1. Introduction
It has long been recognized that vitamin D plays a major role in bone and mineral metabolism, but until recent years, it has also been implicated in immunity and cardiovascular health. The first rate-limiting step in the endogenous synthesis of vitamin D is the skin’s sun exposure, as ultraviolet B (UVB) radiation is necessary to convert 7-dehydrocholesterol into vitamin D3 (cholecalciferol), with estimates of cutaneous synthesis providing between 80% and 100% of the vitamin D requirements of the body [
1]. In the liver, 25-hydroxylase (CYP2R1) converts vitamin D3 into 25-hydroxyvitamin D [25(OH)D], which is then transformed in the kidney by means of 1-alpha-hydroxylase (CYP27B1), into 1,25-dihydroxyvitamin D [1,25(OH)2D3], also known as calcitriol, the hormone’s biologically active form [
2]. The enzyme 1-alpha-hydroxylase is activated by the parathyroid hormone (PTH) and inhibited by fibroblast growth factor-23 (FGF-23). On the other hand, 25-hydroxyvitamin D3 24-hydroxylase (CYP24A1) is involved in calcitriol’s catabolism. This enzyme limits the generation of calcitriol by converting it into hydroxylated derivatives (which will later be excreted), or by converting 25(OH)D3 into 24,25(OH)2D3 or 23,25(OH)2D3, further decreasing its availability for 1-hydroxylation. CYP24A1 is inhibited by PTH and induced by FGF-23 [
3]. By feedback mechanisms, both 1-alpha-hydroxylase and 25-hydroxyvitamin D3 24-hydroxylase regulate 1,25(OH)2D3′s bioavailability as a means to protect against hypercalcemia [
3,
4].
Several risk factors have been described to predispose to vitamin D deficiency. Living in higher latitudes (above 35 degrees) is a well-known risk factor [
5]; however, vitamin D deficiency has been shown to be highly prevalent even in populations living near the equator [
6]. Other risk factors for vitamin D deficiency include skin phototype, sunscreen usage, indoor working environments, minimal outdoor activity, obesity, high body mass index (BMI), and waist circumference (WC), as well as older age, which is accompanied by a 75% decrease in vitamin D synthesis capacity [
1,
6]. Among these factors, the combination of low UVB availability and/or underexposure, in addition to insufficient dietary intake, have been described as the most important [
1].
Notably, nutrition surveillance data from various countries have indicated that vitamin D ingestion is lower than the recommended minimum amount [
1,
7]. There is also evidence that suggests that the recommended intake established by guidelines (600 IU) is actually insufficient and should be increased to at least 800 IU [
5].
Vitamin D also plays a role in immune regulation, as immune cells express 1-alpha-hydroxylase to regulate their own local concentration of calcitriol. This effect has been related to the milieu of cytokines [
8], given that adequate levels of serum 25(OH)D have been associated with increased levels of the anti-inflammatory cytokines interleukins (IL) 4 and 10, and to lower levels of the proinflammatory cytokines IL-1 and IL-6 [
9]. On the contrary, vitamin D deficiency has been associated with chronic inflammatory states and a proinflammatory cytokine profile. Vitamin D deficiency has also been linked with augmented collagen synthesis, oxidative stress, and fibrosis, which are potential mechanisms underlying cardiovascular diseases, such as heart failure [
10].
Heart failure (HF) is one of the most common chronic medical conditions worldwide. It is estimated that as of 2019, more than 6 million Americans are living with this disease [
11,
12,
13]. HF with reduced ejection fraction (HFrEF) is defined by a left ventricular ejection fraction (LVEF) of < 40% [
14]. Most of the risk factors for HFrEF in Western populations have been described in association with ischemic heart disease [
15]. It has been proposed that vitamin D might play a role in disease pathogenesis, as serum 25(OH)D levels have been demonstrated to be lower in patients with HF compared with control subjects [
16]. Specifically, prospective studies have shown that the risk of developing HF is increased in patients with vitamin D deficiency [
17]. One of the mechanistic links regarding this association relies on a proinflammatory cytokine state, including both innate and adaptive immunity [
18,
19,
20].
Regarding innate immunity, vitamin D deficiency has been shown to increase the expression of nuclear-factor kappa B (NF-κB), leading to a higher secretion and release of the main proinflammatory cytokines, such as IL-6 and monocyte chemoattractant protein-1 (MCP-1) [
10]. Cardiomyocytes themselves are activated by hemodynamic stress and are able to secrete several inflammatory cytokines as well [
21]. On the other hand, by activation of mitogen-activated protein kinase (MAPK) phosphatase-1 and subsequent inhibition of p38 MAPK, vitamin D has been demonstrated to switch the cytokine profile into an anti-inflammatory state, with inhibition of the production of tumor necrosis factor-alpha (TNF-α) and IL-6 [
22]. In the case of adaptive immunity, vitamin D has been found to inhibit the CD4+ T-cell course towards the Th17 lineage [
9]. Additionally, vitamin D is able to hinder IL-17 production of those CD4+ T-cells that have been already committed towards the Th17 lineage [
23]. Not only does vitamin D exert its effects through immune regulation, but it is also able to control extracellular matrix metabolism. Vitamin D deficiency has been related to increased expression of matrix metalloproteinase (MMP) 2 and 9 [
10], as well as to augmented tissue macrophage infiltration [
24]. These mechanisms may be associated with some of the structural abnormalities seen in HF, such as ventricular remodeling, tissue fibrosis, and a systemic proinflammatory state. Other mechanisms through which vitamin D deficiency predisposes to HF include: overactivation of the renin-angiotensin-aldosterone system (RAAS); dysfunction of the intracellular calcium handling by the cardiomyocyte; overexpression of procollagen-1, which leads to increased fibrosis; diminished protein kinase A (PKA) levels that result in impaired contractility; and activation of calcineurin signaling, which promotes cardiac hypertrophy [
25,
26,
27,
28].
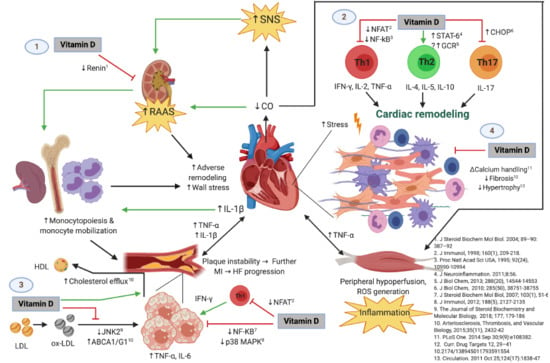
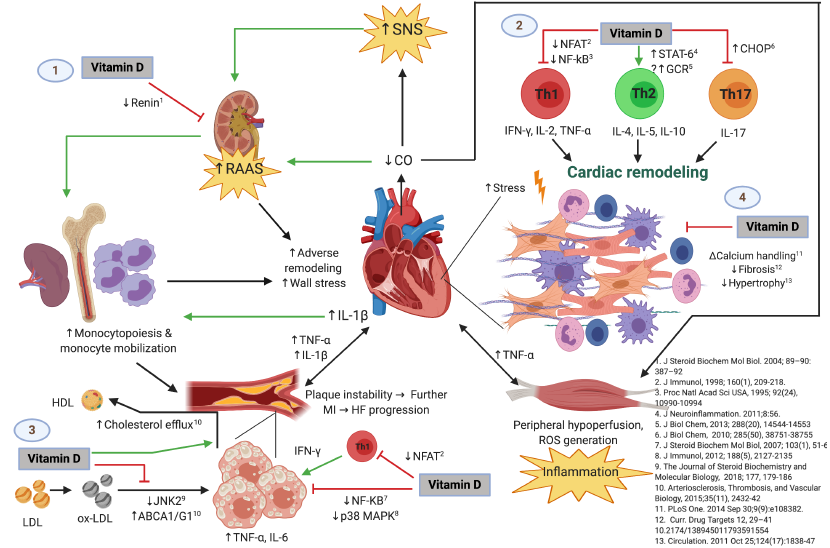
Given the evidence provided relating the mechanisms through which vitamin D deficiency is associated with HF, it is important to study the seasonal variation of vitamin D deficiency and the cytokine profile, as well as the lifestyle factors that exert an influence in patients with HF. Thus, the aim of this study is to describe the seasonal variation of vitamin D deficiency and its association with 13 inflammatory cytokines, biochemical, and lifestyle factors during a 12-month follow-up in a cohort of patients with HFrEF, as well as to describe the molecular inflammatory and atherosclerotic mechanisms related to vitamin D deficiency. An original figure summarizing these mechanisms is also shown.
3. Discussion
3.1. Vitamin D Deficiency in the Setting of Patients with HFrEF
One of the core aspects of recent molecular and clinical research into HFrEF has been to better understand the role of vitamin D and the vitamin D receptor in the initiation and progression of HF. There are several mechanisms underlying this association. Studies in animals and humans have shown that serum 25(OH)D levels are inversely correlated with the RAAS activity, possibly because active vitamin D inhibits renin biosynthesis [
25]. Vitamin D deficiency may, therefore, contribute to a hyper-activated RAAS axis, which then promotes the deleterious hemodynamic consequences of salt and water retention, vasoconstriction, and ventricular remodeling. Another mechanism associating vitamin D deficiency and ventricular dysfunction is intracellular calcium handling by the cardiomyocyte. In a mouse model of cardiac dysfunction, mice lacking calcitriol have been found to exhibit abnormal calcium handling, impaired ventricular functioning, and adverse cardiac remodeling and fibrosis [
26]. Furthermore, activation of the Vitamin D receptor has been shown to regulate the expression of procollagen 1, which in turn may regulate profibrotic activity in the myocardium [
27]. In another animal model, the deletion of the vitamin D receptor gene was also found to activate the calcineurin/nuclear factor of activated T-cells (NFAT) intracellular signaling pathway, a potent pro-hypertrophic signal [
28]. Moreover, myocardial contractility may also be enhanced in the presence of sufficient vitamin D levels, which correlates with increased intracellular concentrations of cyclic adenosine monophosphate (cAMP), PKA, and PKC [
27]. Overall, these findings demonstrate several mechanistic associations in which vitamin D deficiency may participate in HF progression, hemodynamic abnormalities, and structural dysfunction. Our results show that most of the patients with HFrEF in our cohort have low serum 25(OH)D levels and are in line with other studies. A significant high prevalence of vitamin D deficiency in patients with HF, when comparing low and very low vs. normal serum 25(OH)D levels (HR = 1.33 and 2.19, respectively), has been found [
31]. A high prevalence of vitamin D deficiency among patients with HF and an independent association of low 25(OH)D levels with hospitalization and mortality rates have also been described [
32]. On the other hand, in a different type of study to evaluate whether inadequate serum levels of 25(OH)D predict the prevalence of chronic conditions such as cardiovascular disease, type 2 diabetes, and obesity, among others, no association with any chronic condition, including heart failure, was found; this study did not report prevalence of vitamin D deficiency [
33]. Both cellular and hemodynamic mechanisms may be implicated in this association, which not only explains an increased prevalence of vitamin D deficiency among patients with HF, but may also recognize a connection between vitamin D deficiency and higher risk of adverse outcomes in patients with HF. In our cohort of patients with HF, vitamin D deficiency may thus contribute to ventricular dysfunction, cardiac remodeling, hypertrophy, fibrosis, and disease progression by the molecular and cellular signaling mechanisms described above.
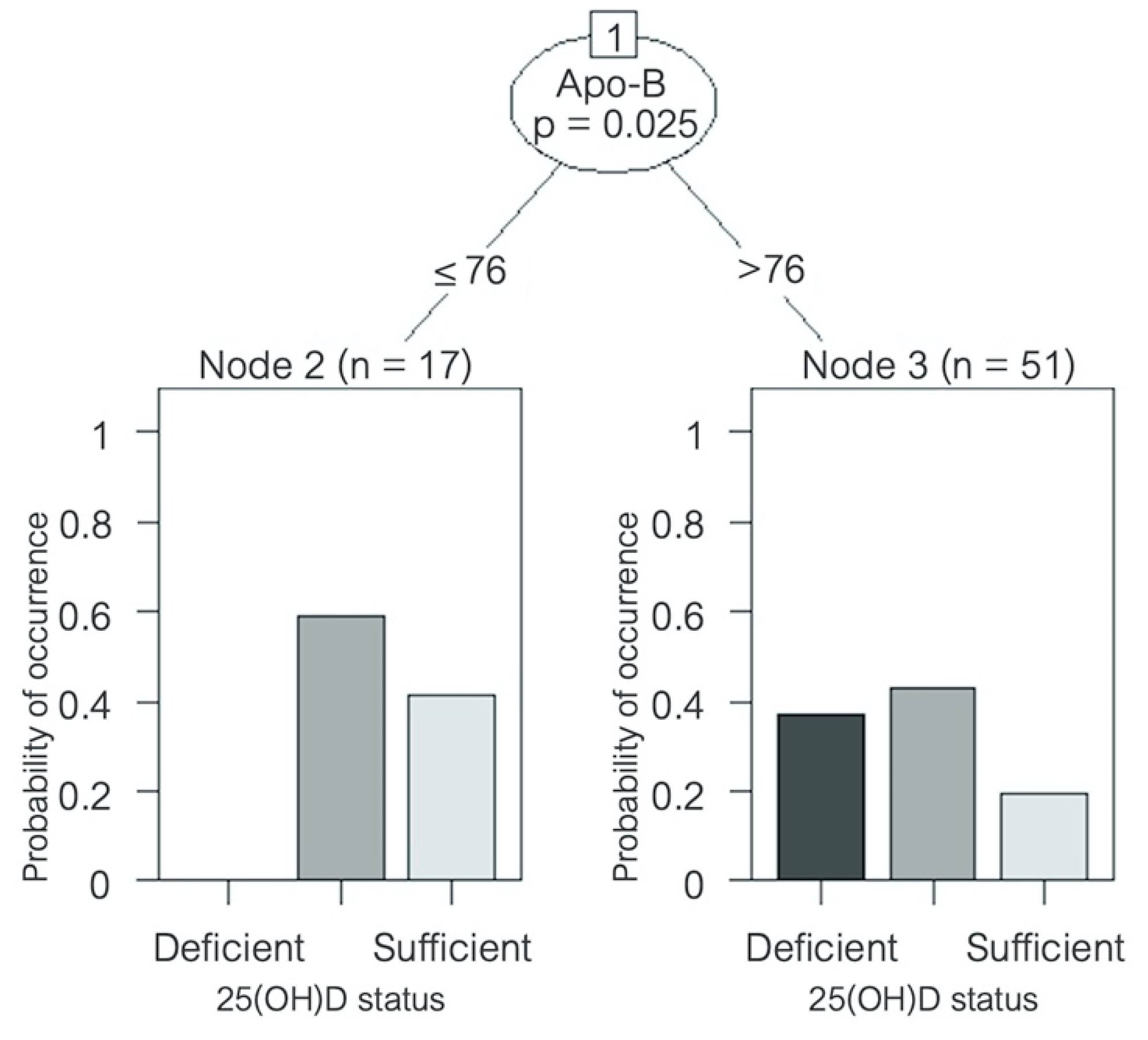
3.2. Correlation of Atherosclerotic and Metabolic Parameters with Vitamin D Deficiency in Patients with HFrEF
Diverse types of dyslipidemias have been recognized as risk factors for the development of HF [
34], and in conjunction with vitamin D deficiency, they may explain some of the mechanisms for disease progression. Atherosclerosis plays a predominant role in the pathophysiology of ischemic heart disease, which by itself, is the leading cause of HF. LDL-cholesterol particles turn into oxidized LDL cholesterol (oxLDL) by reactive oxygen species. Macrophages, which express high-capacity scavenger receptors that are not down-regulated in the presence of high oxLDL concentration, are modified into foam cells [
35]. Clusters of foam cells accumulated in the sub-endothelium, up-regulate the expression of NF-κB, which in turn triggers the membrane expression of vascular cell adhesion protein 1 (VCAM-1) and the endothelial adhesion molecule E-selectin. Another mechanism that increases NF-κB transcription in the endothelial cells is the increased proinflammatory cytokines IL-1, IL-8, and MCP-1 that characterize the inflammatory state in HF [
36]. Adhesion molecules VCAM-1 and E-selectin contribute to increased cellular inflammation in the endothelial surface, as macrophages and T-cells are attracted to these proteins [
35,
36]. This propagates the inflammatory cycle, eventually leading to disturbed blood flow in the vessel’s lumen and progression of atherosclerosis.
Regarding these mechanisms, vitamin D may have lipid-regulating effects. Therefore, adequate levels of 25(OH)D might act as a protective factor in coronary disease and HF, but in the case of vitamin D deficiency, there may be deleterious consequences.
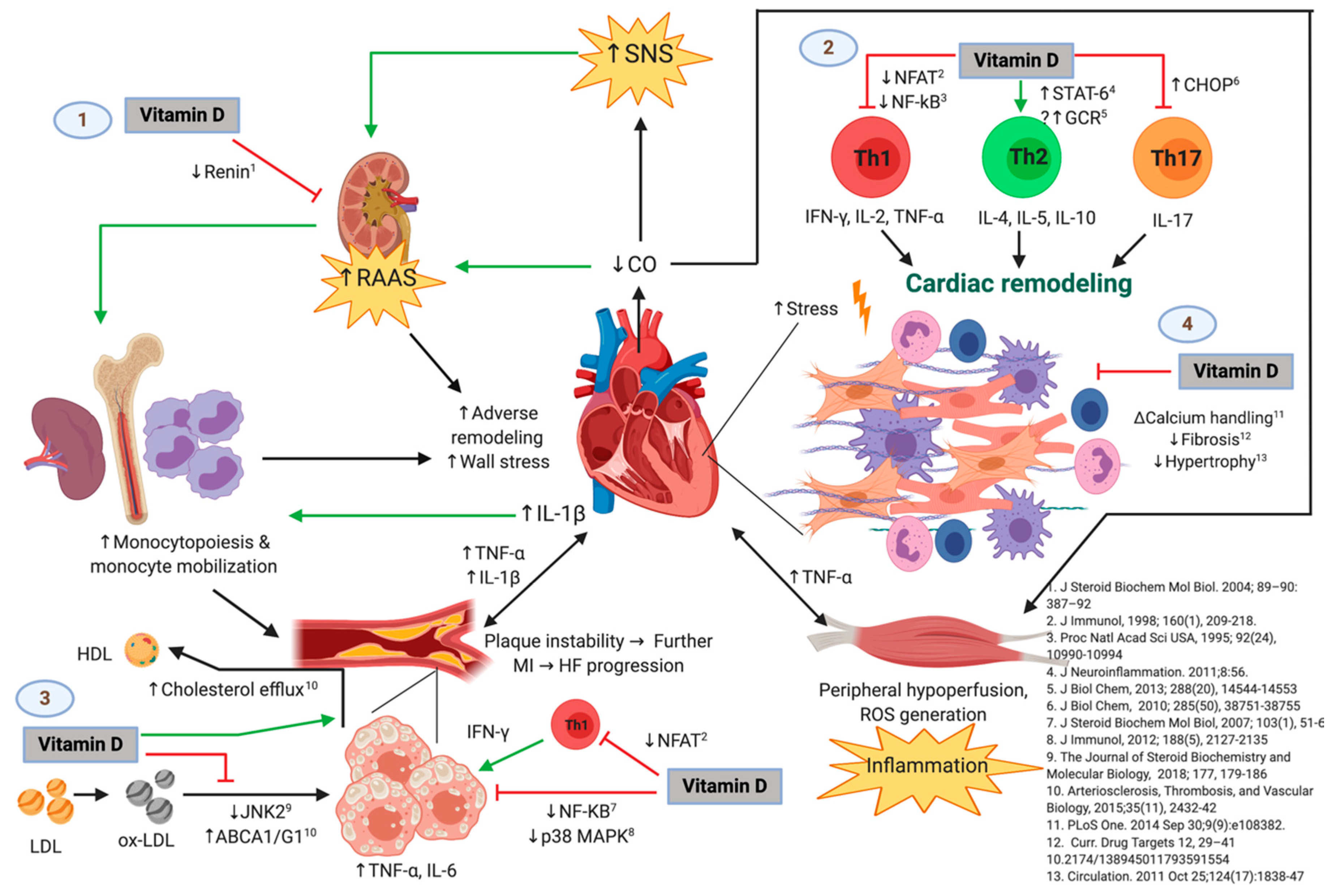
Figure 6 shows the possible immunomodulatory role of vitamin D regarding inflammation in the context of HF progression. In an animal model of hypercholesterolemia, vitamin D was shown to promote cholesterol efflux from macrophage-derived foam cells by augmenting the expression of ATP-binding membrane cassette transporter types A1 and G1 (ABCA1/G1) [
37]. Hepatocyte uptake of cholesterol was also shown to be increased in the presence of higher levels of 25(OH)D concentrations. In addition, vitamin D has also been described to downregulate the c-Jun N-terminal protein kinase 2 (JNK2) cellular pathway in macrophages, which decreases oxLDL uptake and subsequently inhibits transformation into a foam cell phenotype [
38]. Given the high prevalence of vitamin D deficiency throughout the four seasons of the year in our cohort of patients with HF, these protective effects of vitamin D may likely be absent. Furthermore, our results show an inverse correlation between 25(OH)D levels and total cholesterol, LDL cholesterol, non-HDL cholesterol, triglycerides, Apo-B, and Apo-B/Apo-A ratio. These correlations may also reflect the mechanisms described above, pointing towards the lack of the possible protective role of vitamin D in the setting of dyslipidemias. Our results are consistent with the prospective study,
The Atherosclerosis Risk in Communities, in which lower 25(OH)D concentrations were shown to be associated with higher LDL cholesterol and total cholesterol, as well as an overall greater risk of dyslipidemias [
39]. Similarly, Forrest et al. found that vitamin D deficiency was significantly more common in individuals with high LDL cholesterol [
40].
Dysregulation of glucose metabolism has been linked to vitamin D deficiency and the development, as well as progression, of HF. In patients with established HF, the presence of diabetes mellitus has been associated with increased hospitalization rates and overall increased mortality [
34]. Several mechanisms have been described regarding the role of vitamin D in glucose metabolism. In an animal model of vitamin D deficiency, decreased protein kinase B/Akt phosphorylation and reduced expression of glucose transporter 4 (GLUT4) in cardiomyocytes was shown. This, in turn, resulted in insulin resistance by the myocardium, a significant downregulation of the endogenous antioxidant enzymes SOD2 and catalase, and diminished LVEF [
41]. The high prevalence of vitamin D deficiency throughout the 12-month follow-up shown in our cohort of patients with HF may thus worsen or predispose them to alterations in glucose metabolism. Moreover, a significant correlation between 25(OH)D levels < 43 ng/mL and impaired fasting glucose (OR = 3.40) and low insulin sensitivity (defined as HOMA index for insulin sensitivity < 90%) (OR = 2.65) has been reported in healthy adults with a median age of 39.4 years old [
42]. Specifically, in patients with HF, vitamin D deficiency was shown to be correlated with increased rates of diabetes mellitus when compared with a non-vitamin D deficient group (31.4% vs. 22.8%) [
43]. Despite these observations, our results show no correlation between 25(OH)D concentrations and fasting glucose level, insulin concentration, or HOMA index. These findings may be attributed to the low LVEF (below 40%) of all our patients, or to the medications they were taking.
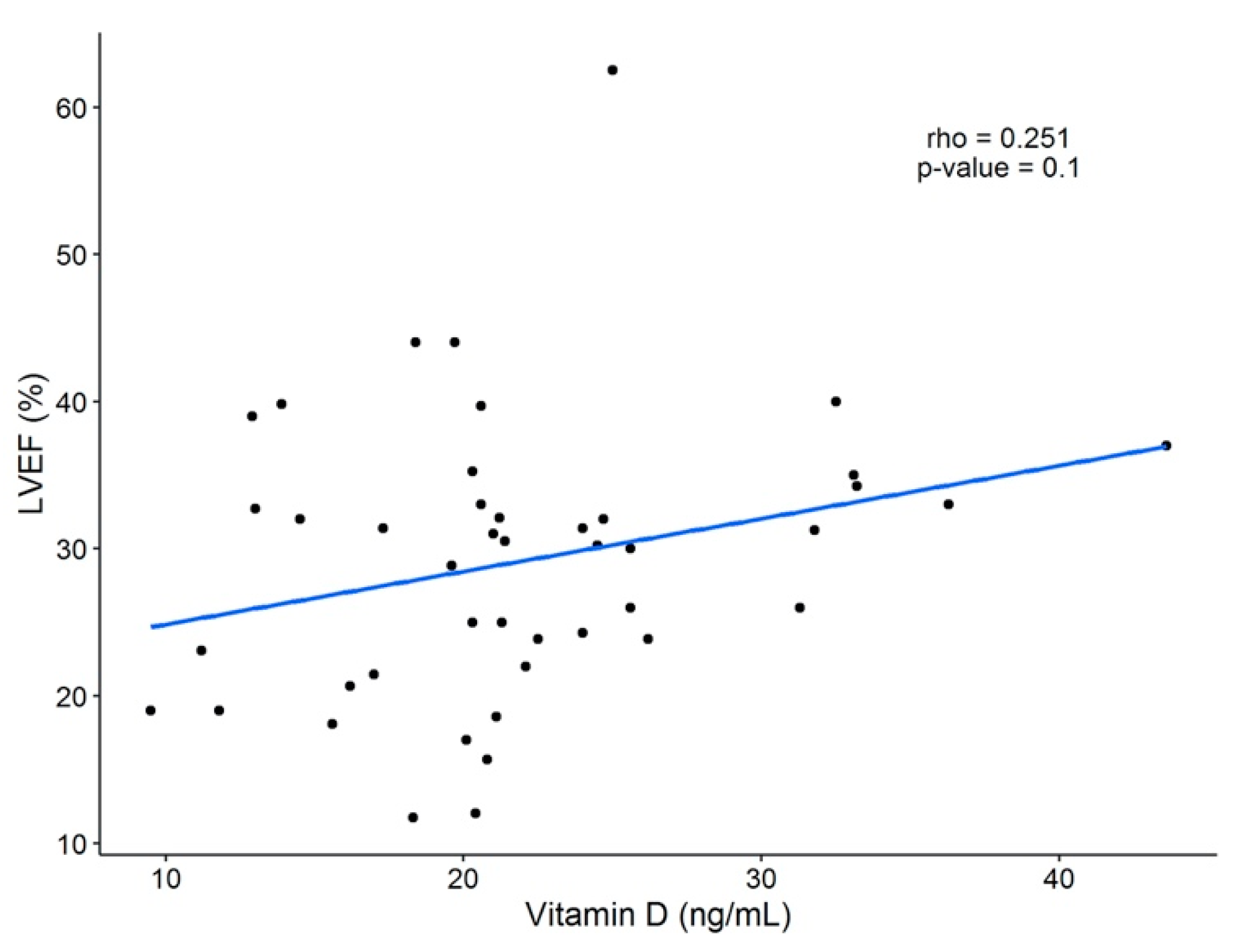
Contrary to what was expected, no correlation was seen between serum 25(OH)D concentrations and LVEF in our cohort. This could be explained by the fact that all of our patients presented abnormally low values of LVEF. Therefore, we could not analyze the whole spectrum of the relationship between vitamin D levels and LVEF.
3.3. Inflammation Pathways Associating Vitamin D Deficiency with HFrEF
Figure 6 shows the possible immunomodulatory role of Vitamin D regarding inflammation in the context of HF progression. HFrEF has long been recognized as a systemic proinflammatory state, with pathways involving activation of both innate and adaptive immunity mechanisms [
19,
20]. Most likely, a crosstalk between the heart and the peripheral organs in which HF promotes inflammation and vice versa might be present [
21]. Hemodynamic stress and volume overload have been described to increment wall stress and cell mechanical damage, which stimulates cardiomyocyte release of proinflammatory cytokines, such as TNF-α, IL-1, MCP-1, and IL-6. On the other hand, peripheral organs respond to these inflammatory signals inducing more deleterious effects, such as: skeletal muscle inflammation (which may itself be secondary to chronic vasoconstriction and hypoperfusion due to decreased cardiac output), adipose tissue inflammation, increased atherogenic progression in the endothelium, augmented monocyte production by the bone marrow, and increased bacterial translocation from the gut [
21]. This chronic proinflammatory cycle has been shown to contribute to the deterioration of ventricular function by inducing myocardial contractile dysfunction, hypertrophy, apoptosis, and fibrosis, with subsequent progression of HF [
51]. Although systemic inflammation is a central pathogenic hallmark of HF [
20], it is currently unclear whether vitamin D deficiency plays a specific role in the pathogenesis of HF through the attributed immunomodulatory functions [
52]. However, it has been shown that vitamin D deficiency is indeed associated with HFrEF and increased markers of ongoing inflammation [
17,
53].
In the context of inflammation, the relationship between vitamin D and the cytokine milieu has been shown to represent a core aspect that leads to a proinflammatory state through several mechanisms. Our results in this cohort of patients with HF show serum 25(OH)D levels to be inversely correlated with the proinflammatory cytokines IL-1β, TNF-α, IL-6, and IL-8, although no correlation with the anti-inflammatory cytokine IL-10 was noted. Vitamin D has been found to inhibit the secretion of proinflammatory cytokines TNF-α and IL-6 by monocytes and macrophages through inhibiting p38 MAPK via induction of MKP1, which results in dephosphorylation of p38 [
22]. The MAPK pathway is stimulated by diverse stressors to induce expression of proinflammatory cytokines, not only in immune cells but also in the myocardium itself [
54]. Since a high prevalence of vitamin D deficiency was found in our study population, induction of this metabolic pathway is highly probable, which might aggravate the inflammatory status that these patients with HFrEF are already in.
Other mechanisms by means of which vitamin D has been associated with inflammation are related to the vitamin D-activated vitamin D receptor/retinoid X receptor (VDR/RXR) heterodimer. VDR/RXR has been reported to interact with transcription factors such as NF-κB, NFAT, and the glucocorticoid receptor (GCR), all of which participate in the transcription of genes involved in inflammatory processes [
52]. NF-κB is involved in the transcription of many proinflammatory cytokines, including TNF-α, IL-1β, and IL-6, among others [
55], which are direct mediators of cardiac dysfunction [
56]. NF-κB expression and activation has been shown to be downregulated by vitamin D [
48,
50], which may function as a protective mechanism that would not be present in the setting of vitamin D deficiency. In its own way, NFAT upregulates the expression of IL-2, a crucial cytokine for T-cell replication, activation, and induction of Th1-mediated inflammatory responses [
47], which appear to be relevant in HF, as evidenced by the higher number of circulating Th1 cells seen in this condition [
57]. NFAT signaling has been shown to be inhibited by vitamin D, preventing this transcription factor from binding to its response elements [
46], which further indicates another immunological pathway by which vitamin D deficiency may be deleterious in HF. On the other hand, GCR upregulates the expression of anti-inflammatory cytokines, while downregulating proinflammatory ones [
58]. Vitamin D has been found to enhance these GCR-mediated anti-inflammatory activities [
59], which may represent an anti-inflammatory mechanism that would be absent in circumstances of vitamin D deficiency. Considering the lines of evidence of the mechanisms and pathways provided, low 25(OH)D levels are expected to be accompanied by increased circulating proinflammatory cytokines, as observed in our cohort, which may possibly contribute to clinical deterioration in these patients.
Regarding one of the main pathways of adaptive immunity, our results show serum 25(OH)D concentrations to be negatively correlated with IL-17A as well. Interestingly, vitamin D has also been shown to inhibit the secretion of the proinflammatory cytokine IL-17 by Th17 cells through a post-transcriptional mechanism [
23]. Others have shown that vitamin D suppresses autoimmunity, not only by decreasing the production of IL-17, but also by inhibiting the ability of naïve CD4+ T-cells to commit to the Th17 lineage [
9]. The anti-Th17 effects of vitamin D could be of pathophysiological relevance in HFrEF, as IL-17 appears to have a role in adverse cardiac remodeling and myocardial fibrosis [
60]. Vitamin D seems to induce a shift from a Th1 towards a Th2 phenotype, as well as to suppress Th17 responses [
52]. Hence, it is reasonable to assume that vitamin D deficiency exacerbates the deleterious generalized proinflammatory state in our patients with HFrEF. In opposition to a previous study in our group with healthy adults [
6], no significant seasonal variation of any cytokine was observed, which is consistent with the permanent state of inflammation that occurs in the context of HF [
19,
20].
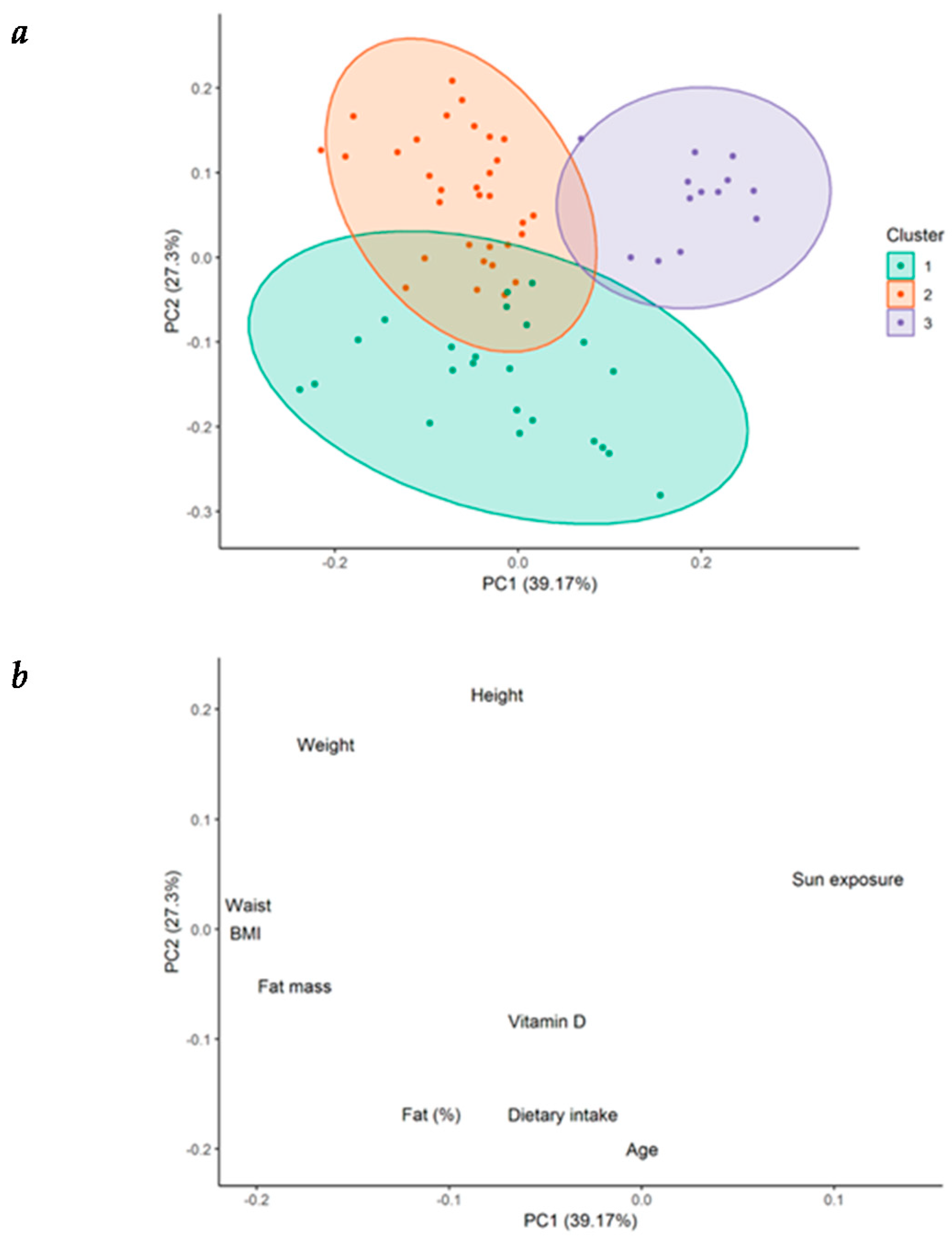
3.4. Cluster Analysis
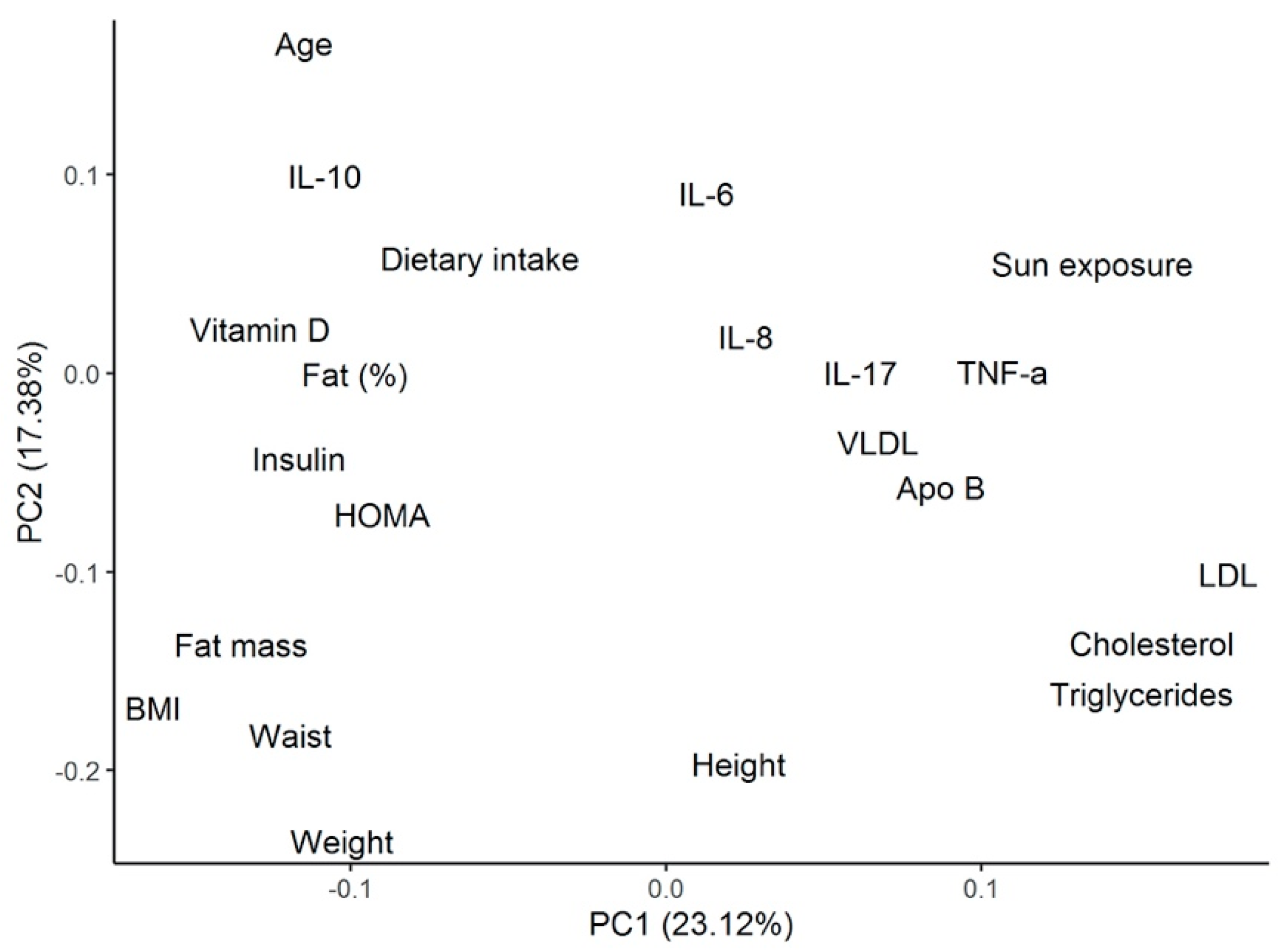
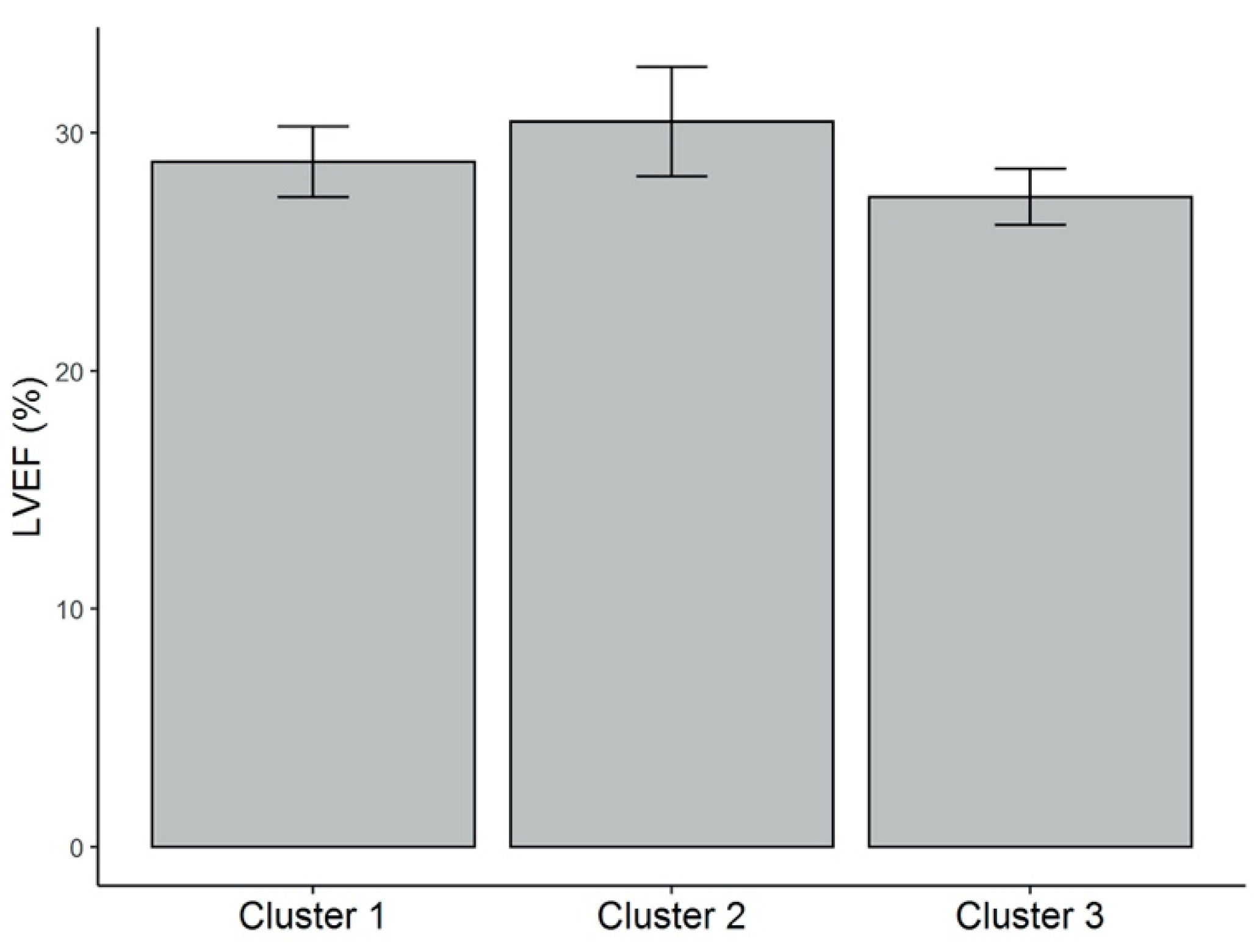
In order to characterize the cytokine profile and the 25(OH)D levels in our patients, we performed a cluster analysis, identifying three clusters. Patients from cluster 1 presented the highest circulating levels of 25(OH)D and the highest dietary intake of vitamin D. They showed the lowest significant levels of the proinflammatory cytokines TNF-α and IL-12p70, and the highest levels of the anti-inflammatory cytokine IL-10, compared with patients from the other two clusters, further reflecting the protective role that vitamin D plays in the cytokine milieu. These patients also showed the lowest concentration of total cholesterol, triglycerides, and Apo-B, representing the possible defensive role of vitamin D in atherosclerosis. Patients from cluster 2 had relatively intermediate levels of vitamin D concentration and vitamin D intake, along with intermediate levels of TNF-α, IL-17A, and IL-10 compared with patients from clusters 1 and 3. Finally, and in opposition, patients from cluster 3 presented the lowest serum 25(OH)D levels. These patients showed the highest concentrations of TNF-α, IL-8, and IL-17A, (significant for TNF-α), and the lowest significant levels of IL-10. They also had the highest levels of total cholesterol, LDL cholesterol, Apo-B, and triglycerides, indicating the proinflammatory and atherosclerotic mechanistic roles of vitamin D deficiency described previously. The role of dietary intake as the main lifestyle factor associated with vitamin D deficiency in this setting of patients may also be considered.
3.5. Correlation of Vitamin D Deficiency with Anthropometric and Lifestyle Parameters in Patients with HFrEF
The relationship between vitamin D and anthropometric parameters in the setting of HF has not been thoroughly explored. In the general population, there seems to be a consensus of a negative correlation between serum 25(OH)D concentrations and BMI, WC, and BF% [
33,
61,
62,
63,
64], mainly attributed to sequestration of vitamin D by adipocytes. However, controversy has been found in patients with HF, as no correlation has been reported in this context between serum 25(OH)D levels and body weight, BMI, mid-arm muscle circumference [
65], arm lean mass, leg lean mass, and grip strength (rather related to muscle mass and strength) [
66]. Similarly, we found no correlation between 25(OH)D levels and BMI, BF%, and fat mass in kg. On the other hand, in a study with 14 HF patients vs. 14 controls, an inverse relationship was found between 25(OH)D levels and BMI [
67]. Our results could be explained by the fact that most of our patients belonged to the overweight BMI and BF% category, and WC was within normal limits. Our sample of patients, like most with HF, are rather not obese because of a higher metabolic waste status due to the disease, and hence do not fully represent each of the BMI or BF% categories.
Likewise, lifestyle factors related to vitamin D status in patients with HF have been understudied. In a questionnaire-based study, patients with HF were less exposed to summer outdoor activity at least once a year compared with controls, but no difference was found in fish consumption (the only food source investigated) or vitamin D supplementation. However, serum 25(OH)D levels were not measured [
67]. In the previously mentioned cross-sectional case-control study, that included 14 patients with HF vs. 14 controls, no significant differences in 25(OH)D concentration was reported. HF patients were shown to present less weekly sun exposure, but no differences in daily dietary vitamin D intake between the groups was found [
67]. In addition, using data from a large population study, no differences were seen among various amounts of vitamin D intake and the incidence (new cases) of HF, but measurement of 25(OH)D levels was not performed either [
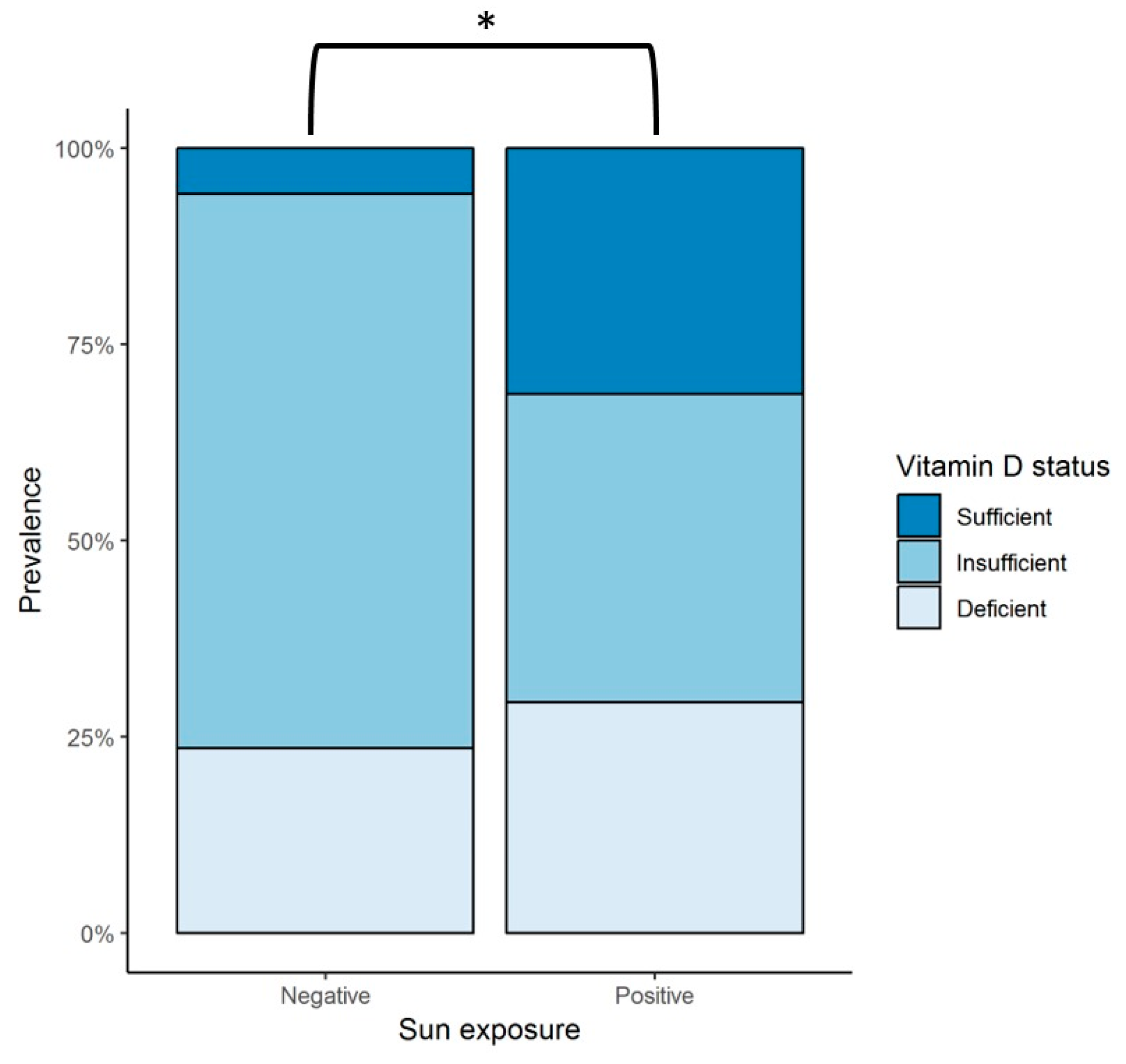
68]. Our results during the 12-month follow up showed a significant positive correlation between dietary intake of vitamin D-rich foods and serum 25(OH)D concentrations. Regarding sun exposure, when the total population was considered, no correlation was found between serum 25(OH)D levels and sun exposure. Nevertheless, in a sub-analysis in which patients were separated into two groups, those with any amount of sun exposure versus those with no exposure at all, the prevalence of vitamin D sufficiency was significantly greater in patients exposed to the sun compared with those that had no exposure. Our group previously studied the seasonal variation of serum 25(OH)D concentration in a cohort of 23 healthy subjects with a follow-up of 12 months, in which seasonal variation was observed [
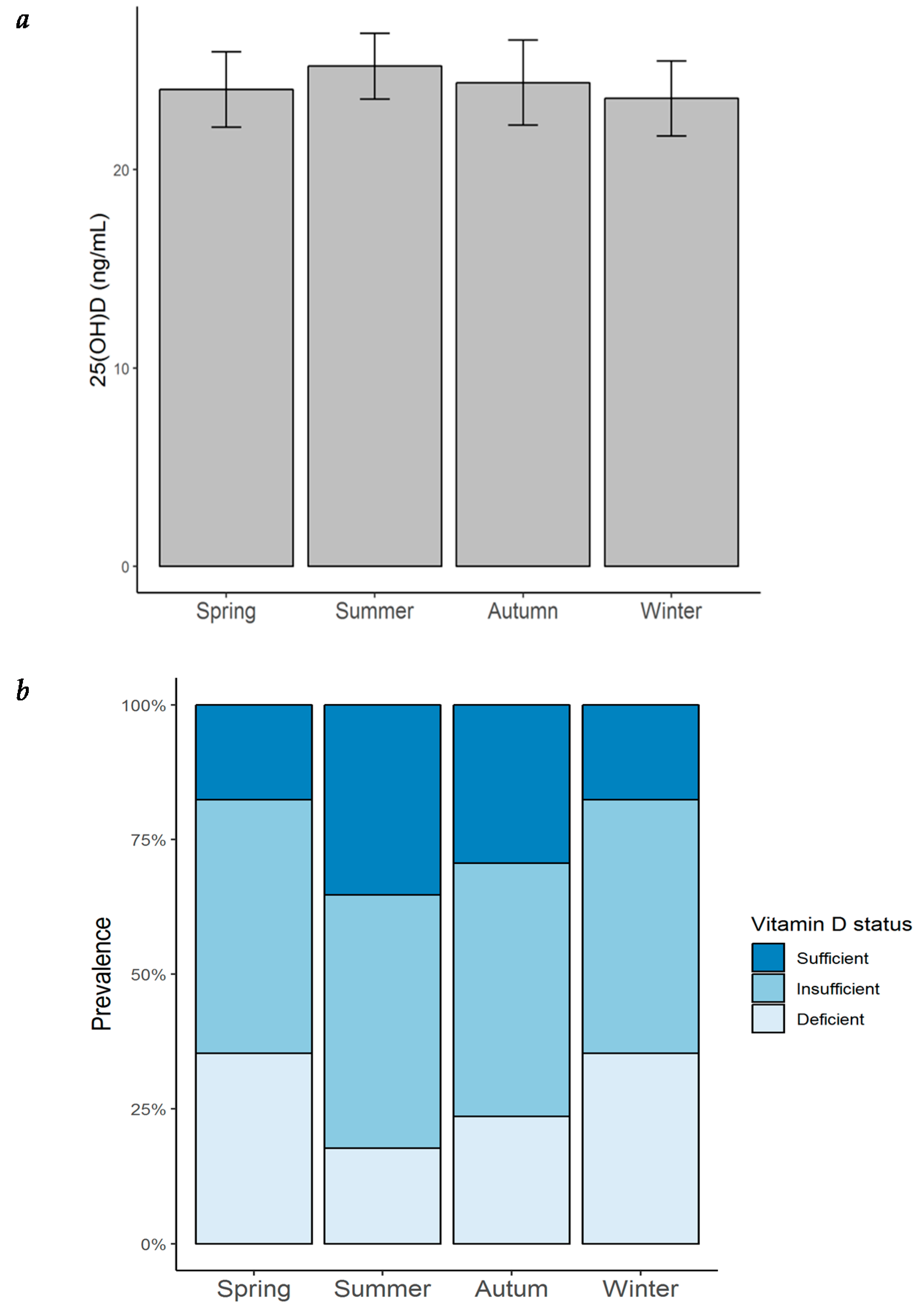
6]. However, no seasonal variation of 25(OH)D levels was found in our present study, suggesting that sun exposure may not play a dominant role in vitamin D status (as opposed to healthy controls), but rather vitamin D intake seems to play a greater role. Consideration has to be given to our patients’ skin phototype. Skin pigmentation represents a significant independent risk factor for vitamin D deficiency because UVB is needed in the initial conversion of 7-dehydrocholesterol into cholecalciferol, which is less available for this reaction in darker skin phototypes, as melanin acts as a light-absorbent protein. Thus, our cohort might be prone to vitamin D deficiency, given that the majority of the subjects belonged to phototypes IV and V.
The recommended daily allowance (RDA) of vitamin D for adults 51–70 years old is 600 IU/day, while that of adults >70 years old is 800 IU/day [
69]. Therefore, none of the patients in our cohort met the nutritional requirements for vitamin D intake, as all of them had an average intake throughout the year of less than 400 IU/day. Although the time of sun exposure required for vitamin D synthesis varies considerably depending on the living latitude and the season of the year, it has been estimated that 5 to 30 min of sun exposure between 10 a.m. and 3 p.m. at least two times per week may be a reasonable amount [
70]. Most of the patients in our cohort (82.3% during summer, autumn, and spring, and 52.9% during winter) obtained at least this level of sun exposure while living at an adequate latitude. Therefore, vitamin D ingestion might be a lifestyle factor as important or even more significant than sun exposure in determining 25(OH)D levels in the setting of our cohort of patients with HF.
The study has some limitations. The sample size is both small and from a Hispanic ethnicity; thus, our results might not be extrapolated to other populations. Hence, results should be cautiously interpreted. Even though data regarding vitamin D-rich foods intake and sun exposure was obtained through a personal interview, the information given by the patients might still be subjective. However, our study has several strengths. This is a 12-month follow up to approach seasonal variation of vitamin D and cytokines. In-vitro and animal model studies concerning the molecular mechanisms were identified to describe and apply these mechanisms to explain our results, given that studies in humans hardly explain them in such detail. We also emphasize these molecular mechanisms in an original figure presented in this article, which could offer insight for further research.
5. Conclusions
Several lines of evidence establish the mechanistic and metabolic roles of vitamin D deficiency contributing to the inflammatory, immunomodulatory, and atherosclerotic status of patients with HF. Our results in this 12-month follow-up of patients with HFrEF are consistent with previous observations of a high prevalence of vitamin D deficiency in these type of patients. We found no seasonal variation of 25(OH)D levels, nor of the proinflammatory cytokine profile. Serum 25(OH)D concentrations correlated with dietary intake of vitamin D-rich foods. When analyzing our total population, no correlation was seen between 25(OH)D levels and sun exposure; nonetheless, when subgrouping the patients into positive (any amount of sun exposure) and negative (no sun exposure), there was a higher prevalence of vitamin D sufficiency in those belonging to the positive group. Considering the inflammatory state observed in the setting of HF, 25(OH)D levels were inversely correlated with the proinflammatory cytokines IL-1β, TNF-α, IL-6, IL-8, IL-17A, IL-18, and IL-33. Total cholesterol, LDL cholesterol, and triglycerides were also negatively correlated with 25(OH)D concentrations. Cluster analysis to better identify different patient profiles related to vitamin D status revealed that patients with the highest levels of 25(OH)D showed the highest dietary intake of vitamin D-rich foods, along with the lowest concentration of TNF-α, IL-17A, total cholesterol, triglycerides, and Apo-B. Overall, these results point towards the lifestyle factors and lipid parameters related to adequate levels of 25(OH)D. In addition, the protective anti-inflammatory role that vitamin D has in the setting of HFrEF was shown in this group of patients. Some of the pathways underlying these mechanisms have been described mostly in in-vitro and animal models: inhibition of renin biosynthesis; less detrimental ventricular remodeling by inhibition of procollagen-1 expression; increased myocardial contractility; and downregulation of JNK2, calcineurin/NFAT, and NF-κB cellular pathways. Induction of cholesterol efflux from foam cells; increased cholesterol uptake by hepatocytes; adequate regulation of GLUT4 expression in cardiomyocytes; and inhibition of the secretion of proinflammatory cytokines by downregulation of the MAPK pathway, have been attributed to vitamin D. Future studies are necessary in patients with HF to establish the molecular mechanisms involved in the pathogenesis of this disease.

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}