Special Issue: The Actin-Myosin Interaction in Muscle: Background and Overview


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
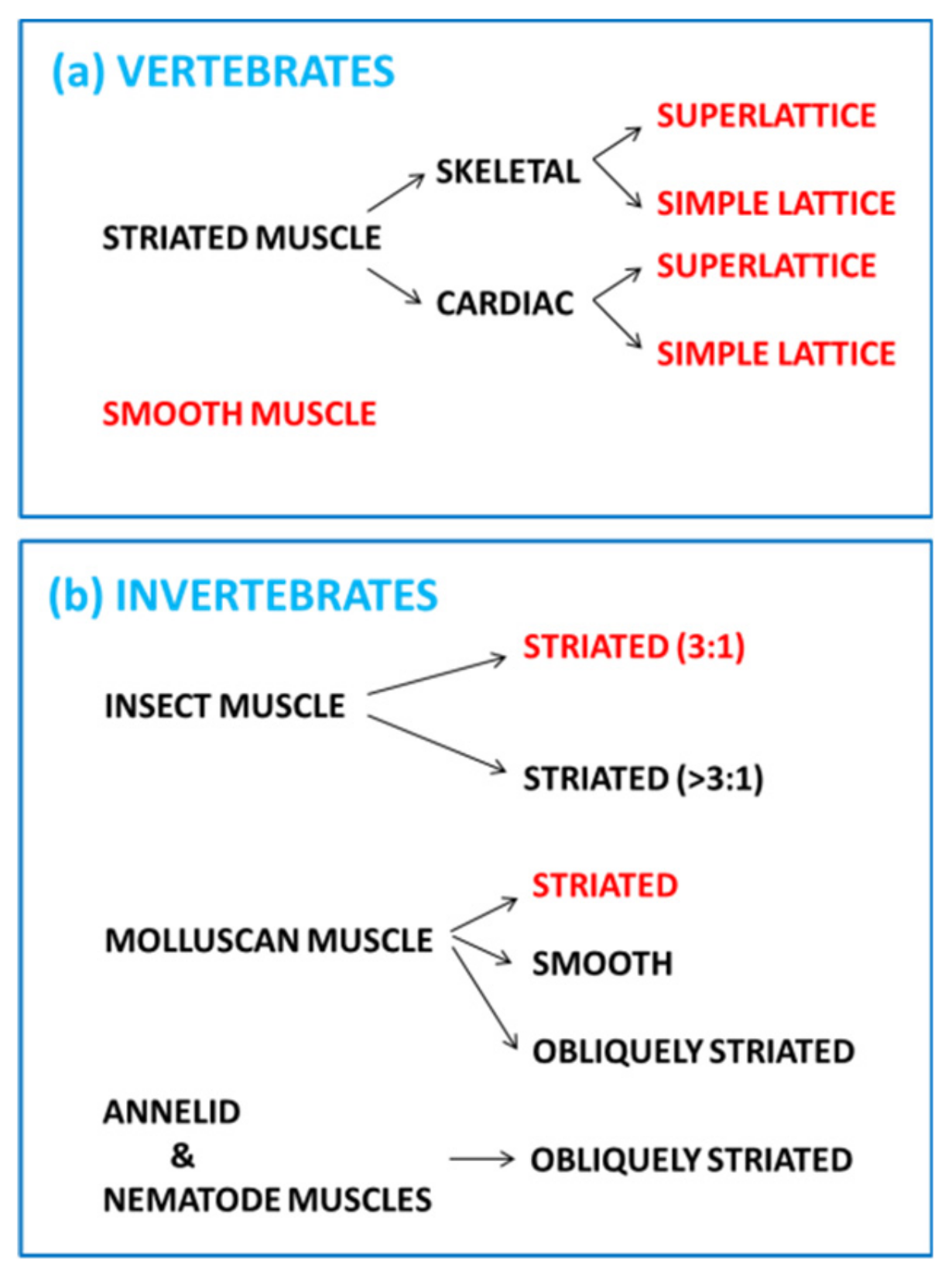
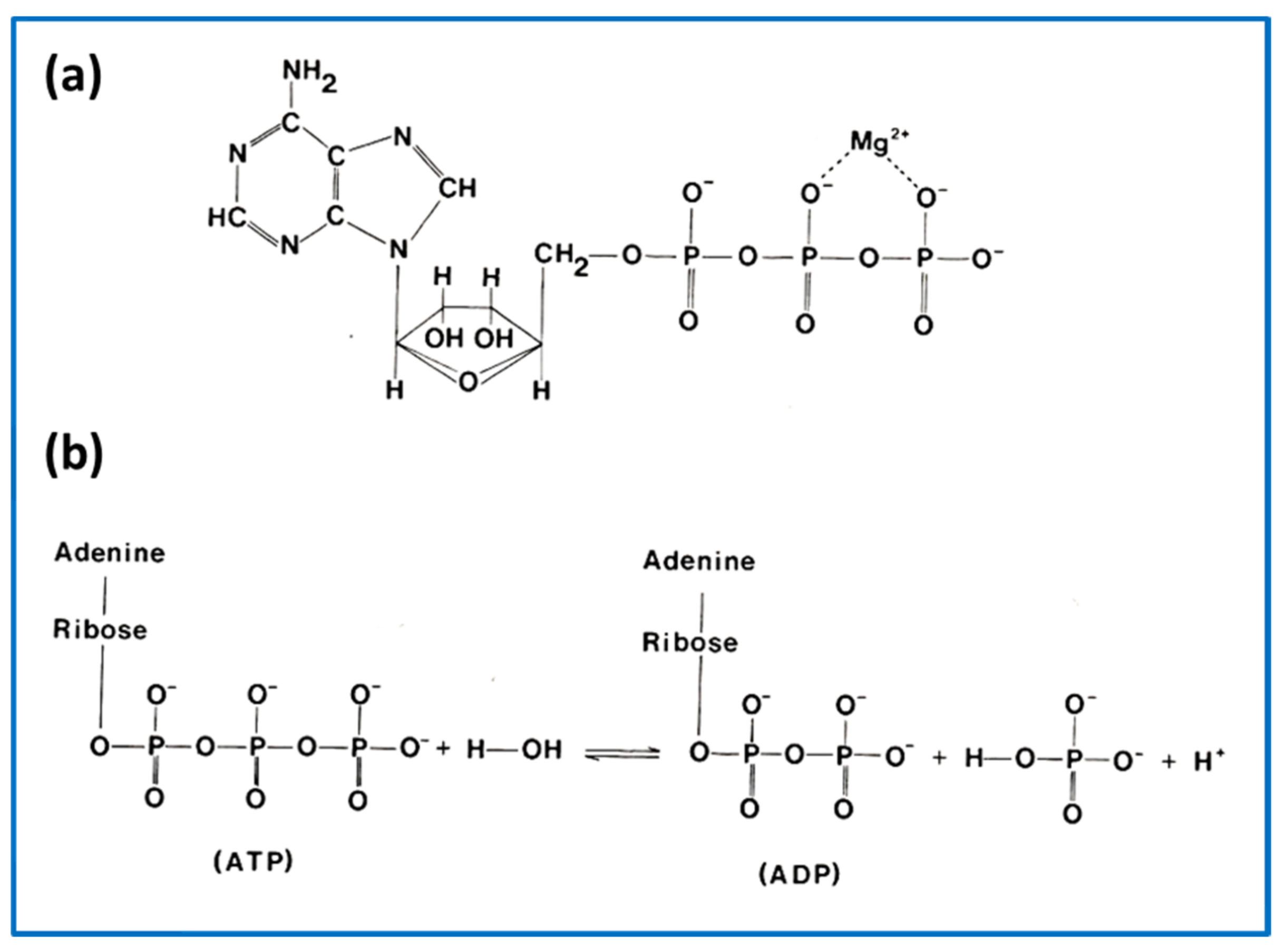
1. Introduction—Nature’s Linear Motors
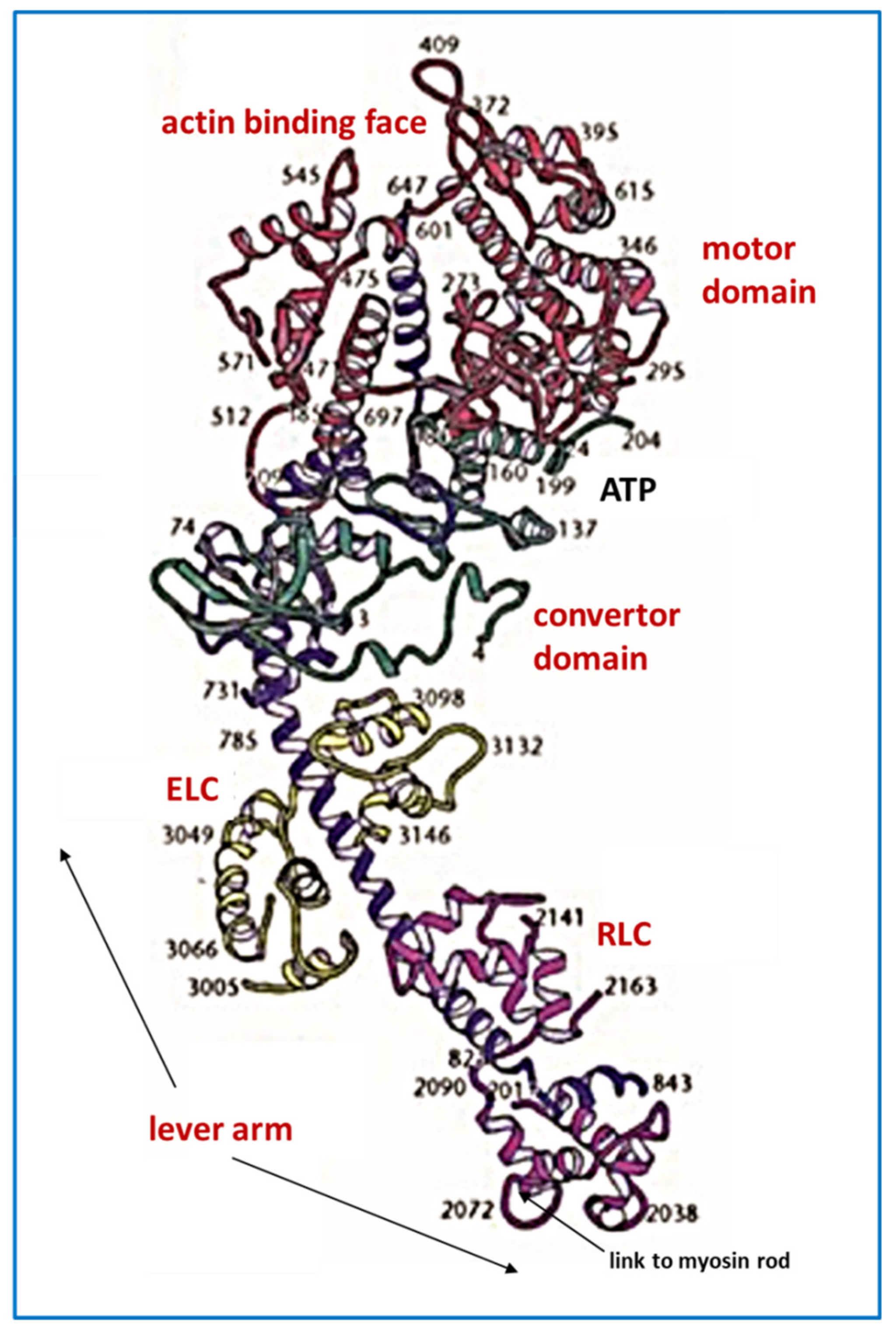
2. Striated Muscle Sarcomeres and the Contractile Mechanism
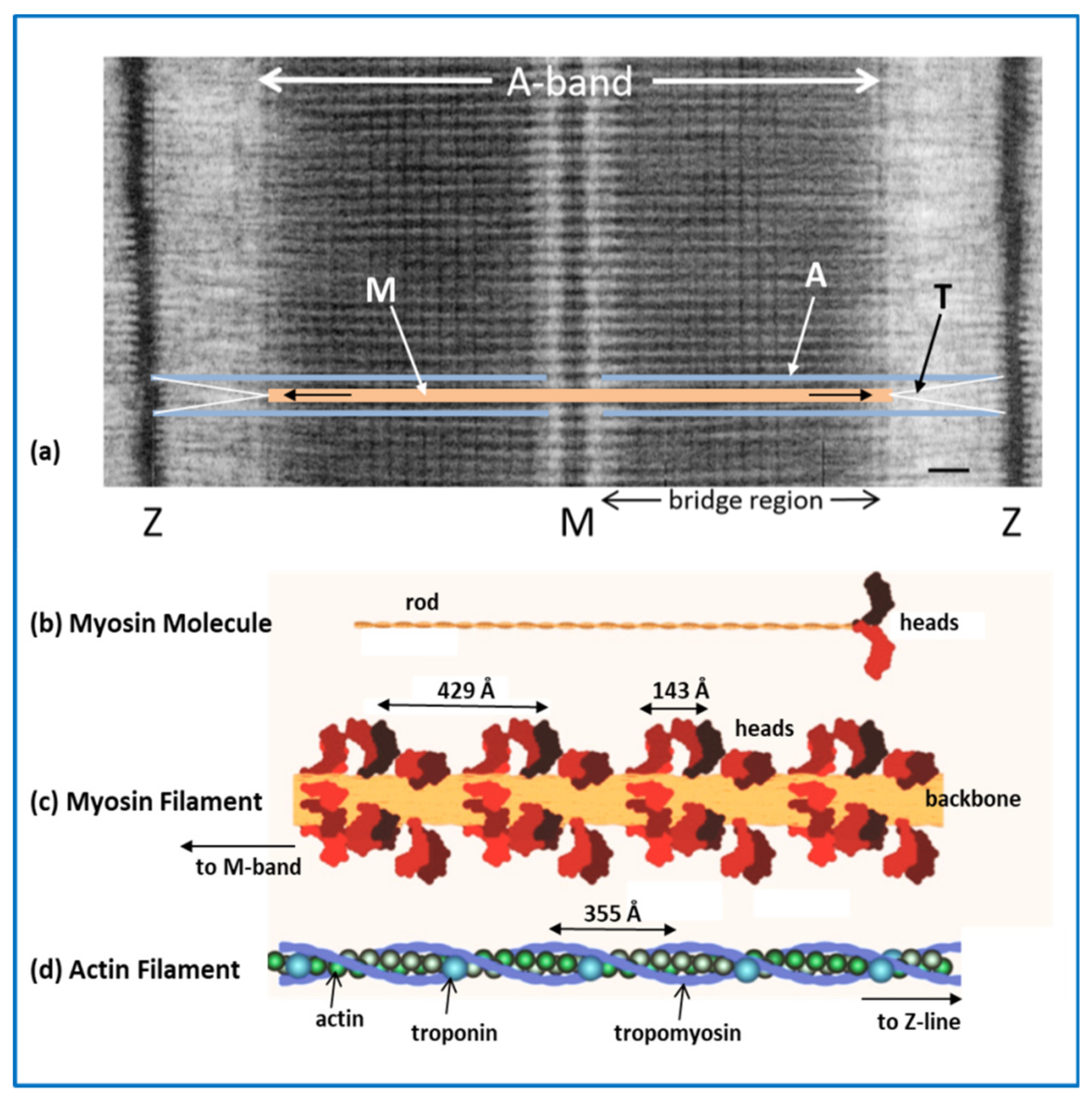
2.1. The Sarcomere
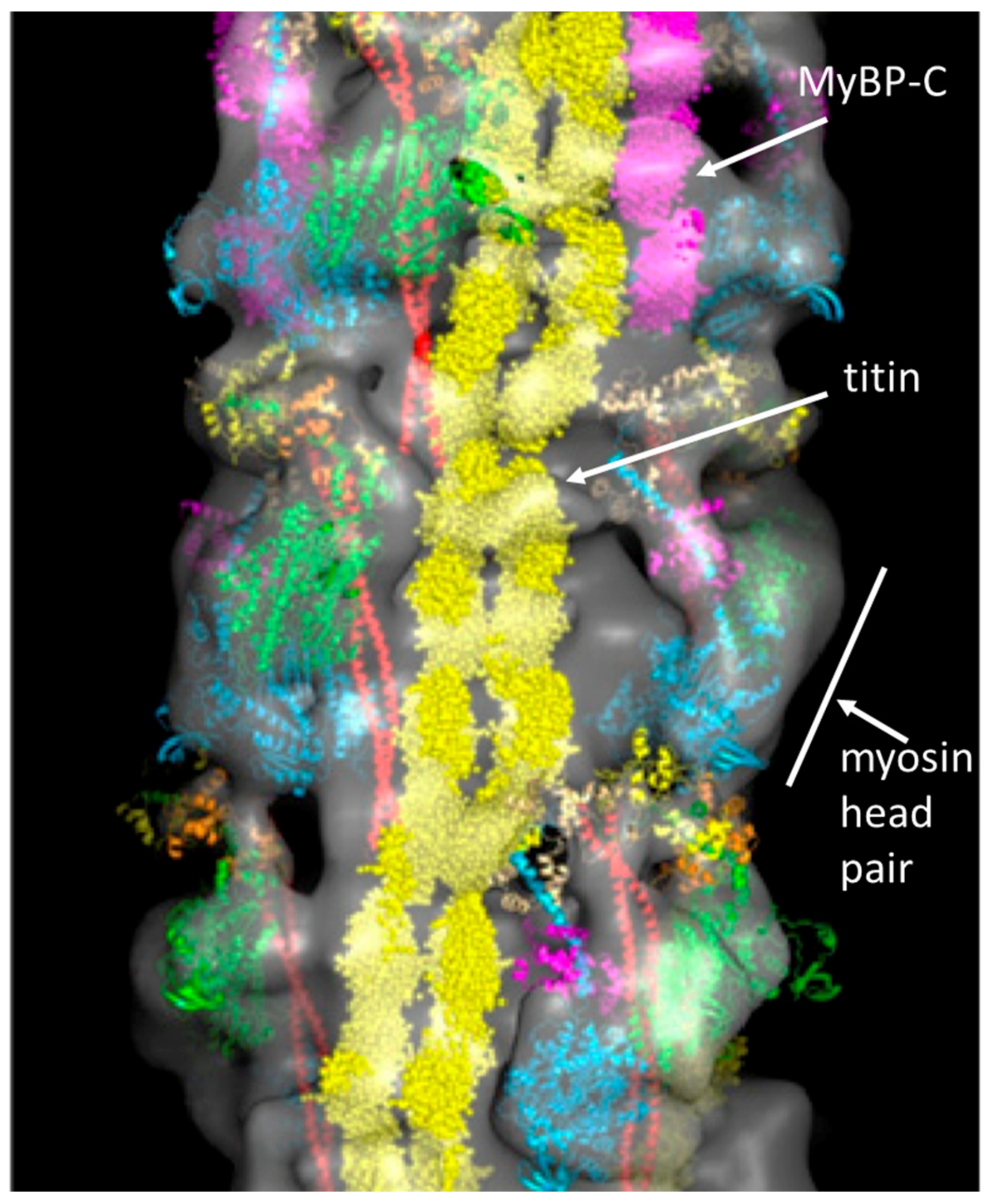
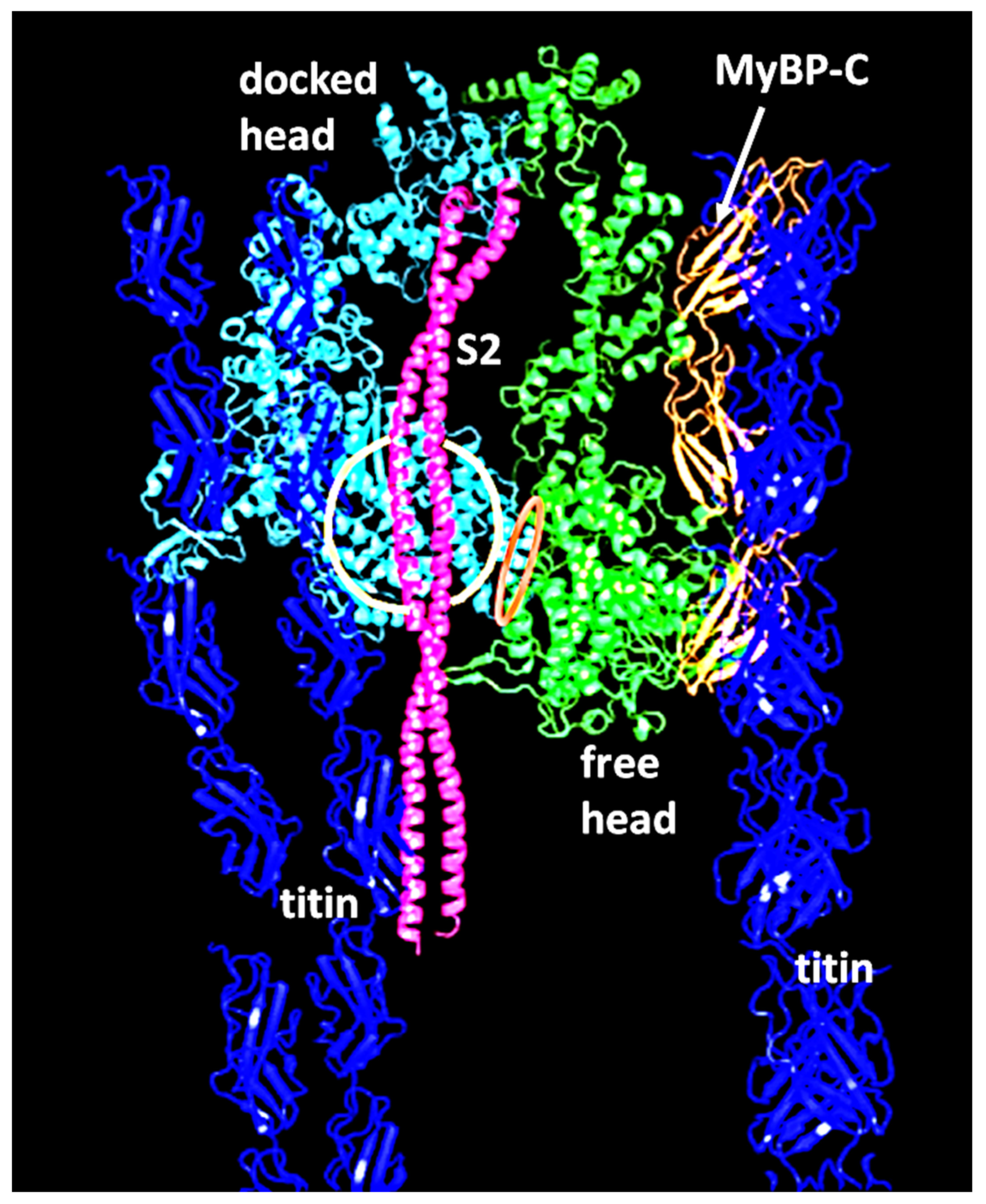
2.2. Myosin Filaments and the M-Band
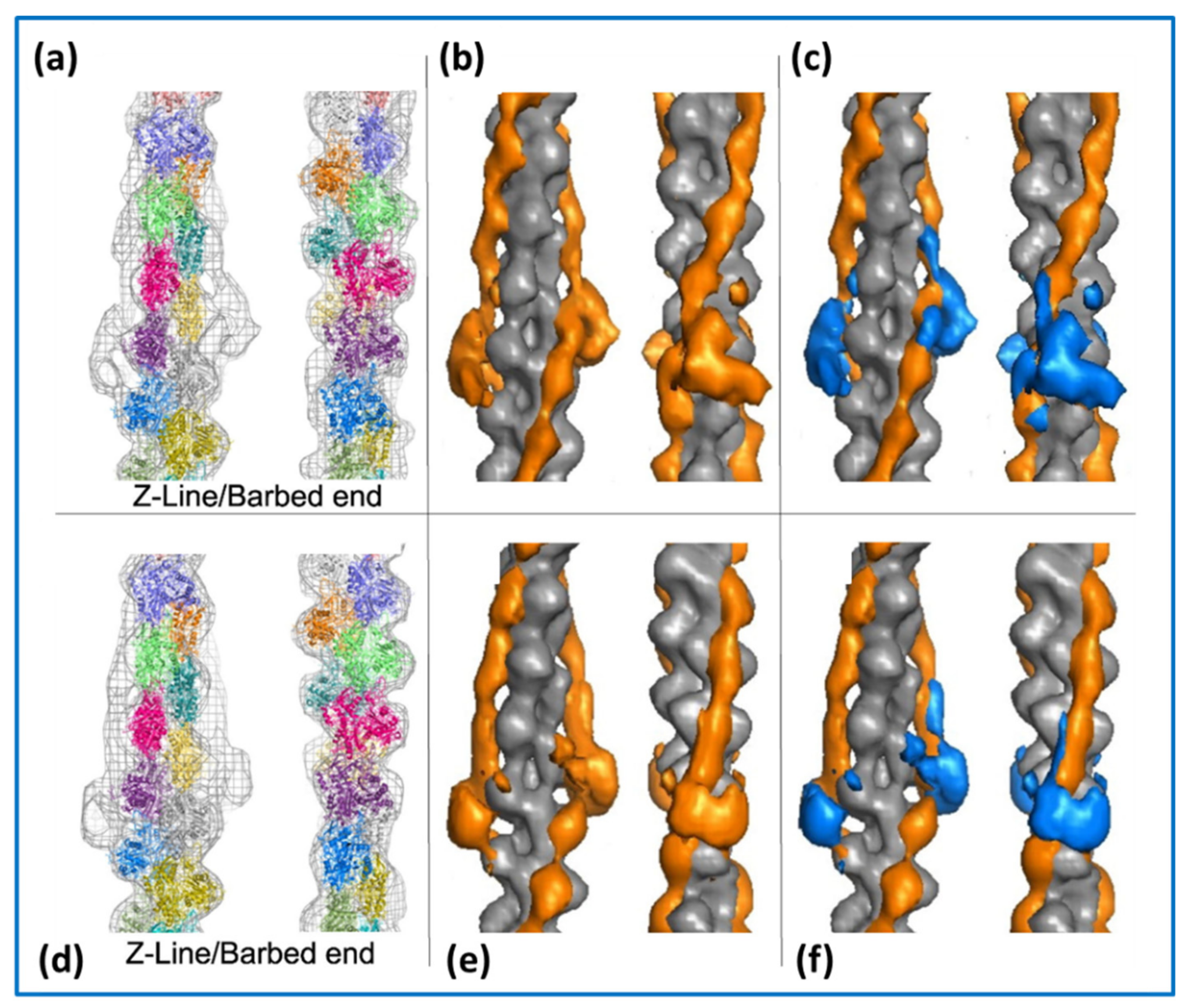
2.3. Actin Filaments and the Z-Line
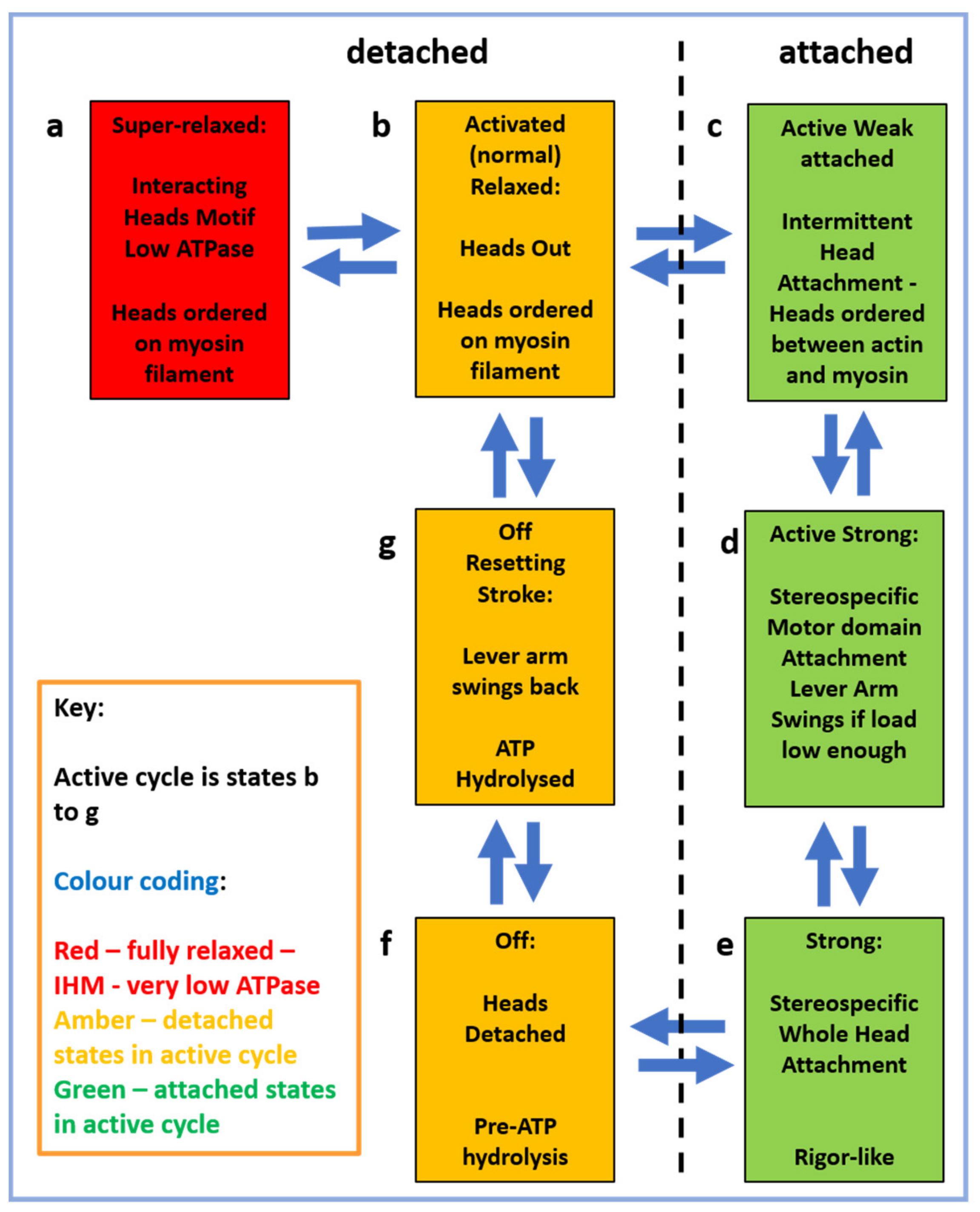
2.4. The Muscle Resting State
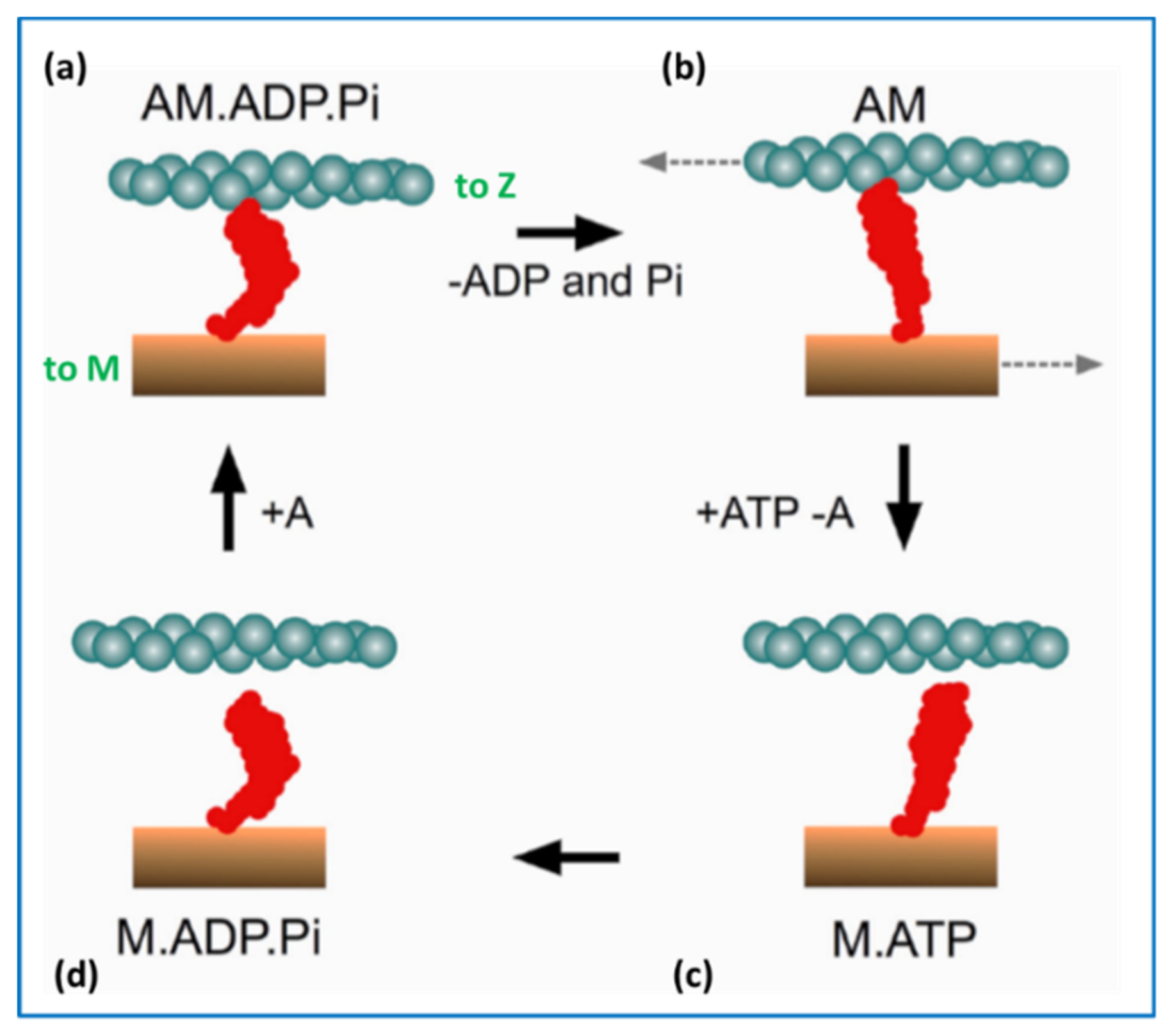
2.5. The Contractile Cycle
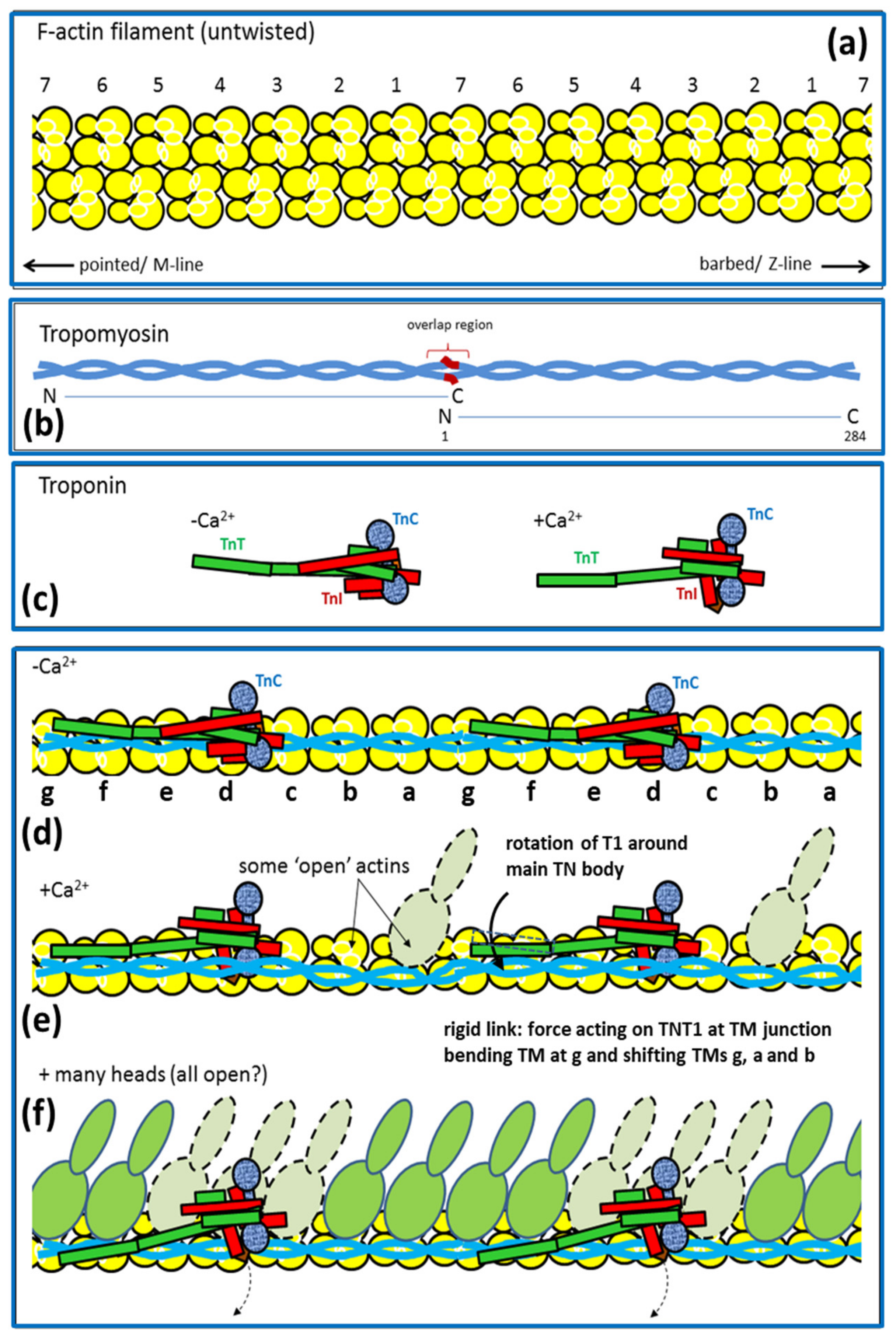
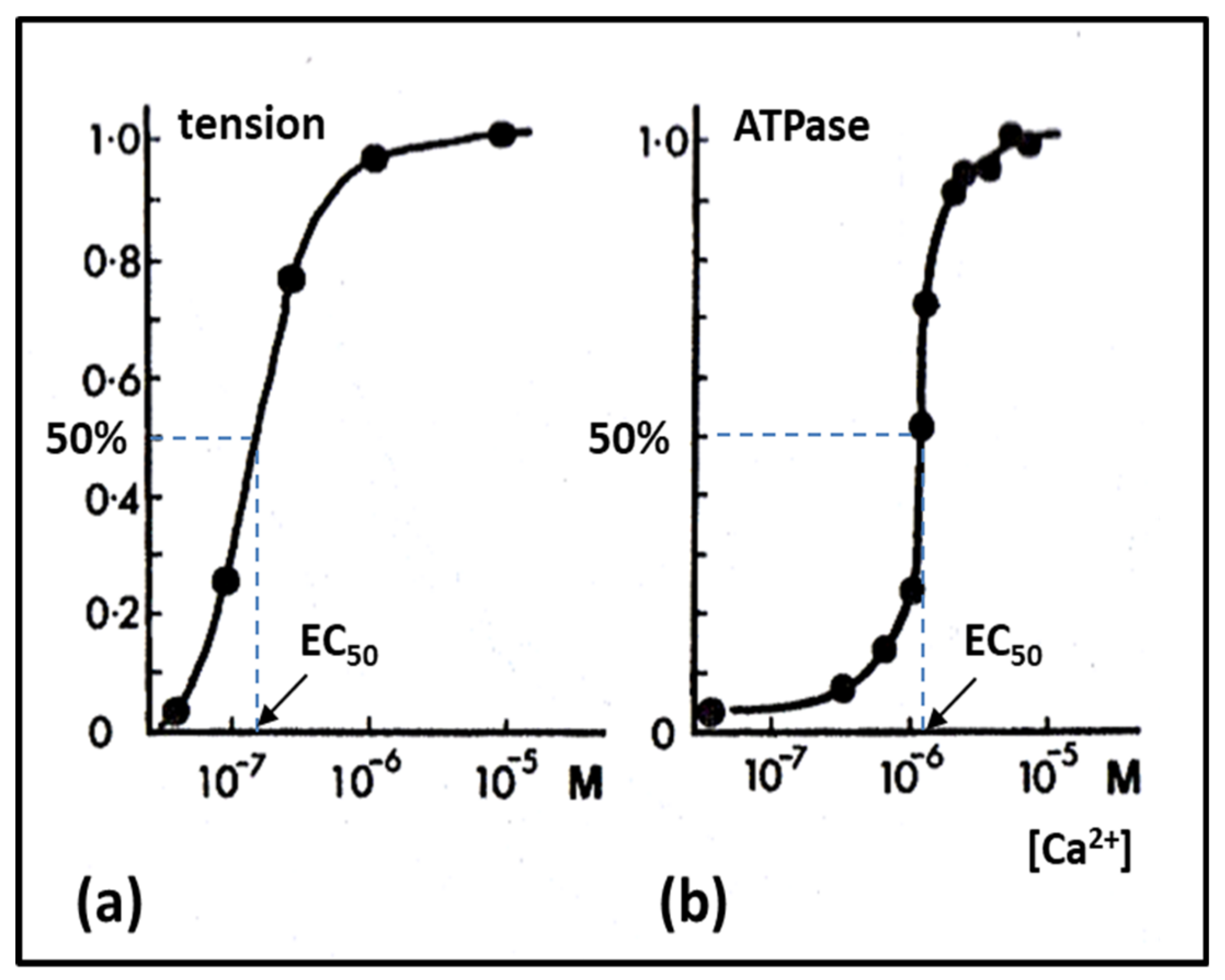
2.6. Muscle Regulation
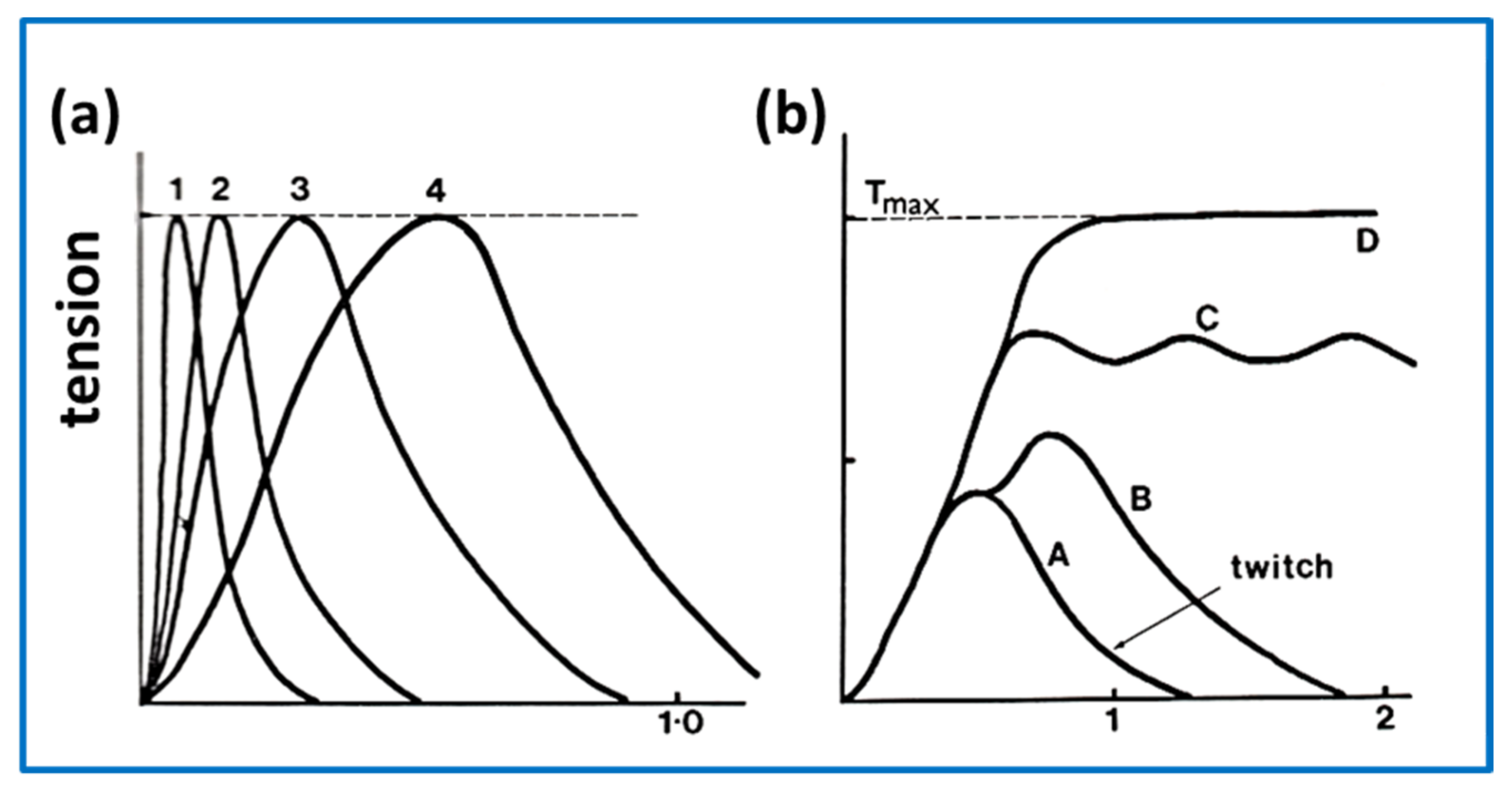
2.7. Muscle Mechanics
2.8. Problems to Be Solved
- (1)
- Is there direct evidence for the lever arm changing its angle on the actin-attached motor domain when force is actually produced?
- (2)
- Is some force generated simply by the process of head attachment to actin in the initial strong AM.ADP.Pi state before phosphate is released?
- (3)
- Is more force generated during the process of phosphate release?
- (4)
- Is additional force generated during the process of ADP release?
- (5)
- Are the preferred end point lever arm angles different in AM.ADP and AM?
- (6)
- How many of the steps between strong states are regulated by troponin/ tropomyosin?
- (7)
- Are the transition rates between strongly attached states sensitive to the load on the muscle?
- (8)
- Is there direct evidence for the reversal of angular change of the lever arm on the motor domain in the recovery step?
- (9)
- Is the super-relaxed state the only ordered state of myosin heads in relaxed muscle? Or do heads just become disordered on Ca2+-activation until they attach to actin to go through the contractile cycle? Or something else?
- (10)
- How much of the compliance of the sarcomere in active muscle is due to the myosin heads and how much to the filament backbones?
- (11)
- What is the maximum extent of lever arm movement produced by the energy released in one ATPase cycle?
- (12)
- How long do myosin heads stay attached to actin in a single cycle in isometric muscle or under different load conditions?
- (13)
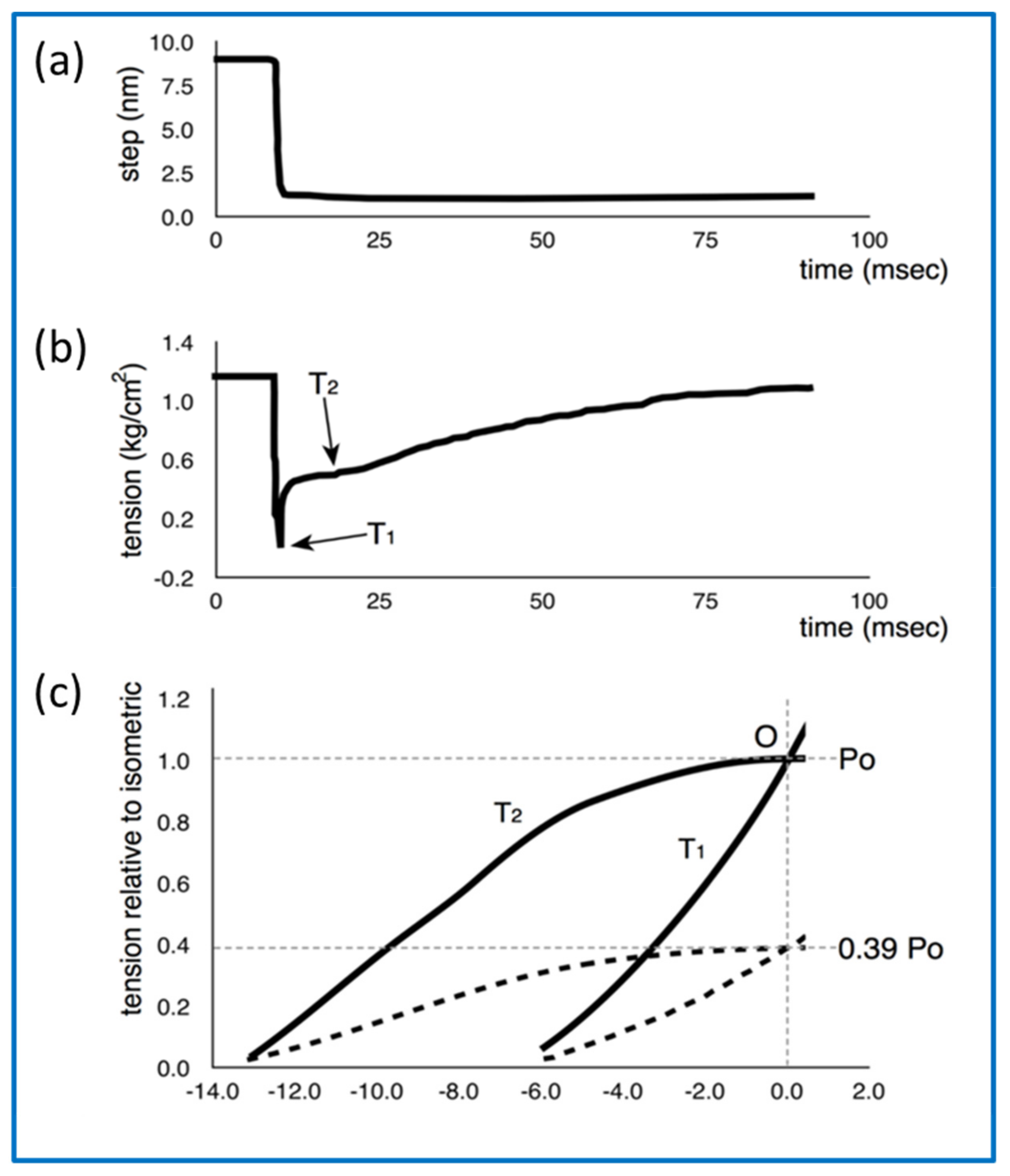
- How can the details of the T2 recovery response (Figure 11b) be explained?
- (14)
- What are the identifiable changes in the molecular structures of the myosin heads in different muscle states?
- (15)
- Can the elastic properties of the myosin head through the contractile cycle be defined?
- (16)
- In an isometric contraction how many heads are in each state?
- (17)
- In isotonic shortening how do the head populations depend on the load on the muscle?
3. Methods of Studying the Crossbridge Cycle
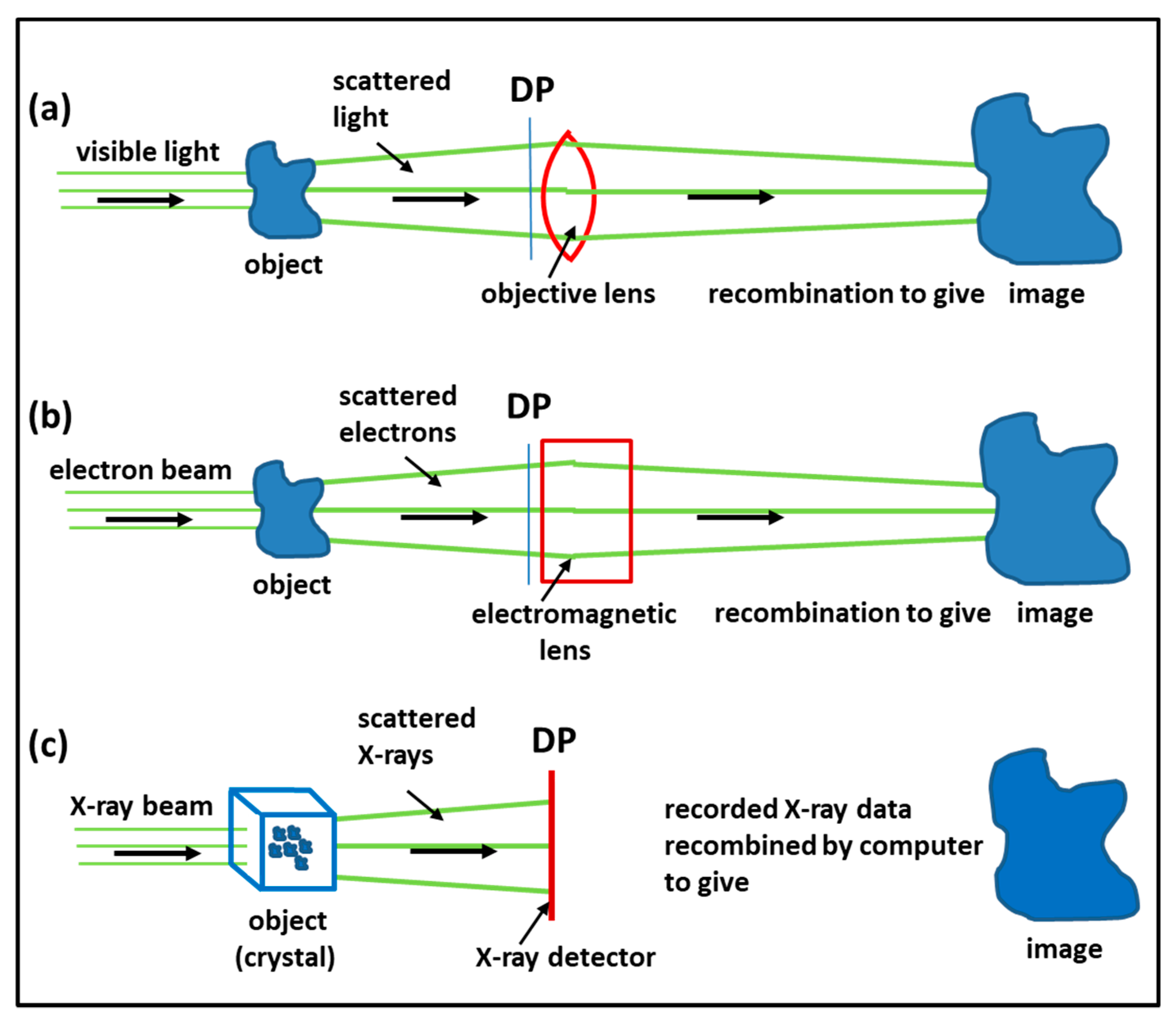
3.1. Imaging Methods: Protein Crystallography, Electron Microscopy, Electron Tomography, Single Particle Analysis
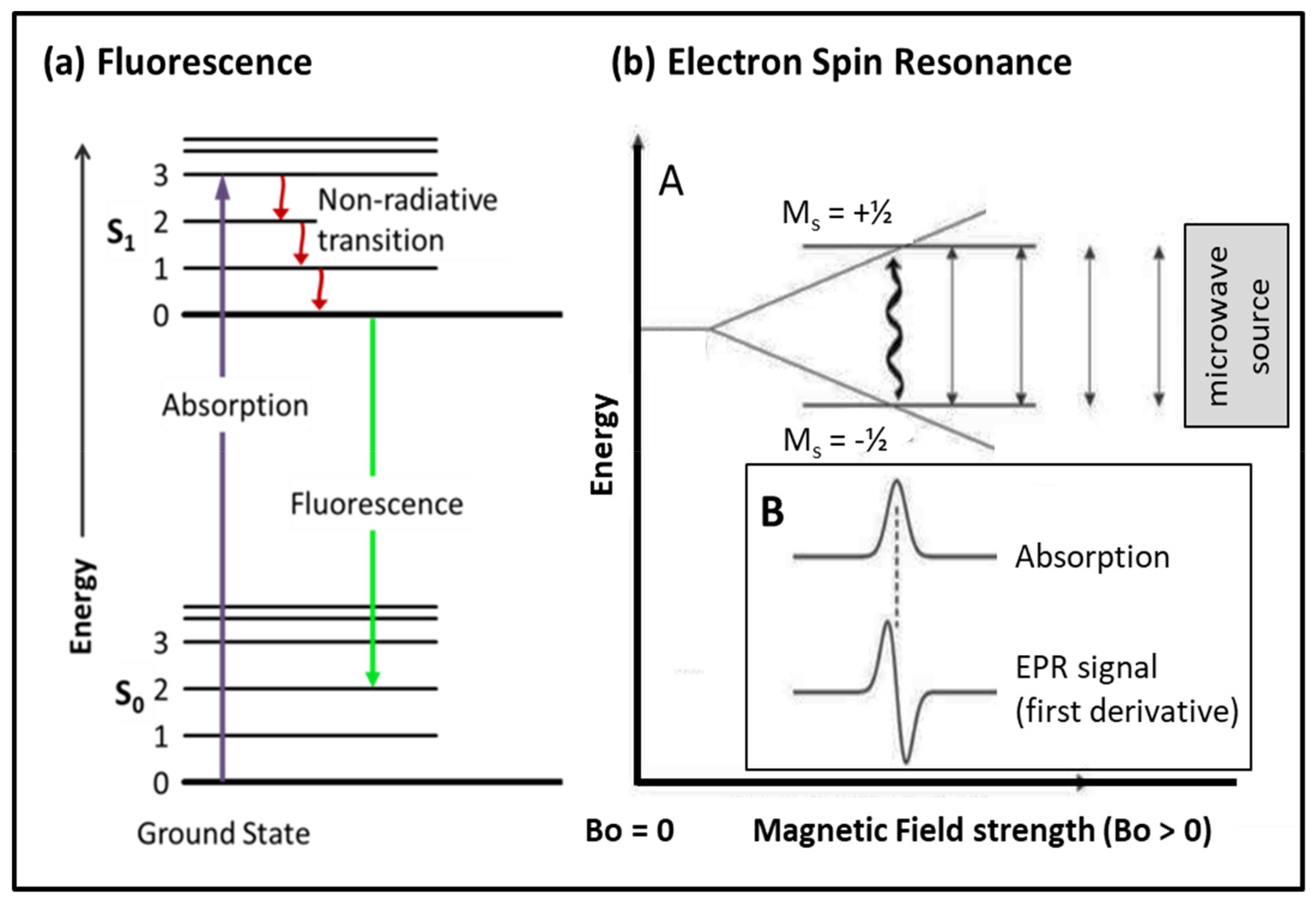
3.2. Probes: Fluorescence, Fluorescence Resonant Energy Transfer (FRET), Spin Probes
3.2.1. Fluorescence
3.2.2. Spin Probes
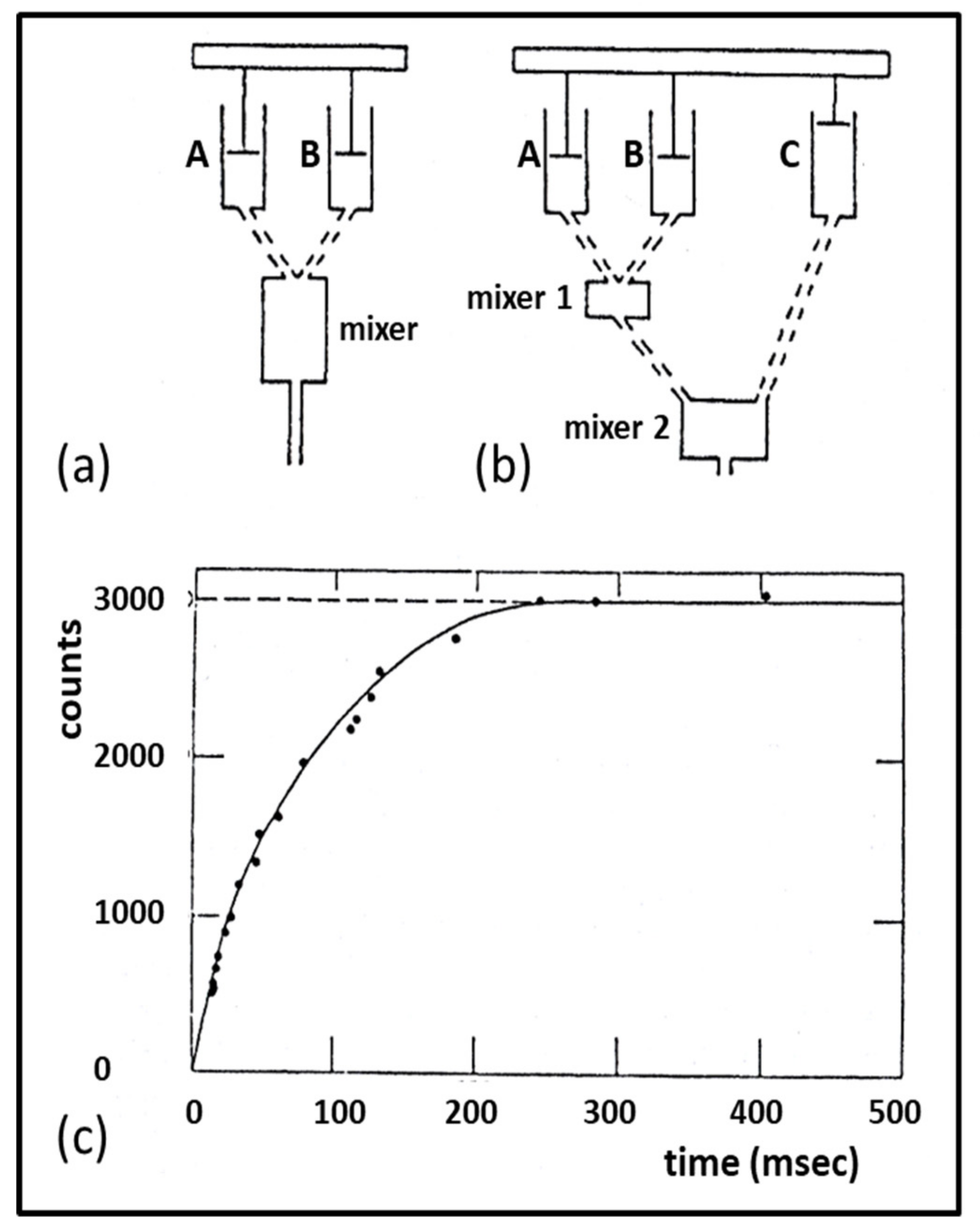
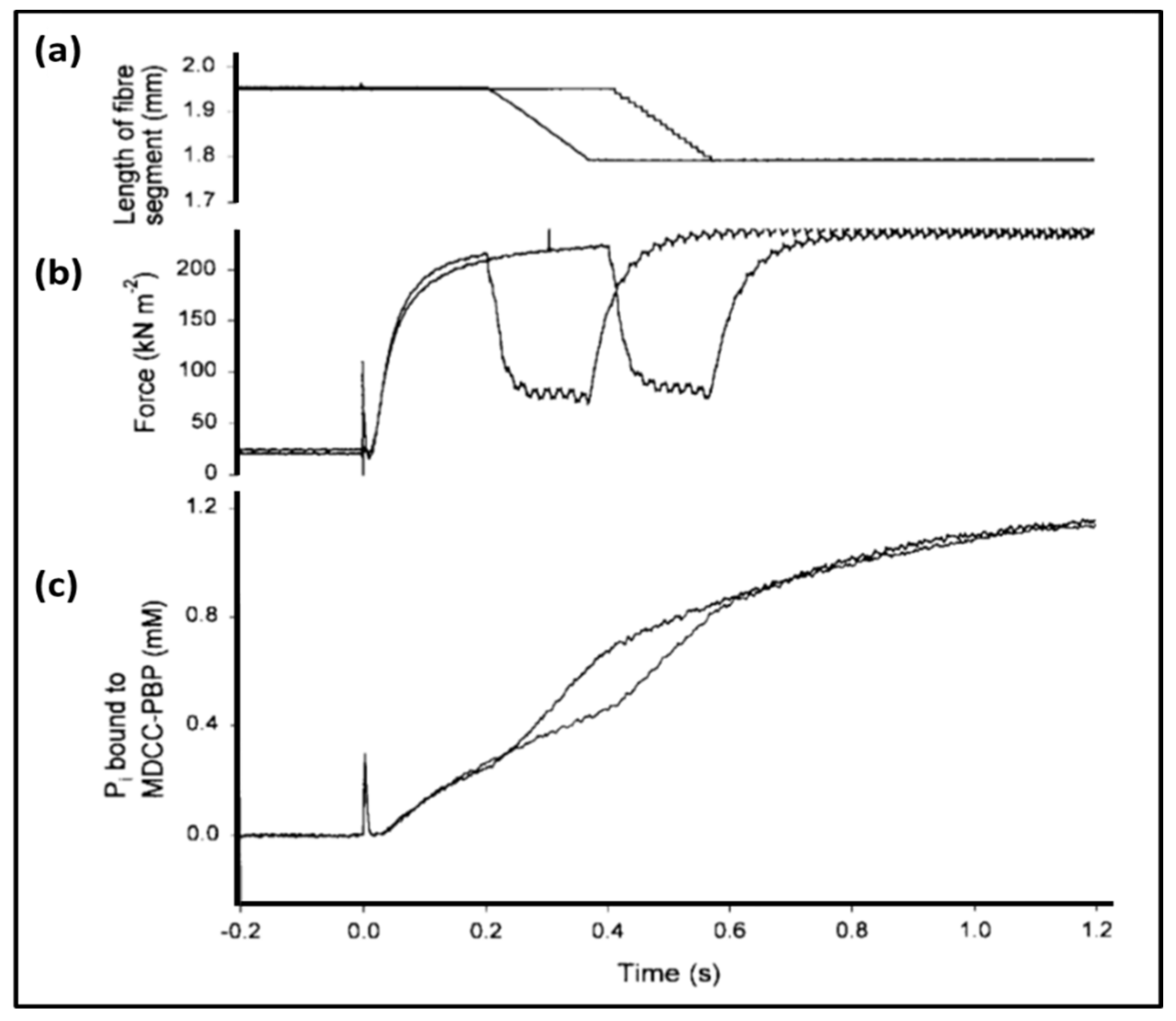
3.3. Biochemical Kinetics and Caged Compounds
3.4. Time-Resolved X-Ray Diffraction
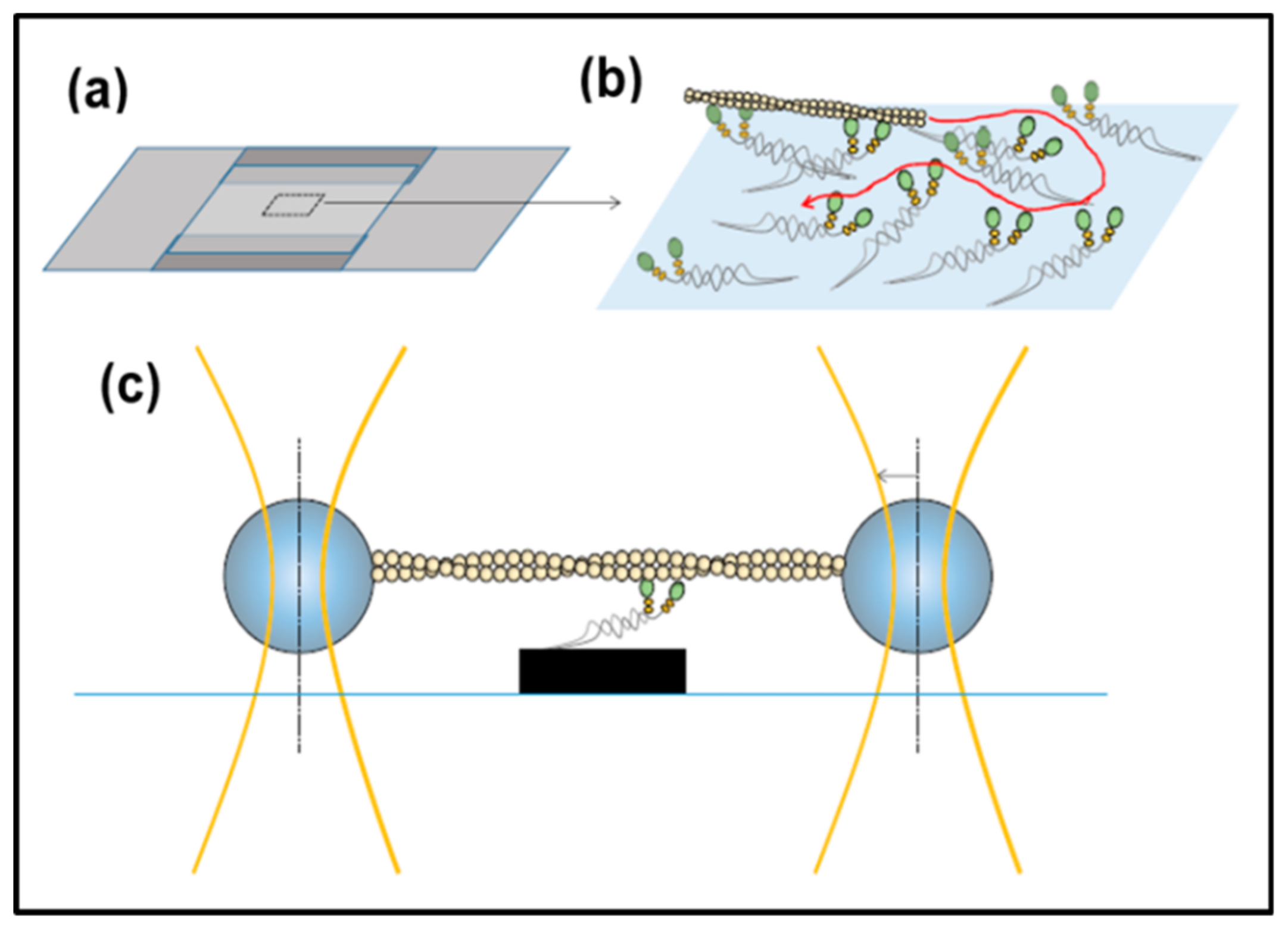
3.5. In Vitro Methods: Motility Assays, Optical Traps
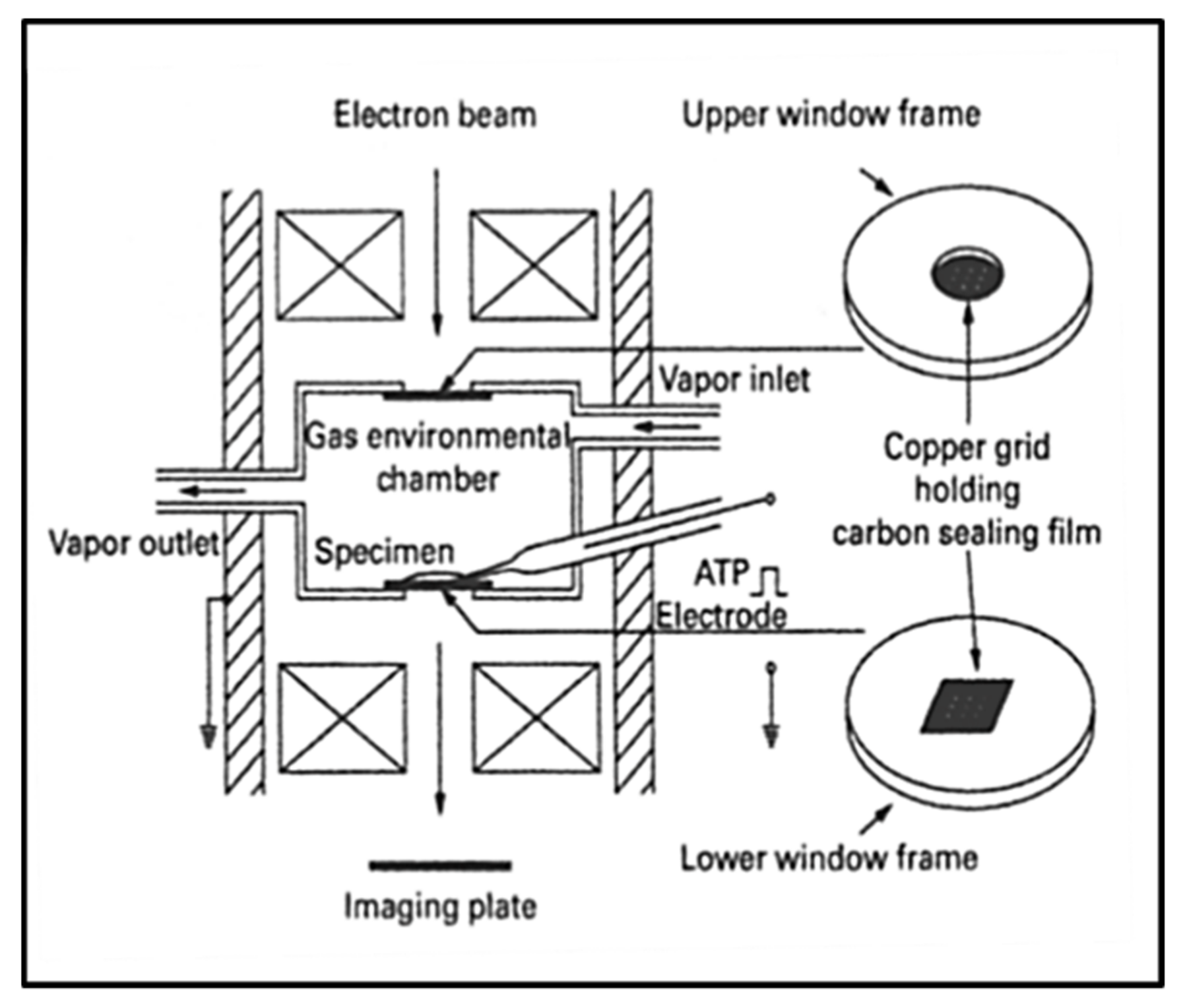
3.6. Electron Microscopy with an Environmental Chamber
4. Structural Biology and Mechanics Insights into the Contractile Mechanism
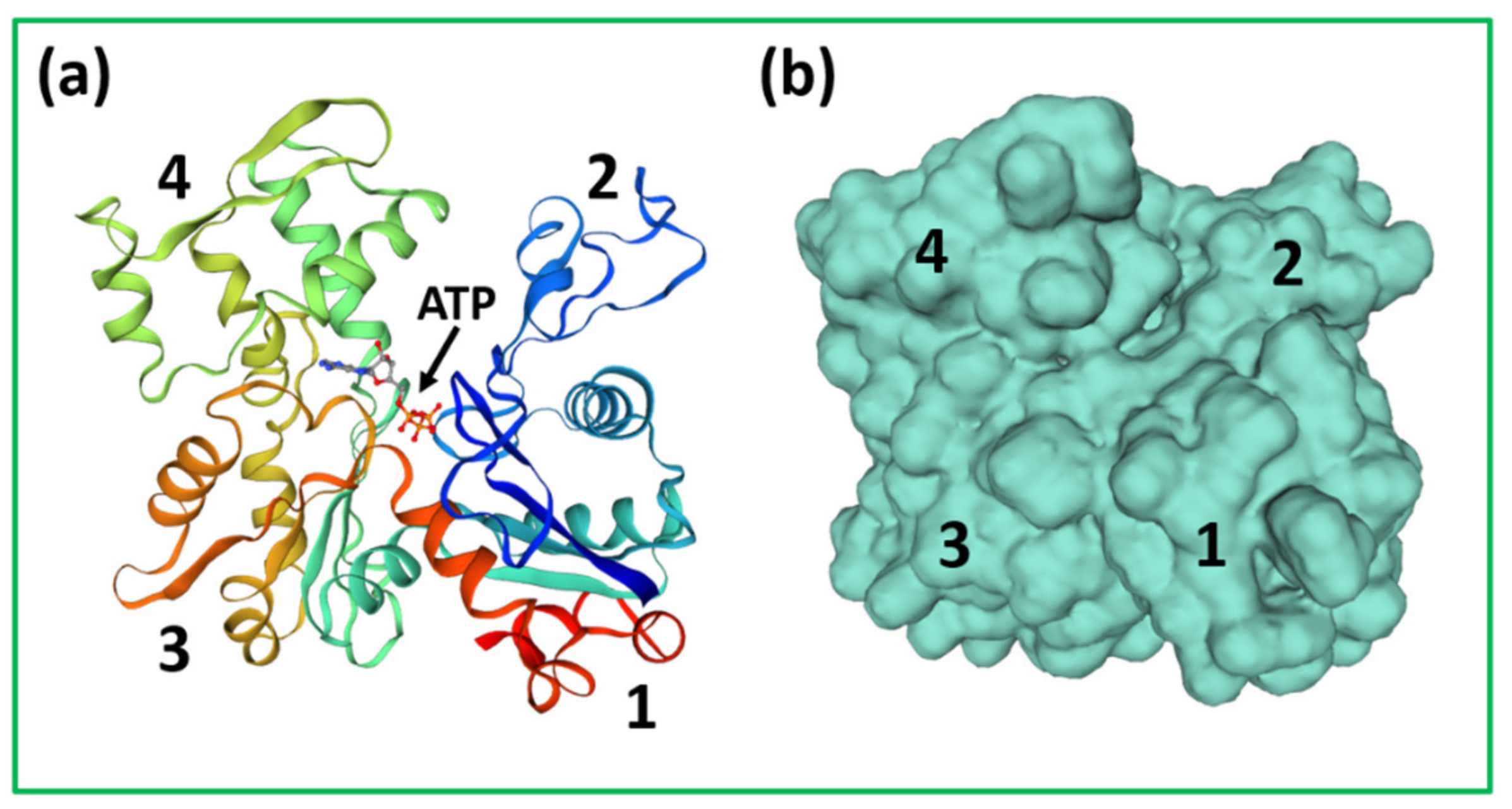
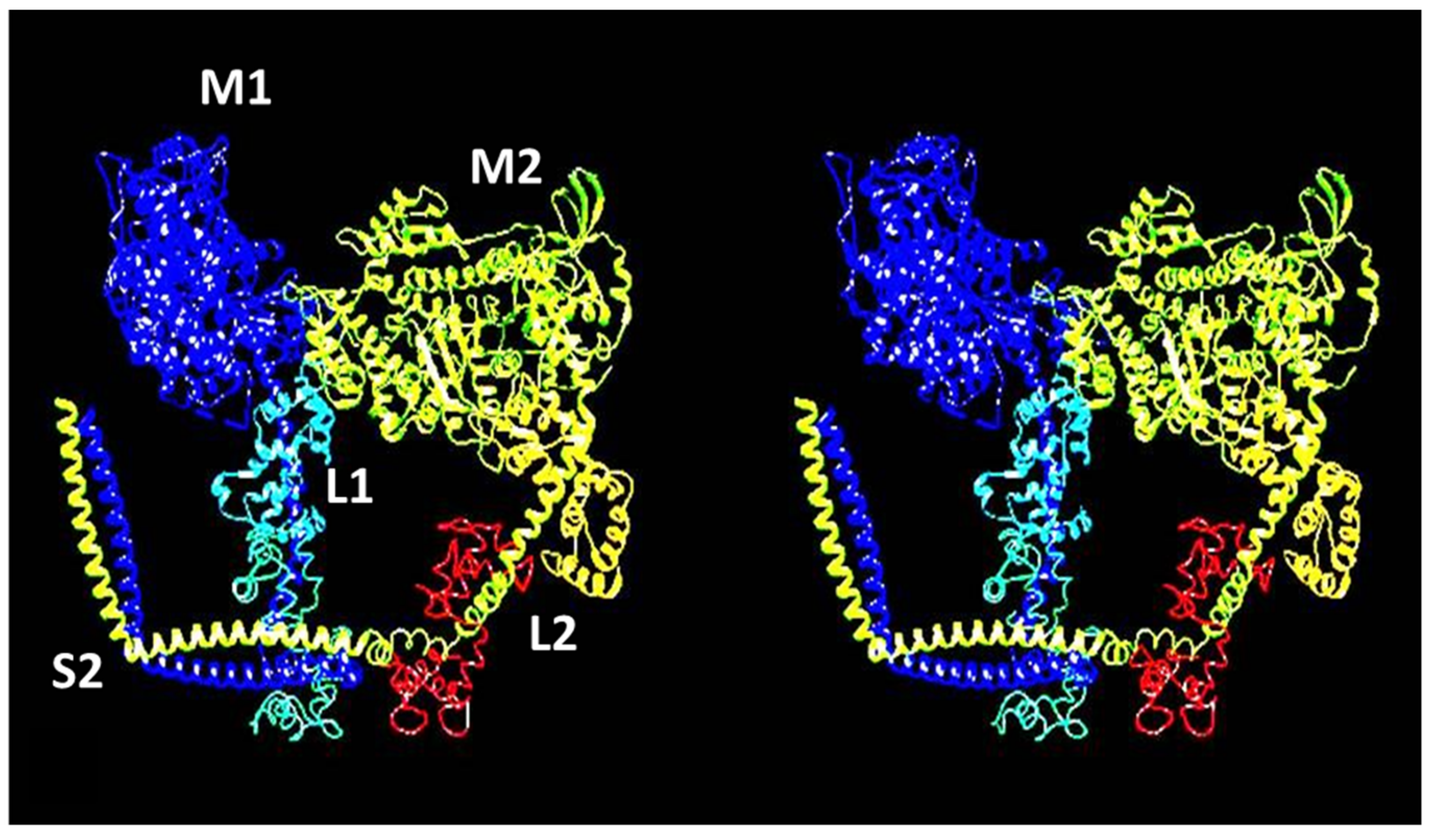
4.1. Protein Crystallography and High-Resolution Electron Microscopy
4.2. Low-Angle X-ray Diffraction: Static Muscle States and Time-Resolved Studies
4.3. Further Aspects of Muscle Mechanics
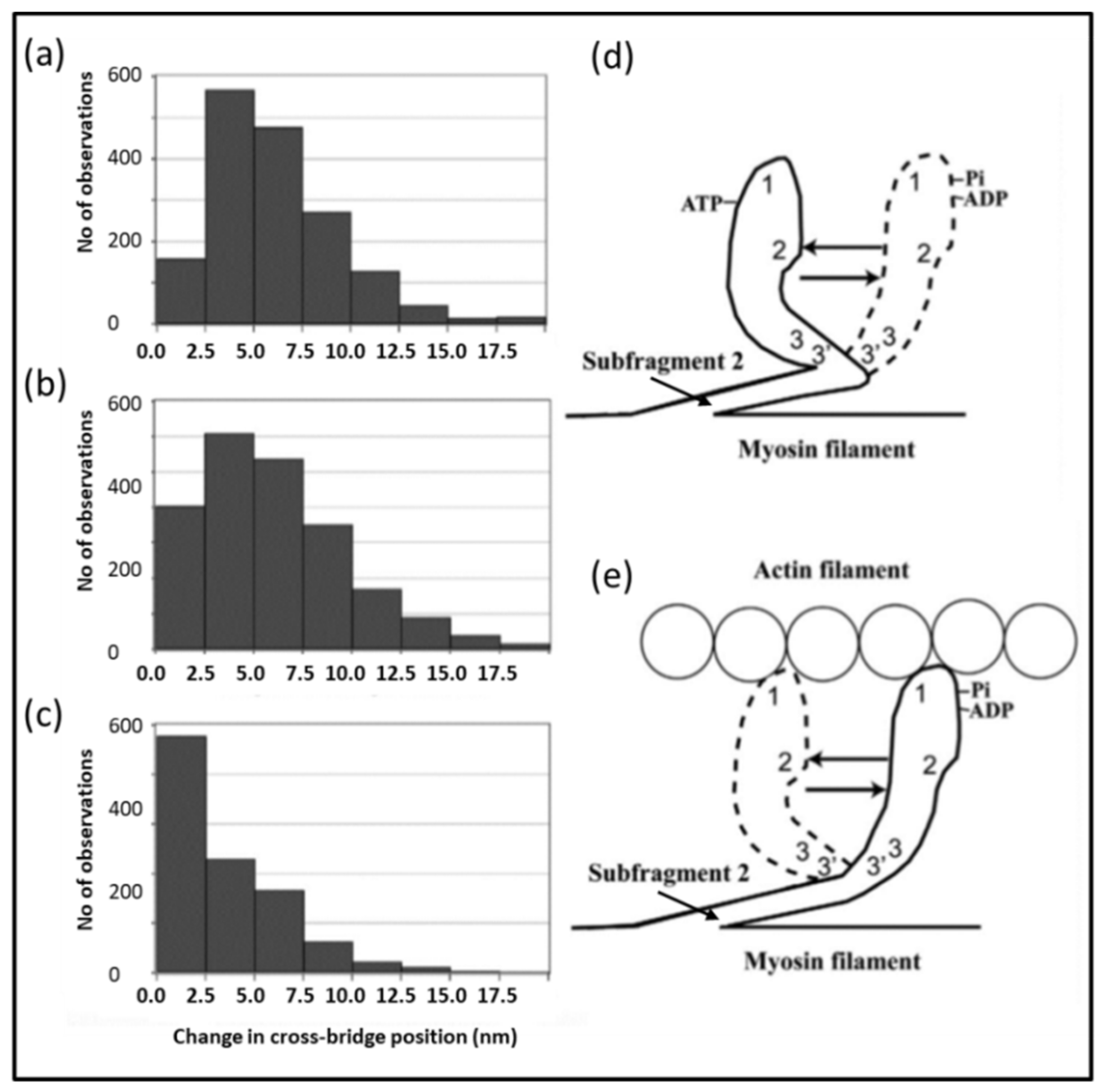
4.4. The Electron Microscopy of Myosin Heads
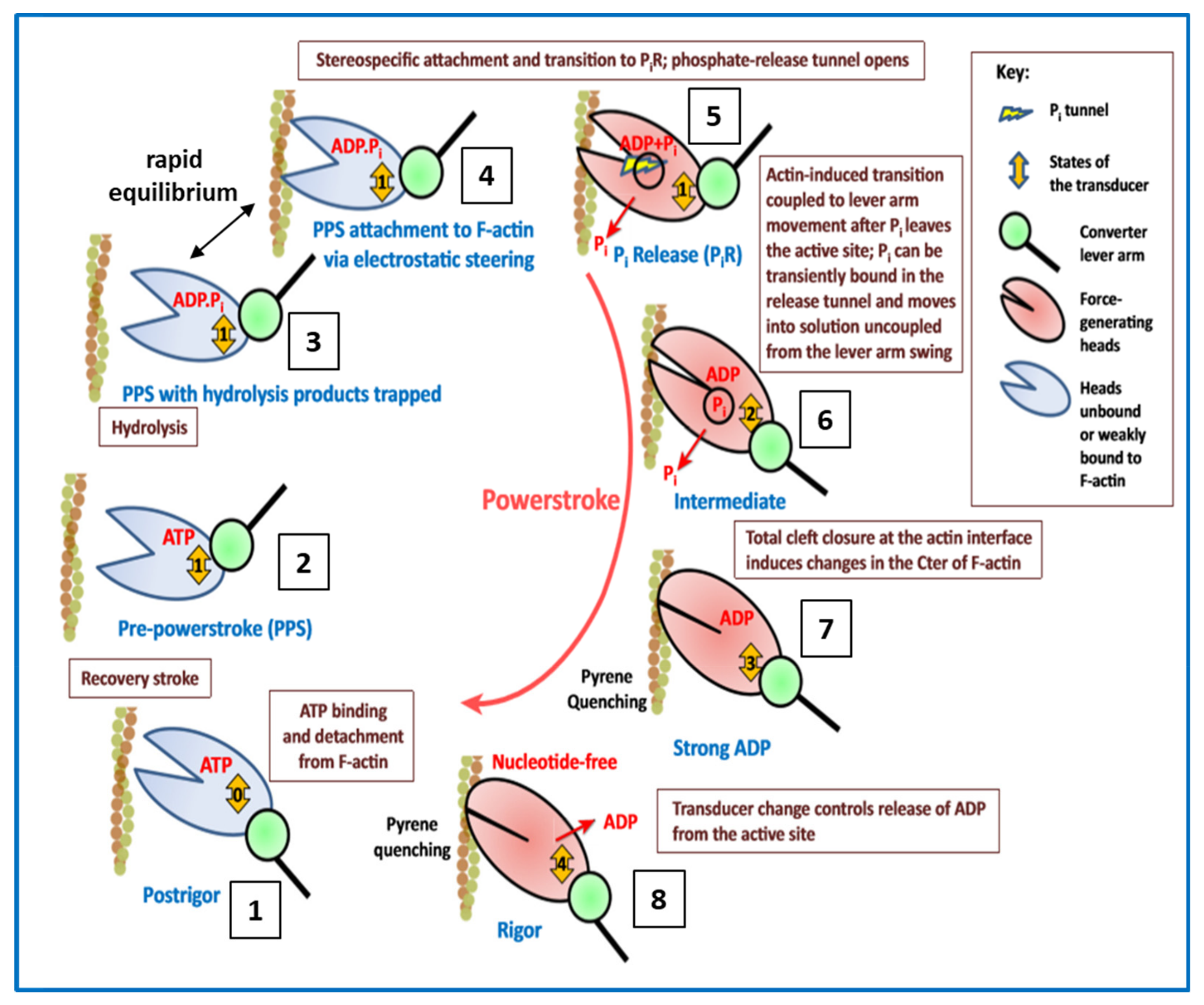
4.5. Conclusions about the Crossbridge Cycle
5. Mutations in the Actin-Myosin Contractile Apparatus: Muscle Diseases
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Huxley, A. Muscle Structure and Theories of Contraction. Prog. Biophys. Biophys. Chem. 1957, 7, 255–318. [Google Scholar] [CrossRef]
- Sweeney, H.L.; Holzbaur, E.L.F. Motor Proteins. Cold Spring Harb. Perspect. Biol. 2018, 10, a021931. [Google Scholar] [CrossRef] [PubMed]
- Squire, J.M. The Structural Basis of Muscular Contraction; Now reprinted; Plenum Publishing Co.: New York, NY, USA, 1981. [Google Scholar]
- Huxley, H.E.; Hanson, J. Changes in the cross-striations of muscle during contraction and stretch and their structural interpretation. Nature 1954, 173, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Huxley, A.F.; Niedergerke, R. Structural changes in muscle during contraction; interference microscopy of living muscle fibres. Nature 1954, 173, 971–973. [Google Scholar] [CrossRef] [PubMed]
- Squire, J.M. Muscle contraction: Sliding filament history, sarcomere dynamics and the two Huxleys. Glob. Cardiol. Sci. Pract. 2016, 2016, e201611. [Google Scholar] [CrossRef] [PubMed]
- Squire, J.M.; Knupp, C. Studies of Muscle Contraction Using X-ray Diffraction. In Muscle Contraction and Cell Motility: Fundamentals and Developments; Sugi, H., Ed.; Pan Stanford Publishing: Singapore, 2017; pp. 35–73. [Google Scholar]
- Small, J.V.; Squire, J.M. Structural basis of contraction in vertebrate smooth muscle. J. Mol. Biol. 1972, 67, 117–149. [Google Scholar] [CrossRef]
- Xu, J.Q.; Harder, B.A.; Uman, P.; Craig, R.M. Myosin filament structure in vertebrate smooth muscle. J. Cell Biol. 1996, 134, 53–66. [Google Scholar] [CrossRef]
- Syamaladevi, D.P.; Spudich, J.A.; Sowdhamini, R. Structural and Functional Insights on the Myosin superfamily. Bioinform. Biol. Insights 2012, 6, 11–21. [Google Scholar] [CrossRef]
- Lowey, S.; Slayter, H.S.; Weeds, A.G.; Baker, H. Substructure of the myosin molecule. I. Subfragments of myosin by enzymic degradation. J. Mol. Biol. 1969, 42, 1–29. [Google Scholar] [CrossRef]
- Rayment, I.; Rypniewski, W.R.; Schmidt-Bäse, K.; Smith, R.; Tomchick, D.R.; Benning, M.M.; Winkelmann, D.A.; Wesenberg, G.; Holden, H.M. Three-dimensional structure of myosin subfragment-1: A molecular motor. Science 1993, 261, 50–58. [Google Scholar] [CrossRef]
- Squire, J.M. General model of myosin filament structure III: Molecular packing arrangements in myosin filaments. J. Mol. Biol. 1973, 77, 291–323. [Google Scholar] [CrossRef]
- Hu, Z.; Taylor, D.W.; Reedy, M.K.; Edwards, R.J.; Taylor, K.A. Structure of myosin filaments from relaxed Lethocerus flight muscle by cryo-EM at 6 Å resolution. Sci. Adv. 2016, 2, e1600058. [Google Scholar] [CrossRef] [PubMed]
- Squire, J.M. General model of myosin filament structure II: Myosin filaments and crossbridge interactions in vertebrate striated and insect flight muscles. J. Mol. Biol. 1972, 72, 125–138. [Google Scholar] [CrossRef]
- Kensler, R.W.; Stewart, M. The relaxed crossbridge pattern in isolated rabbit psoas muscle thick filaments. J. Cell Sci. 1983, 105, 841–848. [Google Scholar]
- Luther, P.K.; Munro, P.M.G.; Squire, J.M. Three-dimensional structure of the vertebrate muscle A-band III: M-region structure and myosin filament symmetry. J. Mol. Biol. 1981, 151, 703–730. [Google Scholar] [CrossRef]
- Huxley, H.; Brown, W. The low-angle X-ray diagram of vertebrate striated muscle and its behaviour during contraction and rigor. J. Mol. Biol. 1967, 30, 383–434. [Google Scholar] [CrossRef]
- Harford, J.J.; Squire, J.M. The ‘crystalline’ myosin cross-bridge array in relaxed bony fish muscles. Biophys. J. 1986, 50, 145–155. [Google Scholar] [CrossRef]
- Al-Khayat, H.A.; Kensler, R.W.; Squire, J.M.; Marston, S.B.; Morris, E.P. Atomic model of the human cardiac muscle myosin filament. Proc. Natl. Acad. Sci. USA 2013, 110, 318–323. [Google Scholar] [CrossRef]
- Wendt, T.; Taylor, D.; Messier, T.; Trybus, K.M.; Taylor, K.A. Visualization of head-head interactions in the inhibited state of smooth muscle myosin. J. Cell Biol. 1999, 147, 1385–1390. [Google Scholar] [CrossRef]
- Knupp, C.; Morris, E.; Squire, J.M. The Interacting Head Motif Structure Does Not Explain the X-Ray Diffraction Patterns in Relaxed Vertebrate (Bony Fish) Skeletal Muscle and Insect (Lethocerus) Flight Muscle. Biology 2019, 8, 67. [Google Scholar] [CrossRef]
- Luther, P.K.; Squire, J.M. Three-dimensional structure of the vertebrate muscle M-region. J. Mol. Biol. 1978, 125, 313–324. [Google Scholar] [CrossRef]
- Pask, H.; Jones, K.L.; Luther, P.K.; Squire, J.M. M-band Structure, M-bridge interactions and contraction speed in vertebrate cardiac muscles. J. Muscle Res. Cell Motil. 1994, 15, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.; Pinotsis, N.; Agarkova, I.; Ehler, E. The M-band: The underestimated part of the sarcomere. Biochim. Biophys. Acta Mol. Cell Res. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Luther, P.K.; Squire, J.M. Three-dimensional structure of the vertebrate muscle A-band. II. The myosin filament superlattice. J. Mol. Biol. 1980, 141, 409–439. [Google Scholar] [CrossRef]
- Luther, P.K.; Squire, J.M. The intriguing dual lattices of the myosin filaments in vertebrate striated muscles: Evolution and advantage. Biology 2014, 3, 846–865. [Google Scholar] [CrossRef]
- Tonino, P.; Kiss, B.; Gohlke, J.; Smith, J.E., 3rd; Granzier, H. Fine mapping titin’s C-zone: Matching cardiac myosin-binding protein C stripes with titin’s super-repeats. J. Mol. Cell. Cardiol. 2019, 133, 47–56. [Google Scholar] [CrossRef]
- Rome, L.C.; Funke, R.P.; Alexander, R.M.; Lutz, G.; Aldridge, H.; Scott, F.; Freadman, M. Why animals have different muscle fibre types. Nature 1988, 335, 824–827. [Google Scholar] [CrossRef]
- Trinick, J.; Knight, P.; Whiting, A. Purification and properties of native titin. J. Mol. Biol. 1984, 180, 331–356. [Google Scholar] [CrossRef]
- Ottenheijm, C.A.; Granzier, H. Role of titin in skeletal muscle function and disease. Adv. Exp. Med. Biol. 2010, 682, 105–122. [Google Scholar]
- Trinick, J.A. End-filaments: A new structural element of vertebrate skeletal muscle thick filaments. J. Mol. Biol. 1981, 151, 309–314. [Google Scholar] [CrossRef]
- Knupp, C.; Luther, P.K.; Squire, J.M. Titin organisation and the 3D architecture of the vertebrate striated muscle I-Band. J. Mol. Biol. 2002, 322, 731–739. [Google Scholar] [CrossRef]
- Offer, G.; Moos, C.; Starr, R. A new protein of the thick filaments of vertebrate skeletal myofibrils. Extractions, purification and characterization. J. Mol. Biol. 1973, 74, 653–676. [Google Scholar] [CrossRef]
- Luther, P.K.; Winkler, H.; Taylor, K.; Zoghbi, M.E.; Craig, R.; Padrón, R.; Squire, J.M.; Liu, J. Direct visualization of myosin-binding protein C bridging myosin and actin filaments in intact muscle. Proc. Natl. Acad. Sci. USA 2011, 108, 11423–11428. [Google Scholar] [CrossRef] [PubMed]
- Robinett, J.C.; Hanft, L.M.; Geist, J.; Kontrogianni-Konstantopoulos, A.; McDonald, K.S. Regulation of myofilament force and loaded shortening by skeletal myosin binding protein C. J. Gen. Physiol. 2019, 151, 645–659. [Google Scholar] [CrossRef]
- Kabsch, W.; Mannherz, H.G.; Suck, D.; Pai, E.F.; Holmes, K.C. Atomic structure of the actin:DNase I complex. Nature 1990, 347, 37–44. [Google Scholar] [CrossRef]
- Holmes, K.C.; Popp, D.; Gebhard, W.; Kabsch, W. Atomic model of the actin filament. Nature 1990, 347, 44–49. [Google Scholar] [CrossRef]
- Paul, D.M.; Squire, J.M.; Morris, E.P. Relaxed and active thin filament structures; a new structural basis for the regulatory mechanism. J. Struct. Biol. 2017, 197, 365–371. [Google Scholar] [CrossRef]
- Hitchcock-DeGregori, S.E.; Barua, B. Tropomyosin Structure, Function, and Interactions: A Dynamic Regulator. Subcell. Biochem. 2017, 82, 253–284. [Google Scholar] [CrossRef]
- Lehman, W.; Moore, J.R.; Campbell, S.G.; Rynkiewicz, M.J. The Effect of Tropomyosin Mutations on Actin-Tropomyosin Binding: In Search of Lost Time. Biophys. J. 2019, 116, 2275–2284. [Google Scholar] [CrossRef]
- Bowman, J.D.; Lindert, S. Computational Studies of Cardiac and Skeletal Troponin. Front. Mol. Biosci. 2019, 6, 68. [Google Scholar] [CrossRef]
- Marston, S. Small molecule studies: The fourth wave of muscle research. J. Muscle Res. Cell Motil. 2019, 40, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Luther, P.K. The vertebrate muscle Z-disc: Sarcomere anchor for structure and signalling. J. Muscle Res. Cell Motil. 2009, 30, 171–185, Erratum in 2011, 31, 383. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, T.; Heumann, J.M.; Morris, E.P.; Knupp, C.; Liu, J.; Reedy, M.K.; Taylor, K.A.; Wang, K.; Luther, P.K. Three-dimensional structure of the basketweave Z-band in midshipman fish sonic muscle. Proc. Natl. Acad. Sci. USA 2019, 116, 15534–15539. [Google Scholar] [CrossRef] [PubMed]
- Gautel, M.; Djinović-Carugo, K. The sarcomeric cytoskeleton: From molecules to motion. J. Exp. Biol. 2016, 219, 135–145. [Google Scholar] [CrossRef]
- Frank, D.; Frey, N. Cardiac Z-disc Signaling Network. J. Biol. Chem. 2011, 286, 9897–9904. [Google Scholar] [CrossRef]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.P.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository—New features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef]
- McNamara, J.W.; Li, A.; Dos Remedios, C.G.; Cooke, R. The role of super-relaxed myosin in skeletal and cardiac muscle. Biophys. Rev. 2015, 7, 5–14. [Google Scholar] [CrossRef]
- Lee, K.H.; Sulbarán, G.; Yang, S.; Mun, J.Y.; Alamo, L.; Pinto, A.; Sato, O.; Ikebe, M.; Liu, X.; Korn, E.D.; et al. Interacting-heads motif has been conserved as a mechanism of myosin II inhibition since before the origin of animals. Proc. Natl. Acad. Sci. USA 2018, 115, E1991–E2000. [Google Scholar] [CrossRef]
- Woodhead, J.L.; Zhao, F.-Q.; Craig, R.; Egelman, E.H.; Alamo, L.; Padron, R. Atomic model of a myosin filament in the relaxed state. Nature 2005, 436, 1195–1199. [Google Scholar] [CrossRef]
- Zoghbi, M.E.; Woodhead, J.L.; Moss, R.L.; Craig, R. Three-dimensional structure of vertebrate cardiac muscle myosin filaments. Proc. Natl. Acad. Sci. USA 2008, 105, 2386–2390. [Google Scholar] [CrossRef]
- AL-Khayat, H.A.; Morris, E.P.; Kensler, R.W.; Squire, J.M. Myosin filament 3D structure in mammalian cardiac muscle. J. Struct. Biol. 2008, 163, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Alamo, L.; Koubassova, N.; Pinto, A.; Gillilan, R.; Tsaturyan, A.; Padrón, R. Lessons from a tarantula: New insights into muscle thick filament and myosin interacting-heads motif structure and function. Biophys. Rev. 2017, 9, 461–480. [Google Scholar] [CrossRef] [PubMed]
- Lymn, R.W.; Taylor, E.W. Mechanism of adenosine triphosphate hydrolysis by actomyosin. Biochemistry 1971, 10, 4617–4624. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.; Chalovich, J.M.; Greene, L.E.; Eisenberg, E.; Schoenberg, M. Stiffness of skinned rabbit psoas fibers in MgATP and MgPPi solution. Biophys. J. 1986, 50, 685–691. [Google Scholar] [CrossRef]
- Huxley, H.E.; Kress, M. Cross-bridge behaviour during muscle contraction. J. Muscle Res. Cell Motil. 1985, 6, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Cecchi, G.; Griffiths, P.J.; Taylor, S. Muscular contraction: Kinetics of crossbridge attachment studied by high-frequency stiffness measurements. Science 1982, 217, 70–72. [Google Scholar] [CrossRef]
- Bagni, M.A.; Cecchi, G.; Colombini, B.; Colomo, F. Sarcomere tension-stiffness relation during the tetanus rise in single frog muscle fibres. J. Muscle Res. Cell Motil. 1999, 20, 469–476. [Google Scholar] [CrossRef]
- Squire, J.M. Muscle Contraction. In eLS; John Wiley and Sons, Ltd.: Chichester, UK, 2011. [Google Scholar] [CrossRef]
- Ebashi, S.; Endo, M. Calcium ion and muscle contraction. Prog. Biophys. Mol. Biol. 1968, 18, 123–183. [Google Scholar] [CrossRef]
- Huxley, H.E. Structural changes in actin- and myosin-containing filaments during contraction. Cold Spring Harb. Symp. Quant. Biol. 1972, 37, 361–376. [Google Scholar] [CrossRef]
- Haselgrove, J.C. X-ray evidence for conformational change in actin filaments of vertebrate skeletal muscle. Cold Spring Harb. Symp. Quant. Biol. 1972, 37, 341–352. [Google Scholar] [CrossRef]
- Parry, D.A.; Squire, J.M. Structural role of tropomyosin in muscle regulation: Analysis of the X-ray diffraction patterns from relaxed and contracting muscles. J. Mol. Biol. 1973, 75, 33–55. [Google Scholar] [CrossRef]
- Vibert, P.; Craig, R.; Lehman, W. Steric-model for activation of muscle thin filaments. J. Mol. Biol. 1997, 266, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Squire, J.M.; Morris, E.P. A new look at thin filament regulation in vertebrate skeletal muscle. FASEB J. 1998, 12, 761–771. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.A.; Walsh, M.P. Regulation of Smooth Muscle Myosin Light Chain Phosphatase by Multisite Phosphorylation of the Myosin Targeting Subunit, MYPT1. Cardiovasc. Hematol. Disord. Drug Targets 2018, 18, 4–13. [Google Scholar] [CrossRef]
- Szent-Györgyi, A.G. Regulation by myosin: How calcium regulates some myosins, past and present. Adv. Exp. Med. Biol. 2007, 592, 253–264. [Google Scholar]
- Frearson, N.; Perry, S.V. Phosphorylation of the light-chain components of myosin from cardiac and red skeletal muscles. Biochem. J. 1975, 151, 99–107. [Google Scholar] [CrossRef]
- Bunda, J.; Gittings, W.; Vandenboom, R. Myosin phosphorylation improves contractile economy of mouse fast skeletal muscle during staircase potentiation. J. Exp. Biol. 2018, 221, jeb167718. [Google Scholar] [CrossRef]
- Ponnam, S.; Sevrieva, I.; Sun, Y.B.; Irving, M.; Kampourakis, T. Site-specific phosphorylation of myosin binding protein-C coordinates thin and thick filament activation in cardiac muscle. Proc. Natl. Acad. Sci. USA 2019, 116, 15485–15494. [Google Scholar] [CrossRef]
- McNamara, J.W.; Sadayappan, S. Skeletal myosin binding protein-C: An increasingly important regulator of striated muscle physiology. Arch. Biochem. Biophys. 2018, 660, 121–128. [Google Scholar] [CrossRef]
- McKillop, D.F.; Geeves, M.A. Regulation of the interaction between actin and myosin subfragment 1: Evidence for three states of the thin filament. Biophys. J. 1993, 65, 693–701. [Google Scholar] [CrossRef]
- Geeves, M.A.; Conibear, P.B. The role of three-state docking of myosin S1 with actin in force generation. Biophys. J. 1995, 68 (Suppl. 4), 194S–199S, discussion 199S–201S. [Google Scholar] [PubMed]
- Wilkie, D.R. Molecular Basis of Motility; Heilmeyer, L.M.C., Jr., Ruegg, J.C., Wicland, T.L., Eds.; Springer: New York, NY, USA, 1976; pp. 69–78. [Google Scholar]
- Huxley, A.F.; Simmons, R.M. Proposed Mechanism of Force Generation in Striated Muscle. Nature 1971, 233, 533–538. [Google Scholar] [CrossRef]
- Ford, L.E.; Huxley, A.F.; Simmons, R.M. Tension responses to sudden length change in stimulated frog muscle fibres near slack length. J. Physiol. 1977, 269, 441–515. [Google Scholar] [CrossRef] [PubMed]
- Ford, L.E.; Huxley, A.F.; Simmons, R.M. The relation between stiffness and filament overlap in stimulated frog muscle fibres. J. Physiol. 1981, 311, 219–249. [Google Scholar] [CrossRef] [PubMed]
- Knupp, C.; Squire, J.M. Myosin Cross-Bridge Behaviour in Contracting Muscle—The T1 Curve of Huxley and Simmons (1971) Revisited. Int. J. Mol. Sci. 2019, 20, 4892. [Google Scholar] [CrossRef] [PubMed]
- Huxley, H.; Stewart, A.; Sosa, H.; Irving, T. X-ray diffraction measurements of the extensibility of actin and myosin filaments in contracting muscle. Biophys. J. 1994, 67, 2411–2421. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Sugimoto, Y.; Tanaka, H.; Ueno, Y.; Takezawa, Y.; Amemiya, Y. X-ray diffraction evidence for the extensibility of actin and myosin filaments during muscle contraction. Biophys. J. 1994, 68, 1196–1197. [Google Scholar] [CrossRef]
- Offer, G.; Ranatunga, K.W. Cross-bridge and filament compliance in muscle: Implications for tension generation and lever arm swing. J. Muscle Res. Cell Motil. 2010, 31, 245–265. [Google Scholar] [CrossRef]
- Reedy, M.K. Ultrastructure of insect flight muscle. I. Screw sense and structural grouping in the rigor cross-bridge lattice. J. Mol. Biol. 1968, 31, 155–176. [Google Scholar] [CrossRef]
- Squire, J.M.; Harford, J.J. Actin filament organisation and myosin head labelling patterns in vertebrate skeletal muscles in the rigor and weak-binding states. J. Muscle Res. Cell Motil. 1988, 9, 344–358. [Google Scholar] [CrossRef]
- Steffen, W.; Smith, D.; Simmons, R.; Sleep, J. Mapping the actin filament with myosin. Proc. Natl. Acad. Sci. USA 2001, 98, 14949–14954. [Google Scholar] [CrossRef] [PubMed]
- Wlodawer, A.; Minor, W.; Dauter, Z.; Jaskolski, M. Protein crystallography for non-crystallographers, or how to get the best (but not more) from published macromolecular structures. FEBS J. 2008, 275, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Grimes, J.M.; Hall, D.R.; Ashton, A.W.; Evans, G.; Owen, R.L.; Wagner, A.; McAuley, K.E.; von Delft, F.; Orville, A.M.; Sorensen, T.; et al. Where is crystallography going? Acta Crystallogr. D Struct. Biol. 2018, 74, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Orlov, I.; Myasnikov, A.G.; Andronov, L.; Natchiar, S.K.; Khatter, H.; Beinsteiner, B.; Ménétret, J.F.; Hazemann, I.; Mohideen, K.; Tazibt, K.; et al. The integrative role of cryo-electron microscopy in molecular and cellular structural biology. Biol. Cell. 2017, 109, 81–93. [Google Scholar] [CrossRef]
- Henderson, R. From Electron Crystallography to Single Particle CryoEM (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 2018, 57, 10804–10825. [Google Scholar] [CrossRef]
- Zhang, P. Advances in cryo-electron tomography and sub-tomogram averaging and classification. Curr. Opin. Struct. Biol. 2019, 58, 249–258. [Google Scholar] [CrossRef]
- Taylor, K.A.; Rahmani, H.; Edwards, R.J.; Reedy, M.K. Insights into Actin-Myosin Interactions within Muscle from 3D Electron Microscopy. Int. J. Mol. Sci. 2019, 20, 1703. [Google Scholar] [CrossRef]
- Dietz, M.S.; Heilemann, M. Optical super-resolution microscopy unravels the molecular composition of functional protein complexes. Nanoscale 2019. [Google Scholar] [CrossRef]
- Stahlberg, H.; Biyani, N.; Engel, A. 3D reconstruction of two-dimensional crystals. Arch. Biochem. Biophys. 2015, 581, 68–77. [Google Scholar] [CrossRef]
- Vinothkumar, K.R.; Henderson, R. Single particle electron cryo-microscopy: Trends, issues and future perspective. Q. Rev. Biophys. 2016, 49, e13. [Google Scholar] [CrossRef]
- Lyumkis, D. Challenges and opportunities in cryo-EM single-particle analysis. J. Biol. Chem. 2019, 294, 5181–5197. [Google Scholar] [CrossRef] [PubMed]
- Nwanochie, E.; Uversky, V.N. Structure Determination by Single-Particle Cryo-Electron Microscopy: Only the Sky (and Intrinsic Disorder) is the Limit. Int. J. Mol. Sci. 2019, 20, 4186. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Grigorieff, N.; Penczek, P.A.; Walz, T. A primer to single-particle cryo-electron microscopy. Cell 2015, 161, 438–449. [Google Scholar] [CrossRef] [PubMed]
- AL-Khayat, H.A.; Morris, E.P.; Squire, J.M. Single Particle Analysis: A new approach to solving the 3D structure of Myosin Filaments. J. Muscle Res. Cell Motil. 2004, 25, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.M.; Squire, J.M.; Morris, E.P. A novel approach to the structural analysis of partially decorated actin based filaments. J. Struct. Biol. 2010, 170, 278–285. [Google Scholar] [CrossRef]
- Weber, G. Fluorescence-polarization spectrum and electronic-energy transfer in tyrosine, tryptophan and related compounds. Biochem. J. 1960, 75, 335–345. [Google Scholar] [CrossRef]
- Kasuya, M.; Takashina, H. Effects of pH and some agents on the fluorescence of myosin A. J. Biochem. 1967, 61, 35–43. [Google Scholar] [CrossRef]
- Aronson, J.F.; Morales, M.F. Polarization of tryptophan fluorescence in muscle. Biochemistry 1969, 8, 4517–4522. [Google Scholar] [CrossRef]
- Nihei, T.; Mendelson, R.A.; Botts, J. Use of fluorescence polarization to observe changes in attitude of S-1 moieties in muscle fibers. Biophys. J. 1974, 14, 236–242. [Google Scholar] [CrossRef][Green Version]
- Corrie, J.E.; Brandmeier, B.D.; Ferguson, R.E.; Trentham, D.R.; Kendrick-Jones, J.; Hopkins, S.C.; van der Heide, U.A.; Goldman, Y.E.; Sabido-David, C.; Dale, R.E.; et al. Dynamic measurement of myosin light-chain-domain tilt and twist in muscle contraction. Nature 1999, 400, 425–430. [Google Scholar] [CrossRef]
- Bajar, B.T.; Wang, E.S.; Zhang, S.; Lin, M.Z.; Chu, J. A Guide to Fluorescent Protein FRET Pairs. Sensors 2016, 16, 1488. [Google Scholar] [CrossRef] [PubMed]
- Bunt, G.; Wouters, F.S. FRET from single to multiplexed signaling events. Biophys. Rev. 2017, 9, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Olveczky, B.P.; Periasamy, N.; Verkman, A.S. Mapping fluorophore distributions in three dimensions by quantitative multiple angle-total internal reflection fluorescence microscopy. Biophys. J. 1997, 73, 2836–2847. [Google Scholar] [CrossRef]
- Guhathakurta, P.; Prochniewicz, E.; Thomas, D.D. Actin-Myosin Interaction: Structure, Function and Drug Discovery. Int. J. Mol. Sci. 2018, 19, 2628. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.D.; Kast, D.; Korman, V.L. Site-directed spectroscopic probes of actomyosin structural dynamics. Annu. Rev. Biophys. 2009, 38, 347–369. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.D.; Seidel, J.C.; Gergely, J.; Hyde, J.S. The quantitative measurement of rotational motion of the subfragment-1 region of myosin by saturation transfer epr spectroscopy. J. Supramol. Struct. 1975, 3, 376–390. [Google Scholar] [CrossRef]
- Savich, Y.; Binder, B.P.; Thompson, A.R.; Thomas, D.D. Myosin lever arm orientation in muscle determined with high angular resolution using bifunctional spin labels. J. Gen. Physiol. 2019, 151, 1007–1016. [Google Scholar] [CrossRef]
- White, D.C.S.; Thorson, J. The kinetics of muscle contraction. Prog. Biophys. 1973, 27, 175–255. [Google Scholar] [CrossRef]
- Maughan, D.W. Kinetics and energetics of the crossbridge cycle. Heart Fail. Rev. 2005, 10, 175–185. [Google Scholar] [CrossRef]
- McCray, J.A.; Trentham, D.R. Properties and uses of photoreactive caged compounds. Annu. Rev. Biophys. Biophys. Chem. 1989, 18, 239–270. [Google Scholar] [CrossRef]
- Ferenczi, M.A.; He, Z.H.; Chillingworth, R.K.; Brune, M.; Corrie, J.E.; Trentham, D.R.; Webb, M.R. A new method for the time-resolved measurement of phosphate release in permeabilized muscle fibers. Biophys. J. 1995, 68 (Suppl. 4), 191S–192S, Discussion 192S–193S. [Google Scholar] [PubMed]
- He, Z.H.; Chillingworth, R.K.; Brune, M.; Corrie, J.E.; Webb, M.R.; Ferenczi, M.A. The efficiency of contraction in rabbit skeletal muscle fibres determined from the rate of release of inorganic phosphate. J Physiol. 1999, 517, 839–854. [Google Scholar] [CrossRef] [PubMed]
- Martin-Fernandez, M.; Bordas, J.; Diakun, G.; Harries, J.; Lowy, J.; Mant, G.; Svensson, A.; Towns-Andrews, E. Time-resolved X-ray diffraction studies of myosin head movements in live frog sartorius muscle during isometric and isotonic contractions. J. Muscle Res. Cell Motil. 1994, 15, 319–348. [Google Scholar] [CrossRef] [PubMed]
- Harford, J.; Squire, J. Evidence for structurally different attached states of myosin cross-bridges on actin during contraction of fish muscle. Biophys. J. 1992, 63, 387–396. [Google Scholar] [CrossRef]
- Knupp, C.; Offer, G.; Ranatunga, K.; Squire, J.M. Probing Muscle Myosin Motor Action: X-Ray (M3 and M6) Interference Measurements Report Motor Domain Not Lever Arm Movement. J. Mol. Boil. 2009, 390, 168–181. [Google Scholar] [CrossRef]
- Harford, J.J.; Squire, J.M. Time-resolved studies of muscle using synchrotron radiation. Rep. Prog. Phys. 1997, 60, 1723–1787. [Google Scholar] [CrossRef]
- Hudson, L.; Harford, J.J.; Denny, R.C.; Squire, J.M. Myosin head configuration in relaxed fish muscle: Resting myosin heads must swing axially by up to 150 Angstroms or turn upside down to reach rigor. J. Mol. Biol. 1997, 273, 440–455. [Google Scholar] [CrossRef]
- AL-Khayat, H.A.; Hudson, L.; Reedy, M.K.; Irving, T.C.; Squire, J.M. Myosin head configuration in relaxed insect flight muscle: X-ray modelled resting cross-bridges in a pre-powerstroke state are poised for actin binding. Biophys. J. 2003, 85, 1063–1079. [Google Scholar] [CrossRef]
- Lewis, R.A.; Helsby, W.; Jones, A.O.; Hall, C.J.; Parker, B.; Sheldon, J.; Clifford, P.; Hillen, M.; Sumner, I.; Fore, N.S.; et al. The “rapid” high rate large area X-ray detector system. Nucl. Instrum. Methods Phys. Res. Sect. A 1997, 92, 32–41. [Google Scholar] [CrossRef]
- Eakins, F.; Pinali, C.; Gleeson, A.; Knupp, C.; Squire, J.M. X-ray Diffraction Evidence for Low Force Actin-Attached and Rigor-Like Cross-Bridges in the Contractile Cycle. Biology 2016, 5, 41. [Google Scholar] [CrossRef]
- Månsson, A.; Usaj, M.; Moretto, L.; Rassier, D.E. Do Actomyosin Single-Molecule Mechanics Data Predict Mechanics of Contracting Muscle? Int. J. Mol. Sci. 2018, 19, 1863. [Google Scholar] [CrossRef] [PubMed]
- Sugi, H.; Chaen, S.; Akimoto, T. Electron Microscopic Recording of the Power and Recovery Strokes of Individual Myosin Heads Coupled with ATP Hydrolysis: Facts and Implications. Int. J. Mol. Sci. 2018, 19, 1368. [Google Scholar] [CrossRef] [PubMed]
- Houdusse, A.; Sweeney, H.L. How Myosin Generates Force on Actin Filaments. Trends Biochem. Sci. 2016, 41, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Behrmann, E.; Muller, M.; Penczek, P.A.; Mennherz, H.G.; Manstein, D.J.; Raunser, S. Structure of the rigor actin-tropomyosin-myosin complex. Cell 2012, 150, 327–338. [Google Scholar] [CrossRef] [PubMed]
- von der Ecken, J.; Heissler, S.M.; Pathan-Chhatbar, S.; Manstein, D.J.; Raunser, S. Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution. Nature 2016, 534, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Llinas, P.; Isabet, T.; Song, L.; Ropars, V.; Zong, B.; Benisty, H.; Sirigu, S.; Morris, C.; Kikuti, C.; Safer, D.; et al. How actin initiates the motor activity of Myosin. Dev. Cell. 2015, 33, 401–412. [Google Scholar] [CrossRef]
- Trivedi, D.V.; Muretta, J.M.; Swenson, A.M.; Davis, J.P.; Thomas, D.D.; Yengo, C.M. Direct measurements of the coordination of lever arm swing and the catalytic cycle in myosin V. Proc. Natl. Acad. Sci. USA 2015, 112, 14593–14598. [Google Scholar] [CrossRef]
- Sequeira, V.; van der Velden, J. The Frank-Starling Law: A jigsaw of titin proportions. Biophys. Rev. 2017, 9, 259–267. [Google Scholar] [CrossRef]
- Eakins, F.; Harford, J.J.; Knupp, C.; Roessle, M.; Squire, J.M. Different Myosin Head Conformations in Bony Fish Muscles Put into Rigor at Different Sarcomere Lengths. Int. J. Mol. Sci. 2018, 19, 2091. [Google Scholar] [CrossRef]
- Li, K.L.; Rieck, D.; Solaro, R.J.; Dong, W. In situ time-resolved FRET reveals effects of sarcomere length on cardiac thin-filament activation. Biophys. J. 2014, 107, 682–693. [Google Scholar] [CrossRef]
- Iwamoto, H. Synchrotron Radiation X-ray Diffraction Techniques Applied to Insect Flight Muscle. Int. J. Mol. Sci. 2018, 19, 1748. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, M.; Farman, G.; Frye, M.; Bekyarova, T.; Gore, D.; Maughan, D.; Irving, T. Molecular dynamics of cyclically contracting insect flight muscle in vivo. Nature 2005, 433, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Ranatunga, K.W. Temperature Effects on Force and Actin–Myosin Interaction in Muscle: A Look Back on Some Experimental Findings. Int. J. Mol. Sci. 2018, 19, 1538. [Google Scholar] [CrossRef] [PubMed]
- Katayama, E.; Kodera, N. Unconventional Imaging Methods to Capture Transient Structures during Actomyosin Interaction. Int. J. Mol. Sci. 2018, 19, 1402. [Google Scholar] [CrossRef] [PubMed]
- Heuser, J.E. Some personal and historical notes on the utility of “deep-etch” electron microscopy for making cell structure/function correlations. Mol. Biol. Cell 2014, 25, 3273–3276. [Google Scholar] [CrossRef] [PubMed]
- Minoda, H.; Okabe, T.; Inayoshi, Y.; Miyakawa, T.; Miyauchi, Y.; Tanokura, M.; Katayama, E.; Wakabayashi, T.; Akimoto, T.; Sugi, H. Electron microscopic evidence for the myosin head lever arm mechanism in hydrated myosin filaments using the gas environmental chamber. Biochem. Biophys. Res. Commun. 2011, 405, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Gong, H.; Irving, T. Myosin Head Configurations in Resting and Contracting Murine Skeletal Muscle. Int. J. Mol. Sci. 2018, 19, 2643. [Google Scholar] [CrossRef]
- Eakins, F.; Knupp, C.; Squire, J.M. Monitoring the myosin crossbridge cycle in contracting muscle: Steps towards ‘Muscle-the Movie’. J. Muscle Res. Cell Motil. 2019, 40, 77–91. [Google Scholar] [CrossRef]
- Reconditi, M.; Linari, M.; Lucii, L.; Stewart, A.; Sun, Y.B.; Boesecke, P.; Narayanan, T. Fischetti, R.F.; Irving, T.; Piazzesi, G. The myosin motor in muscle generates a smaller and slower working stroke at higher load. Nature 2004, 428, 578–581. [Google Scholar] [CrossRef]
- Marston, S. The Molecular Mechanisms of Mutations in Actin and Myosin that Cause Inherited Myopathy. Int. J. Mol. Sci. 2018, 19, 2020. [Google Scholar] [CrossRef]
- Vikhorev, P.G.; Vikhoreva, N.N. Cardiomyopathies and Related Changes in Contractility of Human Heart Muscle. Int. J. Mol. Sci. 2018, 19, 2234. [Google Scholar] [CrossRef] [PubMed]
- Borovikov, Y.S.; Karpicheva, O.E.; Simonyan, A.O.; Avrova, S.V.; Rogozovets, E.A.; Sirenko, V.V.; Redwood, C.S. The Primary Causes of Muscle Dysfunction Associated with the Point Mutations in Tpm3.12; Conformational Analysis of Mutant Proteins as a Tool for Classification of Myopathies. Int. J. Mol. Sci. 2018, 19, 3975. [Google Scholar] [CrossRef] [PubMed]
- Green, E.M.; Wakimoto, H.; Anderson, R.L.; Evanchik, M.J.; Gorham, J.M.; Harrison, B.C.; Henze, M.; Kawas, R.; Oslob, J.D.; Rodriguez, H.M.; et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 2016, 351, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Mamidi, R.; Li, J.; Doh, C.Y.; Verma, S.; Stelzer, J.E. Impact of the Myosin Modulator Mavacamten on Force Generation and Cross-Bridge Behavior in a Murine Model of Hypercontractility. J. Am. Heart Assoc. 2018, 7, e009627. [Google Scholar] [CrossRef] [PubMed]
- Rohde, J.A.; Roopnarine, O.; Thomas, D.D.; Muretta, J.M. Mavacamten stabilizes an autoinhibited state of two-headed cardiac myosin. Proc. Natl. Acad. Sci. USA 2018, 115, E7486–E7494. [Google Scholar] [CrossRef]
- Hellam, D.C.; Podolsky, R.J. Force measurements in skinned muscle fibres. J. Physiol. 1969, 200, 807–819. [Google Scholar] [CrossRef]
- Weber, A.; Herz, R. The binding of calcium to actomyosin systems in relation to their biological activity. J. Biol. Chem. 1963, 238, 599–605. [Google Scholar]
- Squire, J.M. Muscle: Design, Diversity and Disease; Benjamin/Cummings Publishing Co.: Menlo Park, CA, USA, 1986. [Google Scholar]
- Ervasti, J.M. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim. Biophys. Acta 2007, 1772, 108–117. [Google Scholar] [CrossRef]
- Meyers, T.A.; Townsend, D. Cardiac Pathophysiology and the Future of Cardiac Therapies in Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2019, 20, 4098. [Google Scholar] [CrossRef]
- Wells, D.J. What is the level of dystrophin expression required for effective therapy of Duchenne muscular dystrophy? J. Muscle Res. Cell Motil. 2019, 40, 141–150. [Google Scholar] [CrossRef]
- Claeys, K.G. Congenital myopathies: An update. Dev. Med. Child. Neurol. 2019, 12, 165–174. [Google Scholar] [CrossRef] [PubMed]























© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Squire, J. Special Issue: The Actin-Myosin Interaction in Muscle: Background and Overview. Int. J. Mol. Sci. 2019, 20, 5715. https://doi.org/10.3390/ijms20225715
Squire J. Special Issue: The Actin-Myosin Interaction in Muscle: Background and Overview. International Journal of Molecular Sciences. 2019; 20(22):5715. https://doi.org/10.3390/ijms20225715
Chicago/Turabian StyleSquire, John. 2019. "Special Issue: The Actin-Myosin Interaction in Muscle: Background and Overview" International Journal of Molecular Sciences 20, no. 22: 5715. https://doi.org/10.3390/ijms20225715
APA StyleSquire, J. (2019). Special Issue: The Actin-Myosin Interaction in Muscle: Background and Overview. International Journal of Molecular Sciences, 20(22), 5715. https://doi.org/10.3390/ijms20225715