Teaching an Old Molecule New Tricks: Drug Repositioning for Duchenne Muscular Dystrophy

 , and
, and
Abstract
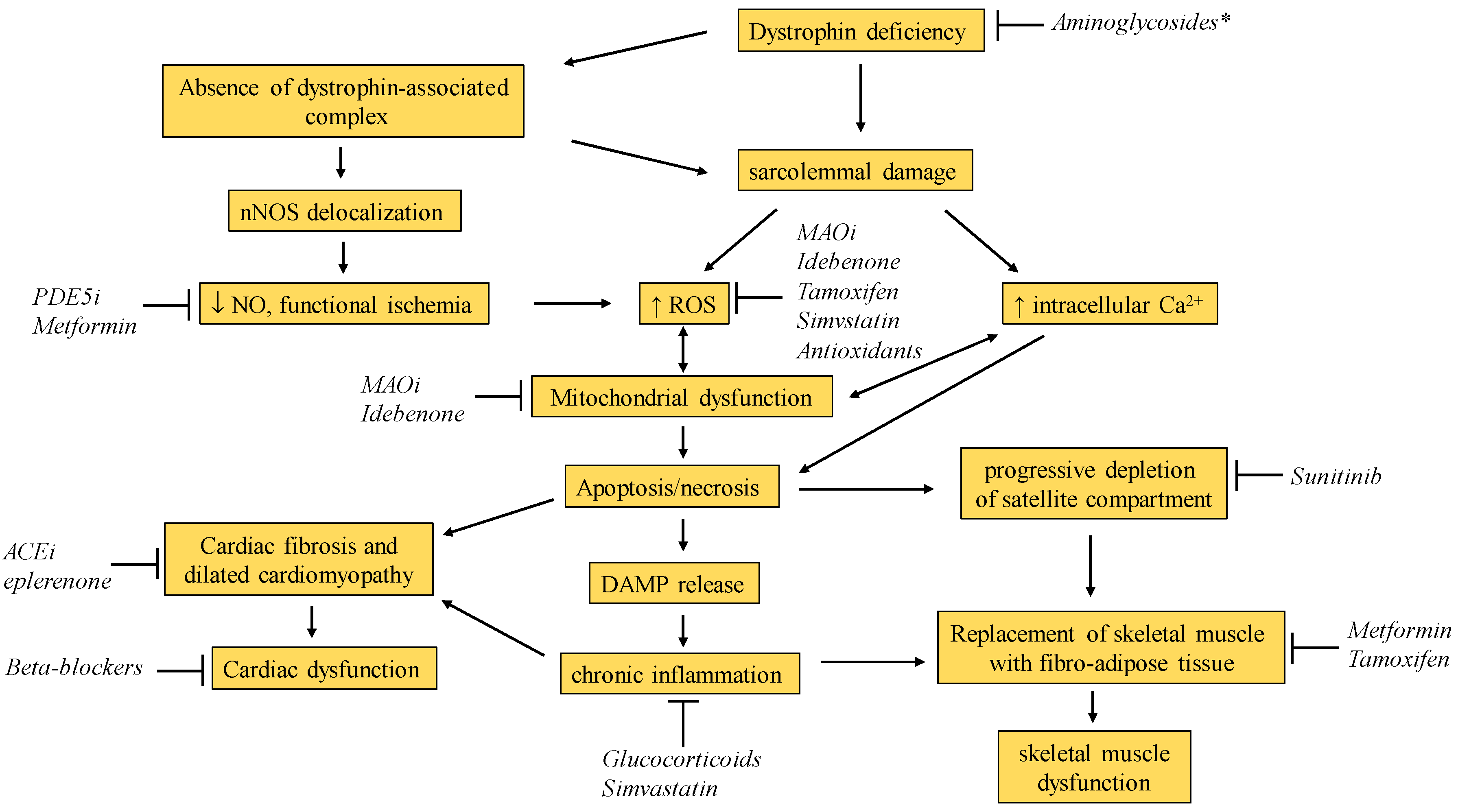
1. Introduction
2. The Gold Standard: Glucocorticoid Corticosteroids
3. Simvastatin
4. N-acetylcysteine and Antioxidants
5. Safinamide and MAO Inhibitors
6. Sunitinib
7. Idebenone
8. Tamoxifen
9. Metformin
10. PDE5 Inhibitors and Nitrate Drugs
11. Food Supplements
12. Aminoglycosides (for Stop Codon Readthrough)
13. Cardiological Drugs (ACE Inhibitors, Beta-Blockers, Eplerenone)
14. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mah, J.K.; Korngut, L.; Dykeman, J.; Day, L.; Pringsheim, T.; Jette, N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul. Disord. 2014, 24, 482–491. [Google Scholar] [CrossRef]
- Ryder, S.; Leadley, R.M.; Armstrong, N.; Westwood, M.; De Kock, S.; Butt, T.; Jain, M.; Kleijnen, J. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: An evidence review. Orphanet J. Rare Dis. 2017, 12, 79. [Google Scholar] [CrossRef]
- Bourke, J.P.; Bueser, T.; Quinlivan, R. Interventions for preventing and treating cardiac complications in Duchenne and Becker muscular dystrophy and X-linked dilated cardiomyopathy. Cochrane Database Syst. Rev. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Goemans, N. A Sequel to the Eteplirsen Saga: Eteplirsen Is Approved in the United States but Was Not Approved in Europe. Nucleic Acid Ther. 2019, 29, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010, 9, 77–93. [Google Scholar] [CrossRef]
- Gloss, D.; Moxley, R.T.; Ashwal, S.; Oskoui, M. Practice guideline update summary: Corticosteroid treatment of Duchenne muscular dystrophy—Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 2016, 86, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Aronson, J. Rare diseases, orphan drugs, and orphan diseases. BMJ 2006, 333, 127. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2018, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.Y.; Loscalzo, J. The emerging paradigm of network medicine in the study of human disease. Circ. Res. 2012, 111, 359–374. [Google Scholar] [CrossRef]
- Piro, R.M. Network medicine: Linking disorders. Hum. Genet. 2012, 131, 1811–1820. [Google Scholar] [CrossRef]
- Guengerich, F.P. Mechanisms of drug toxicity and relevance to pharmaceutical development. Drug Metab. Pharmacokinet. 2011, 26, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Burns, C.M. The History of Cortisone Discovery and Development. Rheum. Dis. Clin. N. Am. 2016, 42, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Drachman, D.B.; Toyka, K.V.; Myer, E. Predisone in Duchenne muscular dystrophy. Lancet 1974, 2, 1409–1412. [Google Scholar] [CrossRef]
- Matthews, E.; Brassington, R.; Kuntzer, T.; Jichi, F.; Manzur, A.Y. Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database Syst. Rev. 2016, 5. [Google Scholar] [CrossRef]
- Bello, L.; Gordish-Dressman, H.; Morgenroth, L.P.; Henricson, E.K.; Duong, T.; Hoffman, E.P.; Cnaan, A.; McDonald, C.M. Prednisone/prednisolone and deflazacort regimens in the CINRG Duchenne Natural History Study. Neurology 2015, 85, 1048–1055. [Google Scholar] [CrossRef]
- McDonald, C.M.; Henricson, E.K.; Abresch, R.T.; Duong, T.; Joyce, N.C.; Hu, F.; Clemens, P.R.; Hoffman, E.P.; Cnaan, A.; Gordish-Dressman, H.; et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: A prospective cohort study. Lancet 2018, 391, 451–461. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Brumbaugh, D.; Case, L.E.; Clemens, P.R.; Hadjiyannakis, S.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef]
- Griggs, R.C.; Herr, B.E.; Reha, A.; Elfring, G.; Atkinson, L.; Cwik, V.; Mccoll, E.; Tawil, R.; Pandya, S.; Mcdermott, M.P.; et al. Corticosteroids in Duchenne muscular dystrophy: Major variations in practice. Muscle Nerve 2013, 48, 27–31. [Google Scholar] [CrossRef]
- Mc Donald, C.M.; Gordish-Dressman, H.; Henricson, E.K.; Duong, T.; Joyce, N.C.; Jhawar, S.; Leinonen, M.; Hsu, F.; Connolly, A.M.; Cnaan, A.; et al. Longitudinal pulmonary function testing outcome measures in Duchenne muscular dystrophy: Long-term natural history with and without glucocorticoids. Neuromuscul. Disord. 2018, 28, 897–909. [Google Scholar] [CrossRef]
- Khan, M.A. Corticosteroid therapy in Duchenne muscular dystrophy. J. Neurol. Sci. 1993, 120, 8–14. [Google Scholar] [CrossRef]
- Anderson, J.E.; Mcintosh, L.M.; Poettcker, R. Deflazacort but not prednisone improves both muscle repair and fiber growth in diaphragm and limb muscle in vivo in the mdx dystrophic mouse. Muscle Nerve 1996, 19, 1576–1585. [Google Scholar] [CrossRef]
- Anderson, J.E.; Weber, M.; Vargas, C. Deflazacort increases laminin expression and myogenic repair, and induces early persistent functional gain in mdx mouse muscular dystrophy. Proc. Cell Transp. 2000, 9, 551–564. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, S.J.G.; Chakkalakal, J.V.; Kolodziejczyk, S.M.; Knudson, J.C.; Jasmin, B.J.; Megeney, L. A Glucocorticoid treatment alleviates dystrophic myofiber pathology by activation of the calcineurin/NF-AT pathway. FASEB J. 2004, 18, 1937–1939. [Google Scholar] [CrossRef] [PubMed]
- Barton-Davis, E.R.; Cordier, L.; Shoturma, D.I.; Leland, S.E.; Sweeney, H.L. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J. Clin. Investig. 1999, 104, 375–381. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Nico, B.; Rolland, J.F.; Cozzoli, A.; Burdi, R.; Mangieri, D.; Giannuzzi, V.; Liantonio, A.; Cippone, V.; De Bellis, M.; et al. Gentamicin treatment in exercised mdx mice: Identification of dystrophin-sensitive pathways and evaluation of efficacy in work-loaded dystrophic muscle. Neurobiol. Dis. 2008, 32, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Politano, L.; Nigro, G.; Nigro, V.; Pilus, G.; Papparella, S.; Paciello, O.; Comi, L.I. Gentamicin administration in Duchenne patients with premature stop codon. Preliminary results. Acta Myol. 2003, 22, 15–21. [Google Scholar]
- Malik, V.; Rodino-Klapac, L.R.; Viollet, L.; Wall, C.; King, W.; Al-Dahhak, R.; Lewis, S.; Shilling, C.J.; Kota, J.; Serrano-Munuera, C.; et al. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann. Neurol. 2010, 67, 771–780. [Google Scholar] [CrossRef]
- Wróbel, M.P.; Marek, B.; Kajdaniuk, D.; Rokicka, D.; Szymborska-Kajanek, A.; Strojek, K. Metformin—A new old drug. Endokrynol. Pol. 2017, 68, 482–496. [Google Scholar] [CrossRef]
- Amin, S.; Lux, A.; O’Callaghan, F. The journey of metformin from glycaemic control to mTOR inhibition and the suppression of tumour growth. Br. J. Clin. Pharmacol. 2019, 85, 37–46. [Google Scholar] [CrossRef]
- Mantuano, P.; Sanarica, F.; Conte, E.; Morgese, M.G.; Capogrosso, R.F.; Cozzoli, A.; Fonzino, A.; Quaranta, A.; Rolland, J.F.; De Bellis, M.; et al. Effect of a long-term treatment with metformin in dystrophic mdx mice: A reconsideration of its potential clinical interest in Duchenne muscular dystrophy. Biochem. Pharmacol. 2018, 154, 89–103. [Google Scholar] [CrossRef]
- Ljubicic, V.; Jasmin, B.J. Metformin increases peroxisome proliferator-activated receptor γ Co-activator-1α and utrophin a expression in dystrophic skeletal muscle. Muscle Nerve 2015, 52, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Casteels, K.; Fieuws, S.; van Helvoirt, M.; Verpoorten, C.; Goemans, N.; Coudyzer, W.; Loeckx, D.; de Zegher, F. Metformin therapy to reduce weight gain and visceral adiposity in children and adolescents with neurogenic or myogenic motor deficit. Pediatr. Diabetes 2010, 11, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Hafner, P.; Bonati, U.; Erne, B.; Schmid, M.; Rubino, D.; Pohlman, U.; Peters, T.; Rutz, E.; Frank, S.; Neuhaus, C.; et al. Improved muscle function in duchenne muscular dystrophy through l-arginine and metformin: An investigator-initiated, open-label, single-center, proof-of-concept-study. PLoS ONE 2016, 11, e0147634. [Google Scholar] [CrossRef] [PubMed]
- Hanff, E.; Hafner, P.; Bollenbach, A.; Bonati, U.; Kayacelebi, A.A.; Fischer, D.; Tsikas, D. Effects of single and combined metformin and l-citrulline supplementation on l-arginine-related pathways in Becker muscular dystrophy patients: Possible biochemical and clinical implications. Amino Acids 2018, 50, 1391–1406. [Google Scholar] [CrossRef] [PubMed]
- Hafner, P.; Bonati, U.; Rubino, D.; Gocheva, V.; Zumbrunn, T.; Gueven, N.; Fischer, D. Treatment with l-citrulline and metformin in Duchenne muscular dystrophy: Study protocol for a single-centre, randomised, placebo-controlled trial. Trials 2016, 17, 389. [Google Scholar] [CrossRef]
- Schönbeck, U.; Libby, P. Inflammation, immunity, and HMG-CoA reductase inhibitors: Statins as antiinflammatory agents? Circulation 2004, 109. [Google Scholar] [CrossRef]
- Pignatelli, P.; Carnevale, R.; Pastori, D.; Cangemi, R.; Napoleone, L.; Bartimoccia, S.; Nocella, C.; Basili, S.; Violi, F. Immediate antioxidant and antiplatelet effect of atorvastatin via inhibition of Nox2. Circulation 2012, 126, 92–103. [Google Scholar] [CrossRef]
- Violi, F.; Pignatelli, P. Statins as Regulators of Redox Signaling in Platelets. Antioxid. Redox Signal. 2014, 20, 1300–1312. [Google Scholar] [CrossRef]
- Whitehead, N.P.; Kim, M.J.; Bible, K.L.; Adams, M.E.; Froehner, S.C. A new therapeutic effect of simvastatin revealed by functional improvement in muscular dystrophy. Proc. Natl. Acad. Sci. USA 2015, 112, 12864–12869. [Google Scholar] [CrossRef]
- Kim, M.J.; Bible, K.L.; Regnier, M.; Adams, M.E.; Froehner, S.C.; Whitehead, N.P. Simvastatin provides long-term improvement of left ventricular function and prevents cardiac fibrosis in muscular dystrophy. Physiol. Rep. 2019, 7, e14018. [Google Scholar] [CrossRef]
- Dorchies, O.M.; Reutenauer-Patte, J.; Dahmane, E.; Ismail, H.M.; Petermann, O.; Patthey-Vuadens, O.; Comyn, S.A.; Gayi, E.; Piacenza, T.; Handa, R.J.; et al. The anticancer drug tamoxifen counteracts the pathology in a mouse model of duchenne muscular dystrophy. Am. J. Pathol. 2013, 182, 485–504. [Google Scholar] [CrossRef] [PubMed]
- Gayi, E.; Neff, L.A.; Massana Muñoz, X.; Ismail, H.M.; Sierra, M.; Mercier, T.; Décosterd, L.A.; Laporte, J.; Cowling, B.S.; Dorchies, O.M.; et al. Tamoxifen prolongs survival and alleviates symptoms in mice with fatal X-linked myotubular myopathy. Nat. Commun. 2018, 9, 4848. [Google Scholar] [CrossRef] [PubMed]
- Gayi, E.; Neff, L.A.; Ismail, H.M.; Ruegg, U.T.; Scapozza, L.; Dorchies, O.M. Repurposing the selective oestrogen receptor modulator tamoxifen for the treatment of Duchenne muscular dystrophy. Proc. Chim. 2018, 182, 485–504. [Google Scholar] [CrossRef] [PubMed]
- Asai, A.; Sahani, N.; Kaneki, M.; Ouchi, Y.; Jeevendra Martyn, J.A.; Yasuhara, S.E. Primary role of functional ischemia, quantitative evidence for the two-hit mechanism, and phosphodiesterase-5 inhibitor therapy in mouse muscular dystrophy. PLoS ONE 2007, 2, e806. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.M.; Rader, E.P.; Crawford, R.W.; Iyengar, N.K.; Thedens, D.R.; Faulkner, J.A.; Parikh, S.V.; Weiss, R.M.; Chamberlain, J.S.; Moore, S.A.; et al. Sarcolemma-localized nNOS is required to maintain activity after mild exercise. Nature 2008, 456, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.D.; Rader, F.; Tang, X.; Tavyev, J.; Nelson, S.F.; Miceli, M.C.; Elashoff, R.M.; Sweeney, H.L.; Victor, R.G. PDE5 inhibition alleviates functional muscle ischemia in boys with Duchenne muscular dystrophy. Neurology 2014, 82, 2085–2091. [Google Scholar] [CrossRef]
- De Arcangelis, V.; Strimpakos, G.; Gabanella, F.; Corbi, N.; Luvisetto, S.; Magrelli, A.; Onori, A.; Passananti, C.; Pisani, C.; Rome, S.; et al. Pathways Implicated in Tadalafil Amelioration of Duchenne Muscular Dystrophy. J. Cell. Physiol. 2016, 231, 224–232. [Google Scholar] [CrossRef]
- Hammers, D.W.; Sleeper, M.M.; Forbes, S.C.; Shima, A.; Walter, G.A.; Sweeney, H.L. Tadalafil Treatment Delays the Onset of Cardiomyopathy in Dystrophin-Deficient Hearts. J. Am. Heart Assoc. 2016, 5, e003911. [Google Scholar] [CrossRef]
- Victor, R.G.; Sweeney, H.L.; Finkel, R.; McDonald, C.M.; Byrne, B.; Eagle, M.; Goemans, N.; Vandenborne, K.; Dubrovsky, A.L.; Topaloglu, H.; et al. A phase 3 randomized placebo-controlled trial of tadalafil for Duchenne muscular dystrophy. Neurology 2017, 89, 1811–1820. [Google Scholar] [CrossRef]
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef]
- Whitehead, N.P.; Pham, C.; Gervasio, O.L.; Allen, D.G. N-Acetylcysteine ameliorates skeletal muscle pathophysiology in mdx mice. J. Physiol. 2008, 586, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Dorchies, O.M.; Wagner, S.; Vuadens, O.; Waldhauser, K.; Buetler, T.M.; Kucera, P.; Ruegg, U.T. Green tea extract and its major polyphenol (−)-epigallocatechin gallate improve muscle function in a mouse model for Duchenne muscular dystrophy. Am. J. Physiol. Cell Physiol. 2006, 290, C616–C625. [Google Scholar] [CrossRef] [PubMed]
- Faivre, S.; Delbaldo, C.; Vera, K.; Robert, C.; Lozahic, S.; Lassau, N.; Bello, C.; Deprimo, S.; Brega, N.; Massimini, G.; et al. Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J. Clin. Oncol. 2006, 24, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Fontelonga, T.M.; Jordan, B.; Nunes, A.M.; Barraza-Flores, P.; Bolden, N.; Wuebbles, R.D.; Griner, L.M.; Hu, X.; Ferrer, M.; Marugan, J.; et al. Sunitinib promotes myogenic regeneration and mitigates disease progression in the mdx mouse model of Duchenne muscular dystrophy. Hum. Mol. Genet. 2019, 28, 2120–2132. [Google Scholar] [CrossRef]
- Sorato, E.; Menazza, S.; Zulian, A.; Sabatelli, P.; Gualandi, F.; Merlini, L.; Bonaldo, P.; Canton, M.; Bernardi, P.; Di Lisa, F. Monoamine oxidase inhibition prevents mitochondrial dysfunction and apoptosis in myoblasts from patients with collagen VI myopathies. Free Radic. Biol. Med. 2014, 75, 40–47. [Google Scholar] [CrossRef]
- Vitiello, L.; Marabita, M.; Sorato, E.; Nogara, L.; Forestan, G.; Mouly, V.; Salviati, L.; Acosta, M.; Blaauw, B.; Canton, M. Drug repurposing for Duchenne muscular dystrophy: The monoamine oxidase B inhibitor safinamide Ameliorates the pathological phenotype in mdx mice and in myogenic cultures From DMD patients. Front. Physiol. 2018, 9, 1087–1096. [Google Scholar] [CrossRef]
- Youdim, M.B.H.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef]
- Buyse, G.M.; Goemans, N.; van den Hauwe, M.; Thijs, D.; de Groot, I.J.M.; Schara, U.; Ceulemans, B.; Meier, T.; Mertens, L. Idebenone as a novel, therapeutic approach for Duchenne muscular dystrophy: Results from a 12 month, double-blind, randomized placebo-controlled trial. Neuromuscul. Disord. 2011, 21, 396–405. [Google Scholar] [CrossRef]
- Buyse, G.M.; Goemans, N.; Van Den Hauwe, M.; Meier, T. Effects of glucocorticoids and idebenone on respiratory function in patients with duchenne muscular dystrophy. Pediatr. Pulmonol. 2013, 48, 912–920. [Google Scholar] [CrossRef]
- Buyse, G.M.; Van der Mieren, G.; Erb, M.; D’hooge, J.; Herijgers, P.; Verbeken, E.; Jara, A.; Van Den Bergh, A.; Mertens, L.; Courdier-Fruh, I.; et al. Long-term blinded placebo-controlled study of SNT-MC17/idebenone in the dystrophin deficient mdx mouse: Cardiac protection and improved exercise performance. Eur. Heart J. 2009, 30, 116–124. [Google Scholar] [CrossRef]
- Buyse, G.M.; Voit, T.; Schara, U.; Straathof, C.S.M.; D’Angelo, M.G.; Bernert, G.; Cuisset, J.M.; Finkel, R.S.; Goemans, N.; McDonald, C.M.; et al. Efficacy of idebenone on respiratory function in patients with Duchenne muscular dystrophy not using glucocorticoids (DELOS): A double-blind randomised placebo-controlled phase 3 trial. Lancet 2015, 385, 1748–1757. [Google Scholar] [CrossRef]
- McDonald, C.; Meier, T.; Voit, T.; Schara, U.; Straathof, C.; D’Angelo, M.; Bernert, G.; Cuisset, J.; Finkel, R.; Goemans, N.; et al. Idebenone reduces respiratory complications in patients with Duchenne muscular dystrophy. Neuromuscul. Disord. 2016, 26, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Mayer, O.H.; Leinonen, M.; Rummey, C.; Meier, T.; Buyse, G.M. Efficacy of Idebenone to Preserve Respiratory Function above Clinically Meaningful Thresholds for Forced Vital Capacity (FVC) in Patients with Duchenne Muscular Dystrophy. J. Neuromuscul. Dis. 2017, 4, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Head, S.I. Antioxidant therapy in a mouse model of Duchenne muscular dystrophy: Some promising results but with a weighty caveat. J. Physiol. 2017, 595, 7015. [Google Scholar] [CrossRef]
- Pinniger, G.J.; Terrill, J.R.; Assan, E.B.; Grounds, M.D.; Arthur, P.G. Pre-clinical evaluation of N-acetylcysteine reveals side effects in the mdx mouse model of Duchenne muscular dystrophy. J. Physiol. 2017, 595, 7093–7107. [Google Scholar] [CrossRef]
- O’Halloran, K.D.; Murphy, K.H.; Burns, D.P. Antioxidant therapy for muscular dystrophy: Caveat lector! J. Physiol. 2018, 596, 737–738. [Google Scholar] [CrossRef]
- Rando, T.A.; Disatnik, M.H.; Yu, Y.; Franco, A. Muscle cells from mdx mice have an increased susceptibility to oxidative stress. Neuromuscul. Disord. 1998, 8, 14–21. [Google Scholar] [CrossRef]
- Disatnik, M.H.; Dhawan, J.; Yu, Y.; Beal, M.F.; Whirl, M.M.; Franco, A.; Rando, T.A. Evidence of oxidative stress in mdx mouse muscle: Studies of the pre-necrotic state. J. Neurol. Sci. 1998, 161, 77–84. [Google Scholar] [CrossRef]
- Irwin, W.A.; Bergamin, N.; Sabatelli, P.; Reggiani, C.; Megighian, A.; Merlini, L.; Braghetta, P.; Columbaro, M.; Volpin, D.; Bressan, G.M.; et al. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat. Genet. 2003, 35, 367–371. [Google Scholar] [CrossRef]
- Palma, E.; Tiepolo, T.; Angelin, A.; Sabatelli, P.; Maraldi, N.M.; Basso, E.; Forte, M.A; Bernardi, P.; Bonaldo, P. Genetic ablation of cyclophilin D rescues mitochondrial defects and prevents muscle apoptosis in collagen VI myopathic mice. Hum. Mol. Genet. 2009, 18, 2024–2031. [Google Scholar] [CrossRef]
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.P.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 2008, 14, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Menazza, S.; Blaauw, B.; Tiepolo, T.; Toniolo, L.; Braghetta, P.; Spolaore, B.; Reggiani, C.; Di Lisa, F.; Bonaldo, P.; Canton, M. Oxidative stress by monoamine oxidases is causally involved in myofiber damage in muscular dystrophy. Hum. Mol. Genet. 2010, 19, 4207–4215. [Google Scholar] [CrossRef] [PubMed]
- Canton, M.; Neverova, I.; Menabò, R.; Van Eyk, J.; Di Lisa, F. Evidence of myofibrillar protein oxidation induced by postischemic reperfusion in isolated rat hearts. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H870–H877. [Google Scholar] [CrossRef] [PubMed]
- Canton, M.; Skyschally, A.; Menabò, R.; Boengler, K.; Gres, P.; Schulz, R.; Haude, M.; Erbel, R.; Di Lisa, F.; Heusch, G. Oxidative modification of tropomyosin and myocardial dysfunction following coronary microembolization. Eur. Heart J. 2006, 27, 875–881. [Google Scholar] [CrossRef]
- Canton, M.; Menazza, S.; Sheeran, F.L.; Polverino De Laureto, P.; Di Lisa, F.; Pepe, S. Oxidation of myofibrillar proteins in human heart failure. J. Am. Coll. Cardiol. 2011, 57, 300–309. [Google Scholar] [CrossRef]
- Finberg, J.P.M.; Rabey, J.M. Inhibitors of MAO-A and MAO-B in psychiatry and neurology. Front. Pharmacol. 2016, 7, 340–348. [Google Scholar] [CrossRef]
- Binda, C.; Newton-Vinson, P.; Hubálek, F.; Edmondson, D.E.; Mattevi, A. Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat. Struct. Biol. 2002, 9, 22–26. [Google Scholar] [CrossRef]
- Binda, C.; Li, M.; Hubalek, F.; Restelli, N.; Edmondson, D.E.; Mattevi, A. Insights into the mode of inhibition of human mitochondrial monoamine oxidase B from high-resolution crystal structures. Proc. Natl. Acad. Sci. USA 2003, 100, 9750–9755. [Google Scholar] [CrossRef]
- De Colibus, L.; Li, M.; Binda, C.; Lustig, A.; Edmondson, D.E.; Mattevi, A. Three-dimensional structure of human monoamine oxidase A (MAO A): Relation to the structures of rat MAO A and human MAO B. Proc. Natl. Acad. Sci. USA 2005, 102, 12684–12689. [Google Scholar] [CrossRef]
- Bello, L.; Hoffman, E.P.; Pegoraro, E. Dystrophinopathies. In Muscular Dystrophy Causes and Management; Angelini, C., Ed.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2013; pp. 69–95. ISBN 978-1-62618-460-2. [Google Scholar]
- Kearney, M.; Orrell, R.W.; Fahey, M.; Pandolfo, M. Antioxidants and other pharmacological treatments for Friedreich ataxia. Cochrane Database Syst. Rev. 2009, 4. [Google Scholar] [CrossRef]
- Schiff, M.; Rustin, P. Idebenone in Friedreich ataxia and Leber’s hereditary optic neuropathy: Close mechanisms, similar therapy? Brain 2016, 139, e39. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.; Bitto, A.; Aguennouz, M.; Mazzeo, A.; Migliorato, A.; Polito, F.; Irrera, N.; Altavilla, D.; Vita, G.L.; Russo, M.; et al. Flavocoxid counteracts muscle necrosis and improves functional properties in mdx mice: A comparison study with methylprednisolone. Exp. Neurol. 2009, 220, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.; Vita, G.L.; Licata, N.; Sframeli, M.; Bitto, A.; Distefano, M.G.; Barcellona, C.; La Rosa, M.; Romeo, S.; Ciranni, A.; et al. Pilot study of flavocoxid in ambulant DMD patients. Neuromuscul. Disord. 2014, 24, 825. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Van Deutekom, J.C.T.; Fokkema, I.F.; Van Ommen, G.J.B.; Den Dunnen, J.T. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Kishor, A.; Fritz, S.E.; Hogg, J.R. Nonsense-mediated mRNA decay: The challenge of telling right from wrong in a complex transcriptome. Wiley Interdiscip. Rev. RNA 2019, 10, e1548. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, M.; Bukowy-Bieryllo, Z.; Zietkiewicz, E. Advances in therapeutic use of a drug-stimulated translational readthrough of premature termination codons. Mol. Med. 2018, 24, 25. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wu, B.; Shah, S.N.; Lu, P.; Lu, Q. Aminoglycoside Enhances the Delivery of Antisense Morpholino Oligonucleotides In Vitro and in mdx Mice. Mol. Ther. Nucleic Acids 2019, 16, 663–674. [Google Scholar] [CrossRef]
- Baradaran-Heravi, A.; Balgi, A.D.; Zimmerman, C.; Choi, K.; Shidmoossavee, F.S.; Tan, J.S.; Bergeaud, C.; Krause, A.; Flibotte, S.; Shimizu, Y.; et al. Novel small molecules potentiate premature termination codon readthrough by aminoglycosides. Nucleic Acids Res. 2016, 44, 6583–6598. [Google Scholar] [CrossRef]
- Kayali, R.; Ku, J.M.; Khitrov, G.; Jung, M.E.; Prikhodko, O.; Bertoni, C. Read-through compound 13 restores dystrophin expression and improves muscle function in the mdx mouse model for Duchenne muscular dystrophy. Hum. Mol. Genet. 2012, 21, 4007–4020. [Google Scholar] [CrossRef]
- Melacini, P. Cardiac problems in DMD. In Muscular Dystrophy: Causes and Management; Angelini, C, Ed.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2013; pp. 367–380. ISBN 978-1-62618-460-2. [Google Scholar]
- Hor, K.N.; Mah, M.L.; Johnston, P.; Cripe, T.P.; Cripe, L.H. Advances in the diagnosis and management of cardiomyopathy in Duchenne muscular dystrophy. Neuromuscul. Disord. 2018, 28, 711–716. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Alman, B.A.; Apkon, S.D.; Blackwell, A.; Case, L.E.; Cripe, L.; Hadjiyannakis, S.; Olson, A.K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018, 17, 347–361. [Google Scholar] [CrossRef]
- Sanders, S.P. Did they lower stress in the trial?: Or was it just wasted energy? J. Am. Coll. Cardiol. 2005, 45, 858–859. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Duboc, D.; Meune, C.; Lerebours, G.; Devaux, J.Y.; Vaksmann, G.; Bécane, H.M. Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J. Am. Coll. Cardiol. 2005, 45, 855–857. [Google Scholar] [CrossRef] [PubMed]
- Duboc, D.; Meune, C.; Pierre, B.; Wahbi, K.; Eymard, B.; Toutain, A.; Berard, C.; Vaksmann, G.; Bécane, H.M. Perindopril preserves left ventricular function in X-linked Duchenne muscular dystrophy. Eur. Hear. J. Suppl. 2007, 9, E20–E24. [Google Scholar] [CrossRef][Green Version]
- Matsumura, T. Beta-blockers in Children with Duchenne Cardiomyopathy. Rev. Recent Clin. Trials 2014, 9, 76–81. [Google Scholar] [CrossRef]
- Viollet, L.; Thrush, P.T.; Flanigan, K.M.; Mendell, J.R.; Allen, H.D. Effects of angiotensin-converting enzyme inhibitors and/or beta blockers on the cardiomyopathy in duchenne muscular dystrophy. Am. J. Cardiol. 2012, 110, 98–102. [Google Scholar] [CrossRef]
- Raman, S.V.; Hor, K.N.; Mazur, W.; Halnon, N.J.; Kissel, J.T.; He, X.; Tran, T.; Smart, S.; McCarthy, B.; Taylor, M.D.; et al. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015, 14, 153–161. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef]
- Shaddy, R.; Canter, C.; Halnon, N.; Kochilas, L.; Rossano, J.; Bonnet, D.; Bush, C.; Zhao, Z.; Kantor, P.; Burch, M.; et al. Design for the sacubitril/valsartan (LCZ696) compared with enalapril study of pediatric patients with heart failure due to systemic left ventricle systolic dysfunction (PANORAMA-HF study). Am. Heart J. 2017, 193, 23–34. [Google Scholar] [CrossRef]
- Buddhe, S.; Cripe, L.; Friedland-Little, J.; Kertesz, N.; Eghtesady, P.; Finder, J.; Hor, K.; Judge, D.P.; Kinnett, K.; McNally, E.M.; et al. Cardiac management of the patient with Duchenne muscular dystrophy. Pediatrics 2018, 142, S72–S81. [Google Scholar] [CrossRef]
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef]
- Verhaart, I.E.C.; Aartsma-Rus, A. Therapeutic developments for Duchenne muscular dystrophy. Nat. Rev. Neurol. 2019, 15, 373–386. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
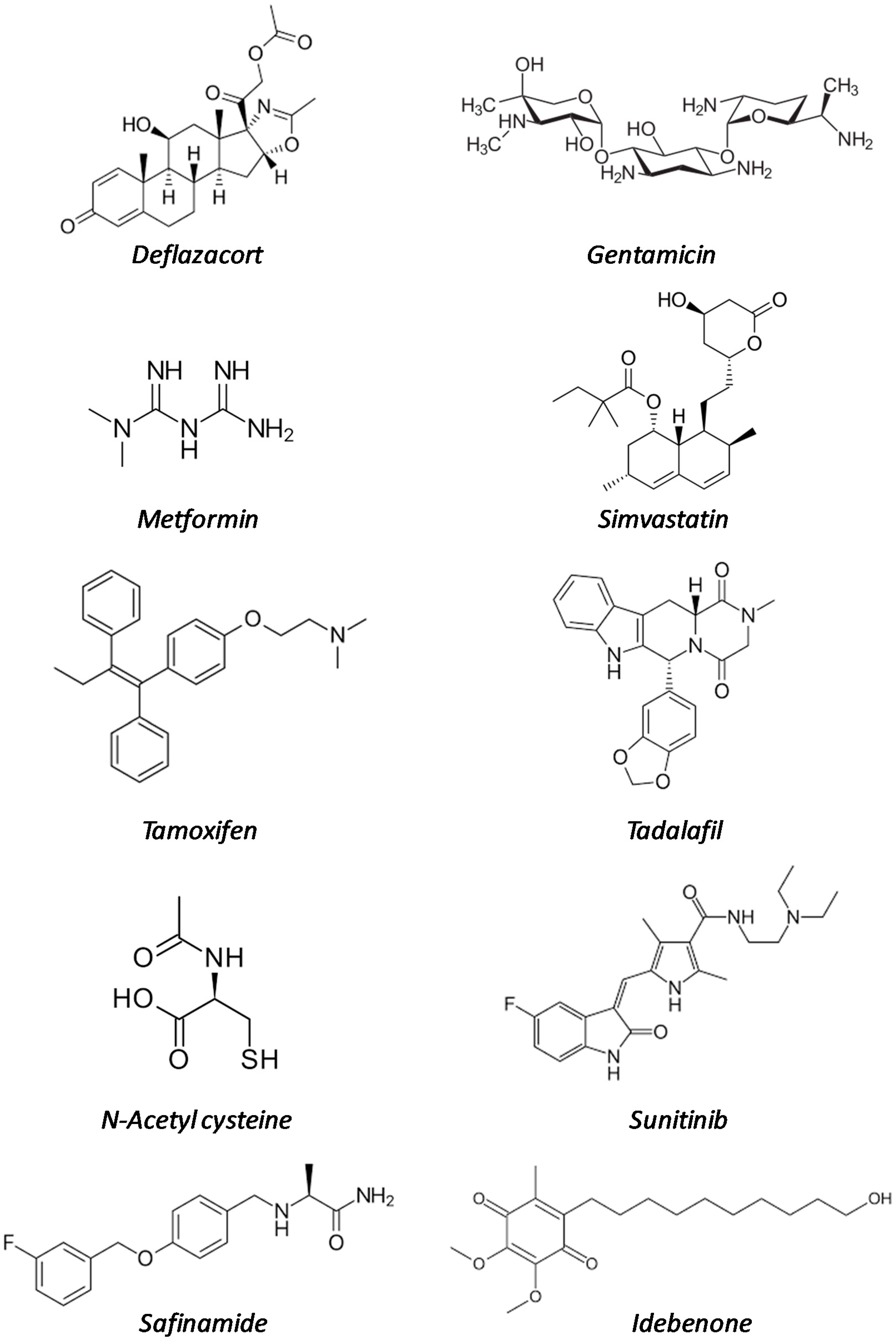
| Drug Name | Original Indication (Date of Approval) | Mode of Action | Development Phase in DMD | References |
|---|---|---|---|---|
| Deflazacort | Rheumatoid arthritis (1955) | Multiple mechanisms | Marketed | [13,14,15,16,17,18,19,20,21,22,23] |
| Gentamicin | Bacterial infections (1987) | Protein synthesis inhibitor | Clinical trials | [24,25,26,27] |
| Metformin | Type II diabetes (1995) | Multiple mechanisms | Clinical trials | [28,29,30,31,32,33,34,35] |
| Simvastatin | Familial hyperlipidaemia (1998) | HMG-CoA reductase inhibitor | Preclinical studies | [36,37,38,39,40] |
| Tamoxifen | ER-positive breast cancer (1998) | Estrogen receptor (ER) modulator | Clinical trials | [41,42,43] |
| Tadalafil | Erectile dysfunction (2003) | PDE5 inhibitor | Clinical trials | [44,45,46,47,48,49] |
| N-acetyl cysteine | Acetaminophen overdose (2004) | Antioxidant | Preclinical studies | [50,51,52] |
| Sunitinib | Renal cell carcinoma and gastrointestinal tumors (2006) | Multireceptor tyrosin kinase (RTK) inhibitor | Preclinical studies | [53,54] |
| Safinamide | Parkinson’s disease (2015) | Monoamine oxidase B inhibitor | Preclinical studies | [55,56,57] |
| Idebenone | Leber neuropathy (2015, not FDA) | coenzyme Q10 analogue | Preclinical studies | [58,59,60,61,62,63] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitiello, L.; Tibaudo, L.; Pegoraro, E.; Bello, L.; Canton, M. Teaching an Old Molecule New Tricks: Drug Repositioning for Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2019, 20, 6053. https://doi.org/10.3390/ijms20236053
Vitiello L, Tibaudo L, Pegoraro E, Bello L, Canton M. Teaching an Old Molecule New Tricks: Drug Repositioning for Duchenne Muscular Dystrophy. International Journal of Molecular Sciences. 2019; 20(23):6053. https://doi.org/10.3390/ijms20236053
Chicago/Turabian StyleVitiello, Libero, Lucia Tibaudo, Elena Pegoraro, Luca Bello, and Marcella Canton. 2019. "Teaching an Old Molecule New Tricks: Drug Repositioning for Duchenne Muscular Dystrophy" International Journal of Molecular Sciences 20, no. 23: 6053. https://doi.org/10.3390/ijms20236053
APA StyleVitiello, L., Tibaudo, L., Pegoraro, E., Bello, L., & Canton, M. (2019). Teaching an Old Molecule New Tricks: Drug Repositioning for Duchenne Muscular Dystrophy. International Journal of Molecular Sciences, 20(23), 6053. https://doi.org/10.3390/ijms20236053