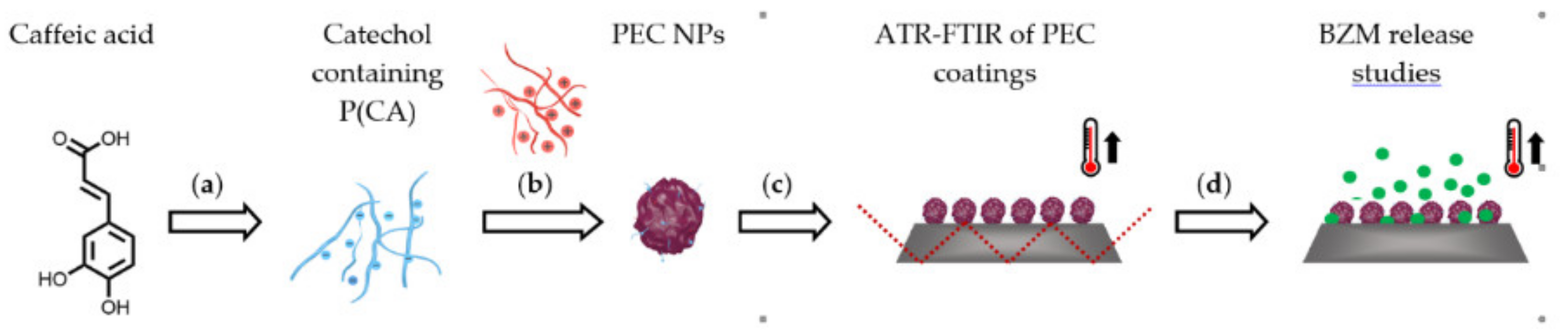
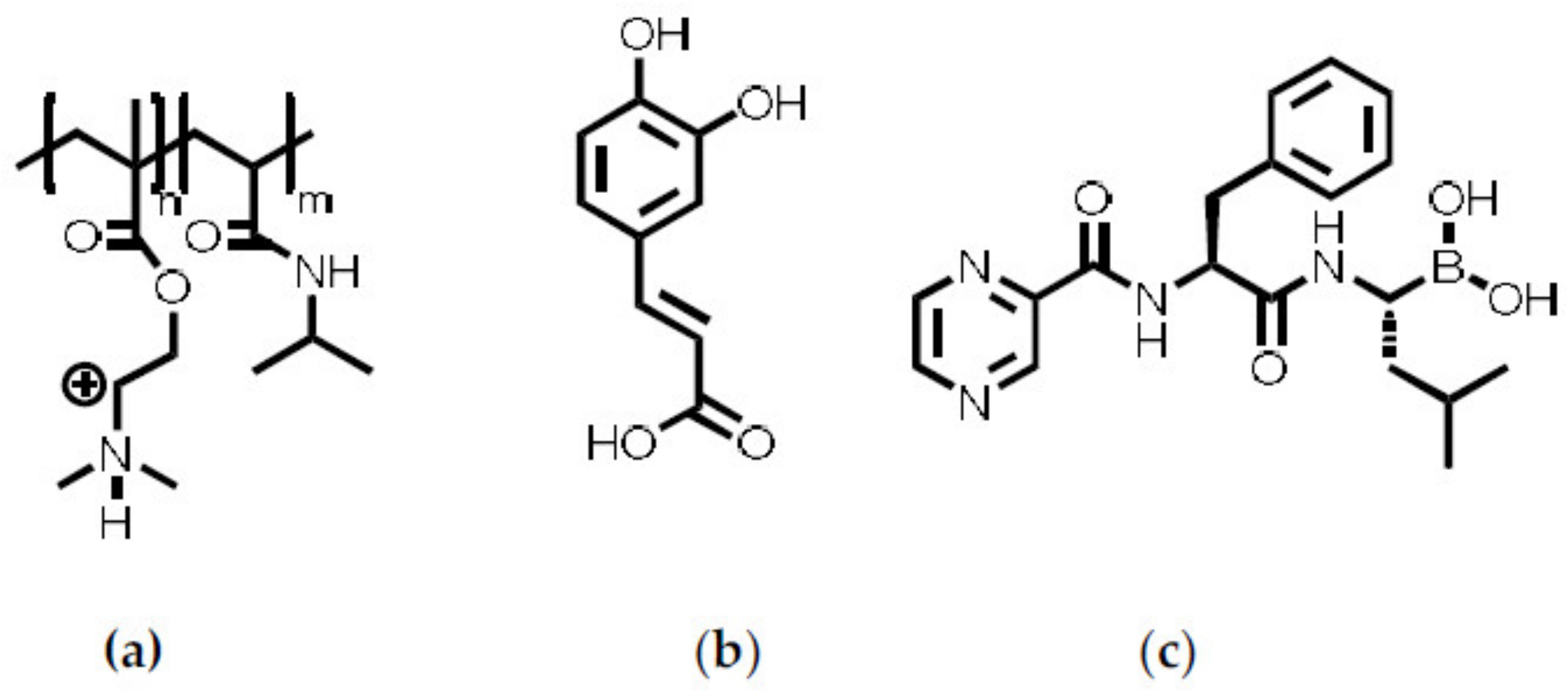
In the following synthesis and molecular structure of PCA, preparation and properties of P(NIPAM-co-DMAEMA)/PCA complexes and finally release of the drug BZM from P(NIPAM-co-DMAEMA)/PCA complex coatings are described.
2.1. Synthesis of Poly(Caffeic Acid) (PCA)
When a caffeic acid (CA) solution was mixed with a NaIO4 solution, the resulting mixture changed its color from clear yellowish to black opaque within 10 s. This indicates an expansion of the π-system via coupling of either the aromatic and/or the conjugated R-C=C-R’ vinyl moieties. The structure of the yielded product was analyzed by various methods including dynamic light scattering (DLS), infrared spectroscopy (FTIR), electrospray ionization mass spectroscopy (ESI-MS), nuclear magnetic resonance (NMR) and colloid titration (PCD), aiming at determination of the molecular weight and closer insights into the coupling mechanism.
Firstly, concerning the molecular weight of PCA, it was observed that during dialysis, using tubes with cut-offs up to 50 kDa, barely any product was eluted. This pointed to a considerable polymerization degree. However, since the related catechol compound poly(dopamine) is known to form aggregates in aqueous solutions, this is not necessarily an indication for high molar mass polymers [
22]. No further insights were obtained by DLS, which revealed no distinct sizes around 2–1000 nm (due to low scattering contrast/intensity).
Secondly, PCD of the PCA product solutions (using low molecular probe polycation) revealed a charge factor of 1.0 ± 0.05, meaning that the quantity of charged groups of PCA is equivalent to the corresponding amount of CA monomers. Note that analogous colloid titration of CA monomer solutions did not reveal any related charge concentration value, since a phase-separated complex did not form, which is required for sensing principle. This observation is a further hint for the formation of a polymer with polymer-bound charges.
Thirdly, the FTIR spectrum (
Figure S1) of the monomer (pH 9.0; under oxygen exclusion) shows four basic functionalities: the vinyl group (δCH 977 cm
−1; νC=C 1635 cm
−1), the aromatic ring structure (νC=C 1433 cm
−1, 1495 cm
−1, 1588 cm
−1), the carboxylate group (νsCOO 1397 cm
−1; νasCOO 1549 cm
−1), which are obviously the origin of the polymer bound charges rationalized by PCD, and the phenolic functionality (νasCCO 1258 cm
−1). All four functionalities are generally preserved during the course of the reaction, whereby especially the carboxylate signals remain widely unchanged. Generally, an increase of the bandwidth for the IR bands of the polymer was obtained, indicating restricted motions compared to the monomer. A strong decrease in intensity combined with a distinct increase of the width is especially observed at δCH 977 cm
−1, suggesting a coupling via the vinyl functionality. The same observation, but less pronounced, can be made for the phenolic and aromatic functionalities. Here, the decrease of their intensities can be explained by the formation of quinones, whose corresponding signal appears at νC=O 1669 cm
−1 as a shoulder [
23]. Further the spectrum of the product does not show any ester signals. This was verified through PCD measurements, which rendered the charge factor of 1.0 ± 0.05 (see above) for the PCA, excluding coupling of the carboxyl and aromatic hydroxyl groups.
Fourthly, for further molecular structure determination,
1H-NMR spectra were recorded (
Figure S2). The variety of broad and narrow peaks obtained point to a heterogeneous mixture of products with varying coupling types along chains of different lengths. The broad peaks of higher order between 6.70 and 8.00 ppm indicate the formation of polymeric structures and support the hypothesis concerning coupling of the π-system (
Figure 2 D2 + T2). The multitude of different coupling positions along the π-system (
Figure 2 T2, exemplary encircled red left monomeric unit) could explain the high number of different subproducts. The peaks in the range of 3.00–4.50 ppm indicate heteroatom substituted aliphatic compounds. As Arakawa et al. reported, the corresponding structures (
Figure 2 D1 + T1) could be formed by an addition-reaction of the diol functionality to the vinyl group [
19]. The loss of the phenolic and vinyl groups are consistent with the decrease of their corresponding IR signals. Since the vinyl group is still clearly visible at 6.45 ppm, this specific coupling mechanism can only partially be held accountable for product formation. By comparing the integral intensities of the 3.00–4.50 ppm signals, deriving from two protons, with the integral of the signal at 6.45 ppm, deriving from one vinylic proton, it becomes clear that about half of the products are formed via the addition mechanism. This is further supported by the remaining phenolic and vinylic IR signals, proving that part of the, for BZM binding essential, diol functionalities outlast the polymerization process.
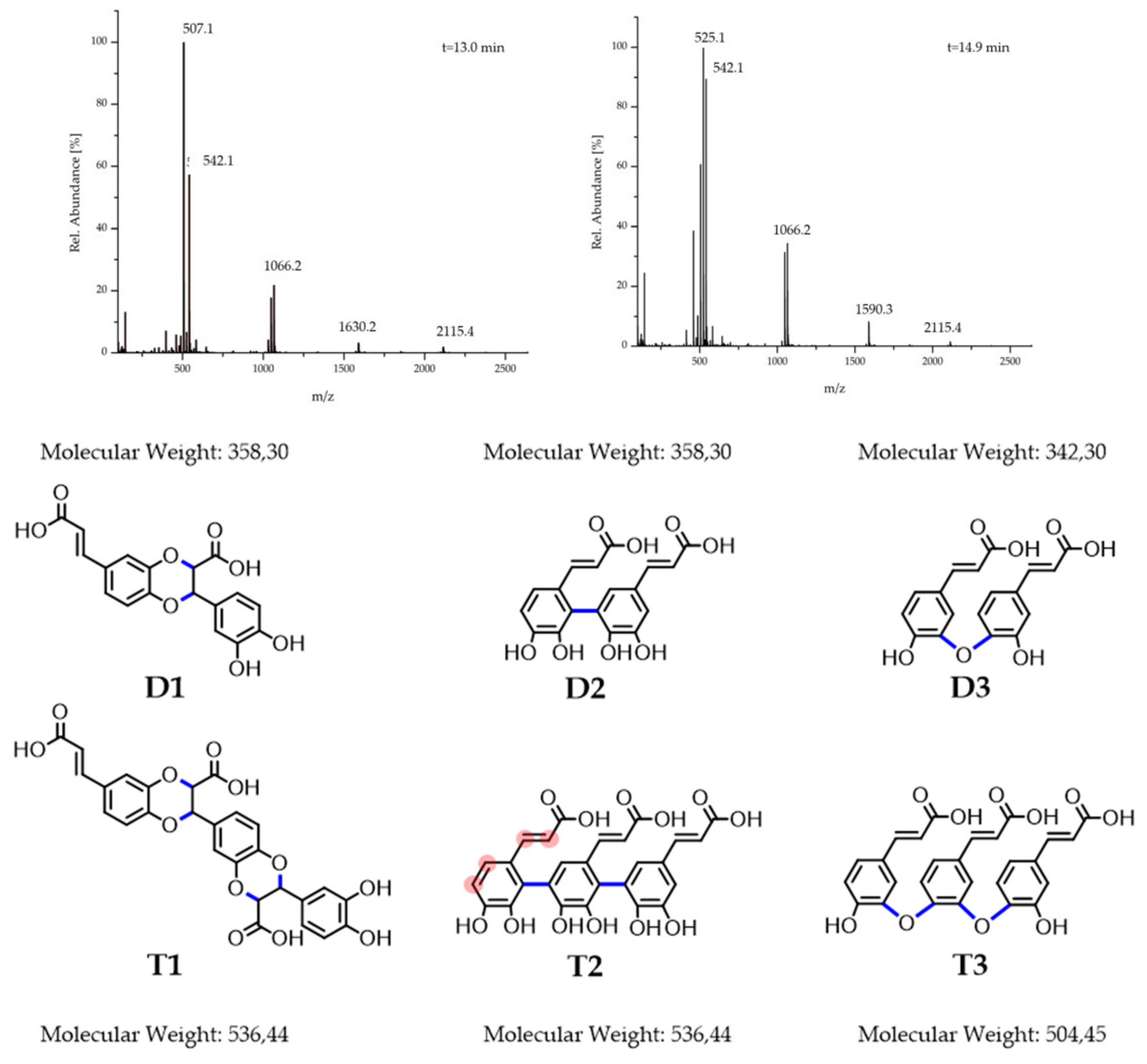
Finally, reverse phase liquid chromatography ESI-MS was applied for further clarification. The chromatogram confirms the formation of a broad spectrum of products (
Figure S3) analogously to the NMR findings. Some single peaks protrude from the underlying wide spectrum, which implements the favored formation of particular products. The ESI mass spectra of the five major formed compounds (elution times 13.0; 14.9; 18.4; 19.2 and 22.0 min) were analyzed. The elution peaks at 18.4 min and 19.2 min are caused by dimeric structures (
Figure S4a,b). The peak at 359
m/z is consistent with the coupling of the double bond and the diol functionality or a π–π coupling, since both possibilities form dimers of 358 g/mol (
Figure 2 D1 + D2) The peak at 341
m/z on the other hand points towards a condensation mechanism with a separation of a water molecule (
Figure 2 D3). This is supported by the peak difference of 18
m/z (360
m/z – 341
m/z – H
+ = 18
m/
z), which matches the molecular weight of water. The elution peak at 22.0 min shows several trimeric units (
Figure S4b). This coincides with the observations from Arakawa et al. who found that trimeric products are generated when CA is oxidized electrochemically at higher voltages [
19].
Of even higher significance are the mass spectra of the elution peaks of 13.0 min and 14.9 min in
Figure 2. Neither of the two spectra shows peaks or peak-differences corresponding to a monomeric unit of 180 g/mol. Instead, all of the lowest peaks are positioned at around 540
m/
z, again indicating the formation of trimeric products. The peaks at higher
m/z ratios are proximate to the many-folds of the trimeric values. Many peaks of the spectra at an elution time of 14.9 min are around 525
m/z apart (e.g., 2115.4 − 1590.3 = 525.1). Since no change of the
m/z value is observed, this may point towards an aggregation of 525.1
m/z trimers. The peak differences that do not correspond to any
m/z peak indicate a chemical reaction and thereby can be attributed to the formation of longer oligomers (t = 13.0 min). For these, a cascade of two different mechanisms is proposed: first, a trimerization of the CA occurs, followed by a subsequent coupling of these trimers to higher oligomers. Even though different coupling types are proposed, it is not distinguishable if all types are responsible for trimerization and the subsequent coupling to longer oligomers. Strikingly, often the spans between neighbored peaks is 18
m/z or 17
m/z (e.g., 525–507 m/z; 542–525 m/z) supporting the condensation mechanism under cleavage of water. Since no ester IR signals or reduction in the charge factor was observed, only ether bonds with IR bands overlapping with the phenolic bands serve as a suitable explanation. Furthermore, if two oligomers with a molar mass of 1066.2 g/mol are coupled via this condensation-mechanism, an oligomer with a corresponding molar mass to the peak at 2115.4
m/z is yielded (2 × 1066.2
m/z – 18
m/z = 2115.4
m/z). This indicates that this mechanism may also be suitable to combine the trimers with each other.
Due to the complexity of the product mixture, further investigations are necessary for profound structural proposals, and the trimeric structures in
Figure 2 should rather be understood as suggestions. However, from the synthetic part given above, it can be concluded that upon oxidation of the CA oligomeric structures were formed under partial preservation of the aromatic diol functionalities, which are important for chemically binding boronic acid-containing drugs, like BZM.
2.2. Preparation and Characterization of P(NIPAM-co-DMAEMA)/PCA Complexes
From our former work it is known, that polyelectrolyte complex nanoparticles (PEC NP) prepared by molar mixing ratios close to 1.0 (see Experimental) show high wet-adhesiveness, which arises from lack of both polymer-bound excess charge and hydrophilicity [
15]. By gradually increasing or decreasing the ratio by 0.1 increments, cationic and anionic PEC NPs were determined, which form a colloidal stable dispersion for at least 5 h. Regardless, the quantity of excess PCA in the anionic PEC system (i.e.,
n−/
n+ > 0, see Experimental) was stable. This is probably due to the high difference in the molar mass of the polyelectrolytes, so that the anionic groups of the significantly shorter PCA oligomers are fully compensated by the cationic groups of the larger P(NIPAM-co-DMAEMA) and thus do not protrude and exhibit an excess charge in the shell region of the PEC NP [
24]. Hence, the lack of such (anionic) excess charge disfavors repulsive forces between the PEC NPs resulting in flocculation even at mixing ratios significantly higher than
n−/
n+ = 1.0 (tested up to 3.0). However, when vice versa the cationic P(NIPAM-co-DMAEMA) is the excess component, longer polycationic sections are protruding at the shell region of PEC NP, which cause interparticle electrostatic repulsion and thus result in stable PEC solutions even at mixing ratios
n−/
n+ = 0.9.
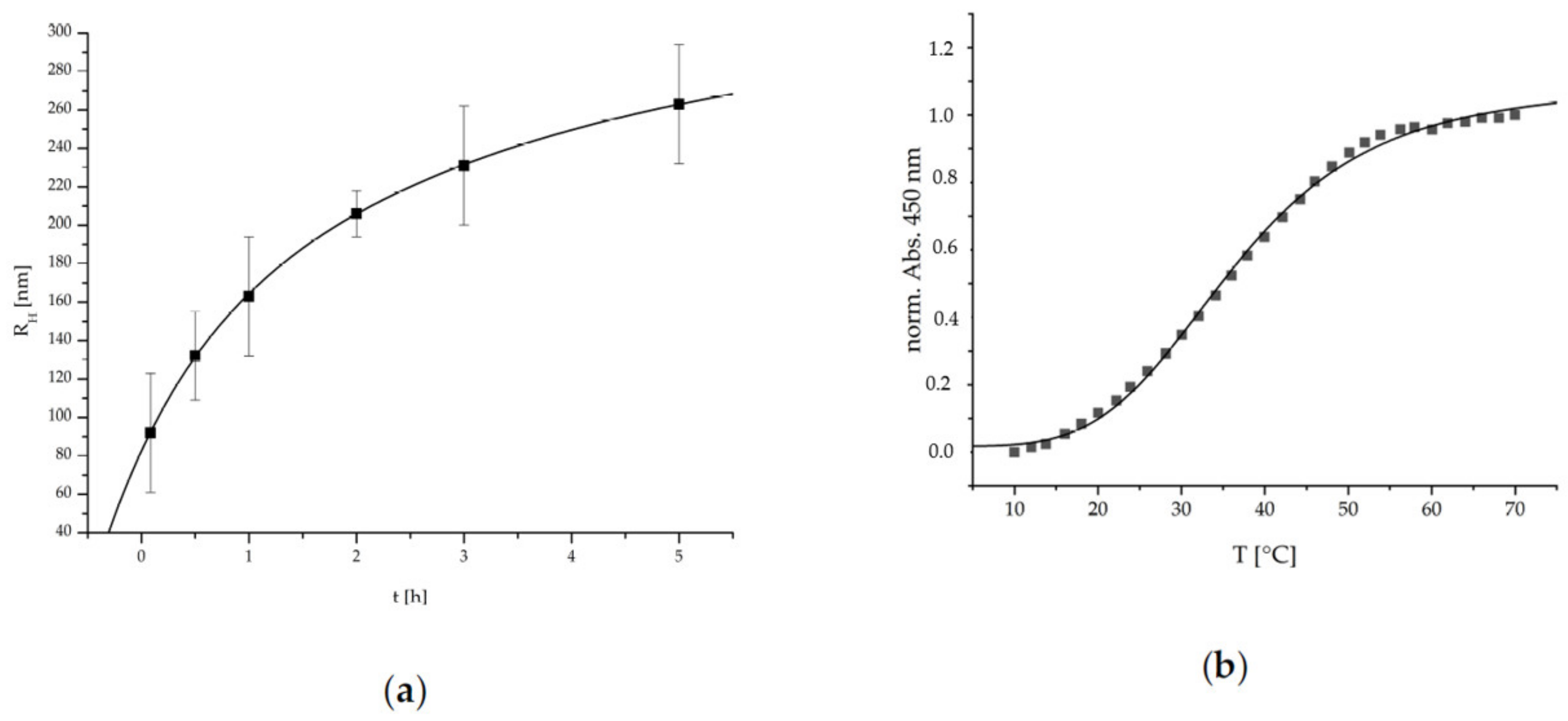
The ripening and growth behavior of such cationic PEC NPs system was analyzed through time-dependent DLS measurements. In
Figure 3, the determined hydrodynamic radii are plotted against the time.
The PEC NPs show a significant exponentially damped (i.e., R
H = R
H0 (1 − exp[−k t])) increase of the hydrodynamic radii R
H over time. This is possibly due to the dynamic competition between long range electrostatic repulsion (see above) and short-range attraction forces between initially formed primary PEC NP like Van der Waals or hydrogen bonding preventing thermodynamic equilibrium and suggesting kinetic control known for other PEC NP systems [
25]. Polyelectrolyte complexation kinetics has been addressed in more detail by Takahashi and Liu based on time-resolved ultra-small-angle X-ray scattering supporting aggregation processes of early formed primary complexes to species termed higher order agglomerates or condensed coacervate droplets [
26,
27]. Practically, to assure reproducible measurements the solutions were always used after 1 h ripening for subsequent analysis or coating generation.
Recently, the thermoresponsive character of PEC systems based on cationic P(NIPAM-co-DMAEMA) and anionic polysaccharides was described [
20]. To evaluate the thermoresponsive properties of the herein used P(NIPAM-co-DMAEMA)/PCA PEC NP system, temperature dependent UV/Vis measurements were conducted. In
Figure 3, the normalized absorption at 450 nm is plotted against the temperature. The error due to increasing turbidity caused by the particle ripening was limited to 5% by time-dependent UV/Vis measurement at RT.
The P(NIPAM-co-DMAEMA)/PCA PEC NP show a typical S-shaped function, which can be represented by a sigmoidal fitting function given therein [
20]. Via this fit a conversion temperature (T
c) of 36.4 ± 0.4 °C and a cooperativity factor (P) of 4.2 ± 0.2 were determined. Concerning the intended application, this LCST behavior is well-suited to match the requirements since it is located in the physiological temperature range. The relatively low P-factor is an indicator for a wide transition span, the advantage being that the complete range of physiological temperatures exhibits a thermoresponsive transition. Additionally, this system is also suitable if medicinal means are utilized that enable the application of higher temperatures such as hyperthermia therapies, etc.
2.2.1. Wet Adhesive Properties
In the preceeding section, thermoresponsiveness was observed for the bulk P(NIPAM-co-DMAEMA) PEC system in dispersion. To check interfacial thermal properties, this PEC system was deposited onto germanium crystals, which are both suitable analytical substrates of choice for surface sensitive ATR-FTIR measurements (see Experimental Section, Immobilization of PEC NPs) as well as model substrates for a series of oxide and hydroxide layer terminated metal and semiconductor materials (titania, steel, magnesium etc.) used as BSM.
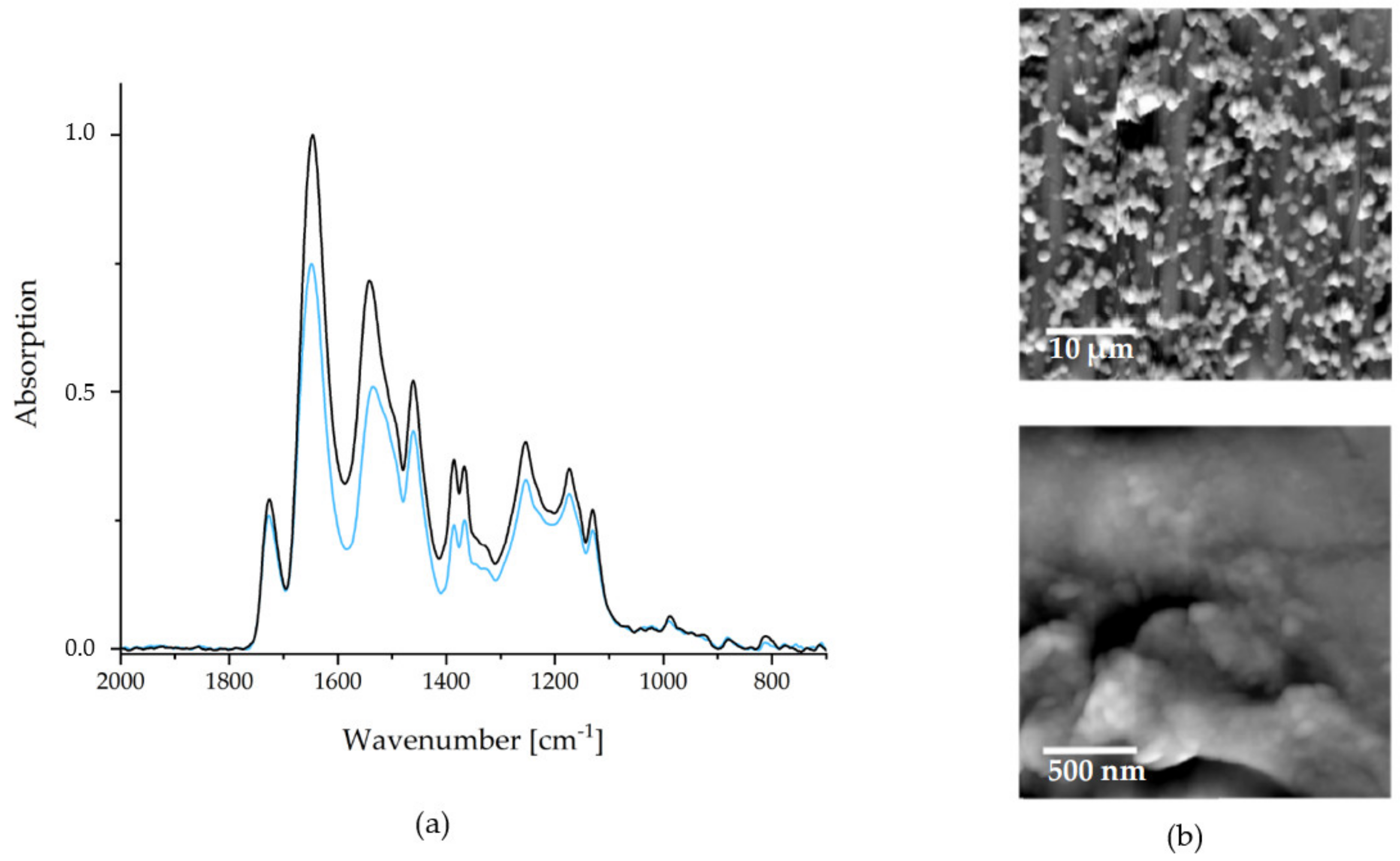
As mentioned above, the wet adhesiveness of the PEC NPs towards a substrate is one of the most significant mandatory properties of biofunctional and biomedical organic coatings of BSM concerning future applications. As a simple test for wet-adhesiveness, FTIR spectroscopy was used, whereby the integrals of the IR signals before (A
BEFORE) and after intense rinsing (A
AFTER) in selected aqueous media are compared. Wet adhesiveness in that sense can be quantified calculating the percentage ratio R
ADHES = (A
AFTER)/(A
BEFORE) × 100%. Typical FTIR recorded before (black) and after (blue) are depicted in
Figure 4.
Ultrapure water was applied as rinse medium, since it was empirically found to be the most erosive for PEC NP coatings. An average wet adhesiveness of the P(NIPAM-co-DMAEMA)/PCA PEC coating of RADHES = 79 ± 10% was determined. The deviation to 100% can be explained by a small amount of unbound excess polycation (see comments above). Consequently, coated implants and bone substitution materials are ready for use after one short rinsing step.
Due to its impact on the release kinetics, the surface morphology is also evident for the quality of a drug delivery coating. In
Figure 4, SFM images are depicted, with the larger section (microscale) on the upper and the smaller section (nanoscale) on the lower hand.
As it can be seen on the microscale, the PCA system strongly tends to cluster formation. These clusters coagulate to an island-like structure, rendering a larger surface area than a smooth coating would. The advantage of a high surface area is, that more, easily accessible, interaction sites for drug binding are present. This also depends on the nanostructures of the films, which are depicted in the lower image in
Figure 4. The NPs, which were observed in solution via DLS measurements, are recognizable. Furthermore, agglomeration and coalescence of these NPs can be observed. The average diameters of individually appearing NPs (range 60–180 nm) were significantly lower than the corresponding values in dispersion (
Figure 4). This indicates a shrinking of the particles during the drying process and suggests a certain swelling of the PEC NP in the dispersed state.
2.2.2. Thermoresponsive Properties
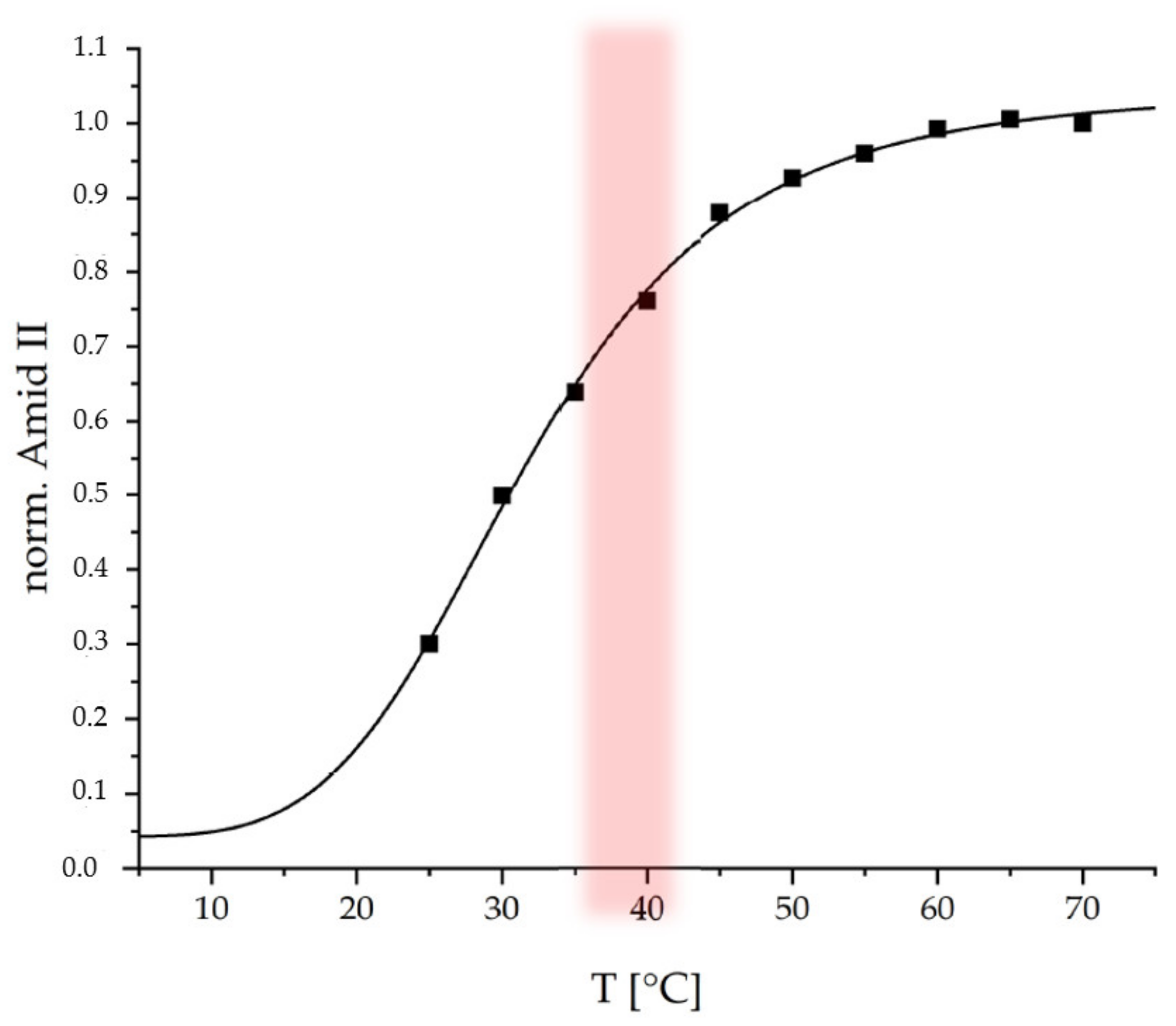
Subsequently the coating was tested regarding its thermoresponsive properties at a physiological pH of 7.4. Temperature-dependent ATR-FTIR spectroscopy was used. In the
Figure 5 the normalized integrals (with respect to end value) of the Amide II band from ATR-FTIR spectra (
Figure S5) recorded at 25 °C to 70 °C are plotted versus temperature.
The Amide II band is used, since it is not interfered by the δ(OH) band of water around 1640 cm
−1, which is the case for the Amide I band. An increase of the Amide II (1560 cm
−1) integral with increasing temperature is observed for the thermoresponsive PEC coating, which is similar to that observed for the thermoresponsive PEC NP dispersion (
Figure 3b). On the one hand, increasing ATR-FTIR signals may originate from increasing thickness within a range up to the penetration depth (d
P < 500 nm dependent on refractive indices of contributing media and incident angle of ATR crystal) of the evanescent wave [
28]. On the other hand, increasing ATR-FTIR signals may originate from increasing density or polymer segment concentration c
POL. Since it is known that PNIPAM systems rather undergo a shrinking process upon temperature increase, the curve in
Figure 5 rather monitors c
POL. This curve was fitted by the same sigmoidal function used to fit the dispersion data in
Figure 3b. For the PEC NP coating, a conversion temperature T
C = 31.7 ± 1.5 °C and a cooperativity factor of P = 4.4 ± 0.6 were determined. Both values for the thermoresponsive PEC coating are close to those for the PEC NP dispersion suggesting that the immobilization process does not have a significant effect on the response behavior. The slightly lower T
C for the coating compared to the dispersion might be caused by the lower water accessibility (hydratability) in case of the coating due to lower surface area and removal of hydrophilic excess polycation.
2.3. Release of BZM
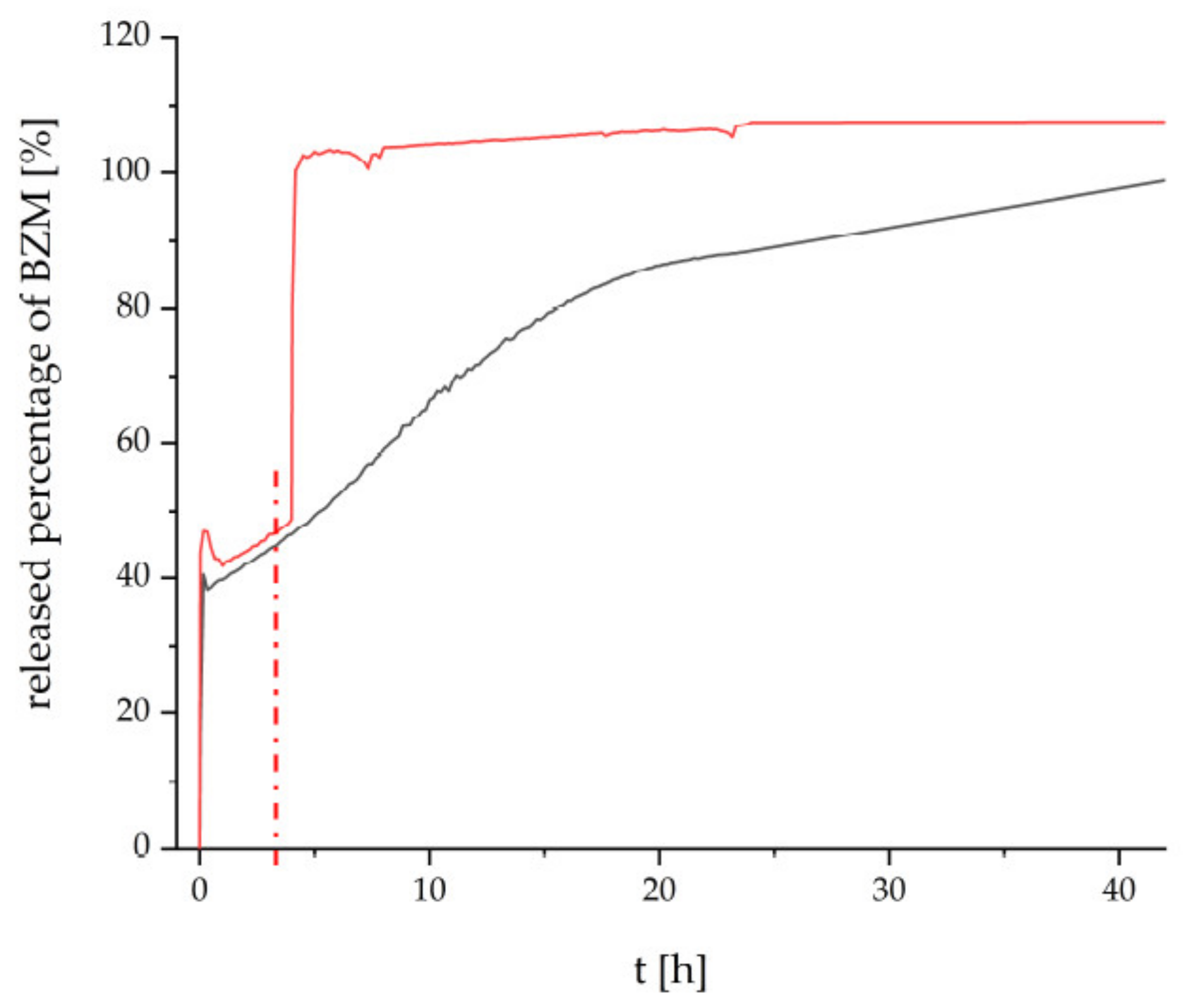
Finally, the thermoresponsive PEC NP (P(NIPAM-co-DMAEMA)/PCA) coating were checked for their drug delivery properties. Bortezomib (BZM) a potent drug for multiple myeloma (MM) described in the introduction was used, which bears a boronic acid moiety. BZM was loaded after preparation of the PEC NP coating. This “postloading” concept (section “BZM loading and release”) features two major advantages. Firstly, it is fast and easy to apply, which makes it appealing for commercial use. Secondly, the drying step is supposed to withdraw water from the equilibrium between the boric acid and the boric acid ester and to promote the ester formation. The dry coating with the bond BZM was subsequently placed into the release media, which was constantly monitored via UV/Vis measurements. In
Figure 6, the released percentage of BZM is plotted versus time.
The release at room temperature (RT) from the PCA based coating is shown in
Figure 6 by the black curve. The beginning is marked by an initial burst release of about 40%, which is most probably caused by loosely bound BZM without chemical bonding to the coating. The following slight decrease might be caused by reabsorption due to swelling of the coating. Thereafter the remaining BZM is gradually released over a time period until 42 h. We assume that the high tendency to form esters between the aromatic diol functionalities and the BZM is mainly accountable for this retardation of the release. The heterogeneous surface morphology, which is destined to provide a distinct number of interaction sites, supposedly amplifies the retention mechanism. Additionally, ATR-FTIR spectra were recorded on the loading and releasing process at an equivalent P(NIPAM-co-DMAEMA)/PCA coating at Ge substrate, which are given in
Figure S6 of the Supplementary. There it can be rationalized that BZM is uptaken at this thermoresponsive PEC coating and it could not be completely rinsed out after rinsing in pure water. However, this data cannot give further information on the location of loaded BZM (shell, core or between particles) within the PEC coating.
The release beginning with RT and after switching to T = 42 °C is given by the red curve in
Figure 6. Obviously, at RT, the PEC NP coating elutes BZM with the same kinetics for 4 h. However after switching to T = 42 °C (switching point is denoted as red dashed line) the BZM is released much faster compared to RT (black line) up to 42 h. Analogously to other PNIPAM-based systems (e.g., pure polymer, crosslinked microgels), the known coil-to-globule transition of the PNIPAM segments causes conformation changes not only within the cationic P(NIPAM-co-DMAEMA), but also within anionic PCA [
29]. Consequently, either physically entrapped BZM may leave the thermoresponsive PEC NP phase on new routes or residual water molecules may access to BZM-diol ester sites, which were previously shielded.



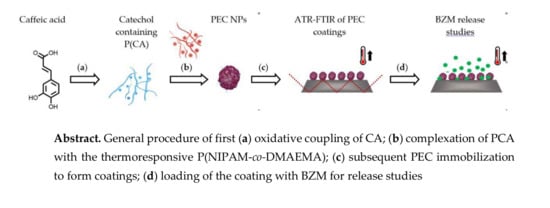
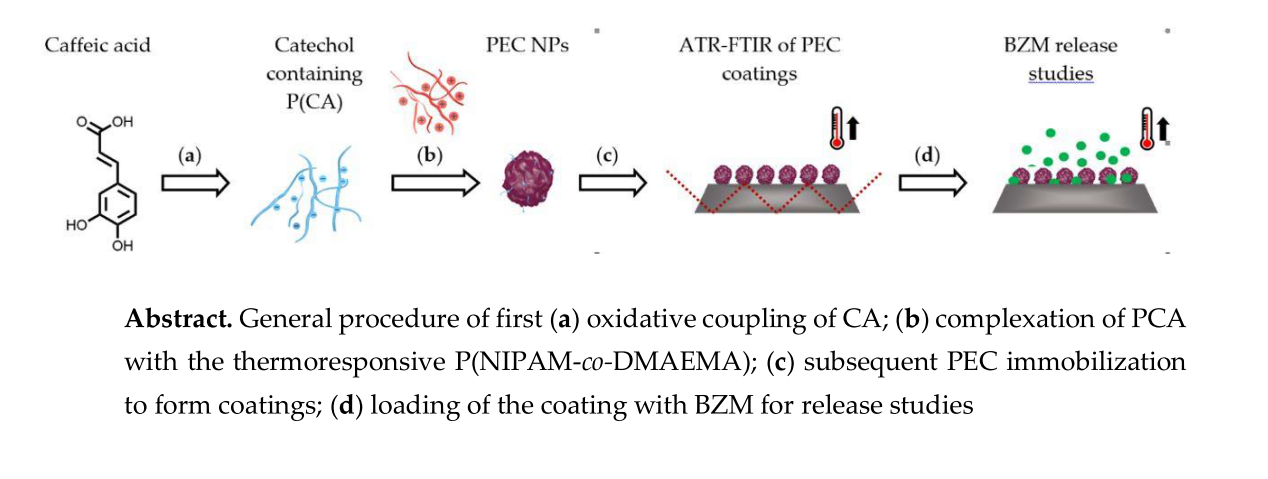{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






