MSP1 | 1, 3 |
| TraesCS2A01G423900 | 1.95 | 2.32 | 91.01 | 82.45 | 11.51 | 5.97 | QRT3 | 6 |
| Enzyme D | Enzyme Code * | S1-Log2 (NWMS1/WT) ** | S2-Log2 (NWMS1/WT) | S3-Log2 (NWMS1/WT) |
|---|---|---|---|---|
| glycogen phosphorylase | 2.4.1.1 | − | −1.2 | −5.64 |
| sucrose synthase | 2.4.1.13 | − | −2 | 5.07 & −2.0 |
| sucrose-phosphate synthase | 2.4.1.14 | − | −2.5 | −1.24 |
| trehalose 6-phosphate synthase | 2.4.1.15 | − | 1.2 & −1.5 | 4.48 & −2.86 |
| 1,4-alpha-glucan branching enzyme | 2.4.1.18 | − | − | 1.43 & −3.96 |
| starch synthase | 2.4.1.21 | − | −1.2 | 1.24 & −1.79 |
| 4-alpha-glucanotransferase | 2.4.1.25 | − | −1.23 | −2.72 |
| Hexokinase | 2.7.1.1 | − | 3.2 & −1.0 | 5.14 & −3.74 |
| Fructokinase | 2.7.1.4 | − | 1.2 & −1.5 | 3.62 & −2.58 |
| glucose-1-phosphate adenylyltransferase | 2.7.7.27 | − | −1.2 | −3.56 |
| UTP-glucose-1-phosphate uridylyltransferase | 2.7.7.9 | − | − | −8.72 |
| trehalose 6-phosphate phosphatase | 3.1.3.12 | 1.67 & −10.25 | 3.0 & −1.5 | 3.16 & −1.68 |
| alpha-amylase | 3.2.1.1 | − | −2.78 | 9.25 & −1.30 |
| beta-amylase | 3.2.1.2 | − | 1 | 2.10 & −3.89 |
| alpha-glucosidase | 3.2.1.20 | − | 1.8 | 1.99 & −2.03 |
| beta-glucosidase | 3.2.1.21 | 1.47 | − | 2.54 & −2.65 |
| eta-fructofuranosidase | 3.2.1.26 | −1.01 | 1.1 & −2.1 | 1.67 & −3.28 |
| alpha, alpha-trehalase | 3.2.1.28 | − | − | 2.46 |
| glucan endo-1,3-beta-glucosidase 1/2/3 | 3.2.1.39 | −1.88 | 1.2 & −1.7 | 1.92 & −1.56 |
| Endoglucanase | 3.2.1.4 | − | 2 | −6.85 |
| ADP-sugar diphosphatase | 3.6.1.21 | − | − | −2.75 |
| Phosphoglucomutase | 5.4.2.2 | − | − | −1.71 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Zhang, J.; Li, H.; Niu, H.; Xu, Q.; Jiao, Z.; An, J.; Jiang, Y.; Li, Q.; Niu, J. The Major Factors Causing the Microspore Abortion of Genic Male Sterile Mutant NWMS1 in Wheat (Triticum aestivum L.). Int. J. Mol. Sci. 2019, 20, 6252. https://doi.org/10.3390/ijms20246252
Li J, Zhang J, Li H, Niu H, Xu Q, Jiao Z, An J, Jiang Y, Li Q, Niu J. The Major Factors Causing the Microspore Abortion of Genic Male Sterile Mutant NWMS1 in Wheat (Triticum aestivum L.). International Journal of Molecular Sciences. 2019; 20(24):6252. https://doi.org/10.3390/ijms20246252
Chicago/Turabian StyleLi, Junchang, Jing Zhang, Huijuan Li, Hao Niu, Qiaoqiao Xu, Zhixin Jiao, Junhang An, Yumei Jiang, Qiaoyun Li, and Jishan Niu. 2019. "The Major Factors Causing the Microspore Abortion of Genic Male Sterile Mutant NWMS1 in Wheat (Triticum aestivum L.)" International Journal of Molecular Sciences 20, no. 24: 6252. https://doi.org/10.3390/ijms20246252
APA StyleLi, J., Zhang, J., Li, H., Niu, H., Xu, Q., Jiao, Z., An, J., Jiang, Y., Li, Q., & Niu, J. (2019). The Major Factors Causing the Microspore Abortion of Genic Male Sterile Mutant NWMS1 in Wheat (Triticum aestivum L.). International Journal of Molecular Sciences, 20(24), 6252. https://doi.org/10.3390/ijms20246252