Secondary Structural Model of Human MALAT1 Reveals Multiple Structure–Function Relationships



Abstract
1. Introduction
2. Results
2.1. A Secondary Structural Model of Human MALAT1 Was Built Using Multiple RNA Structural Probing Datasets and MFE Calculations
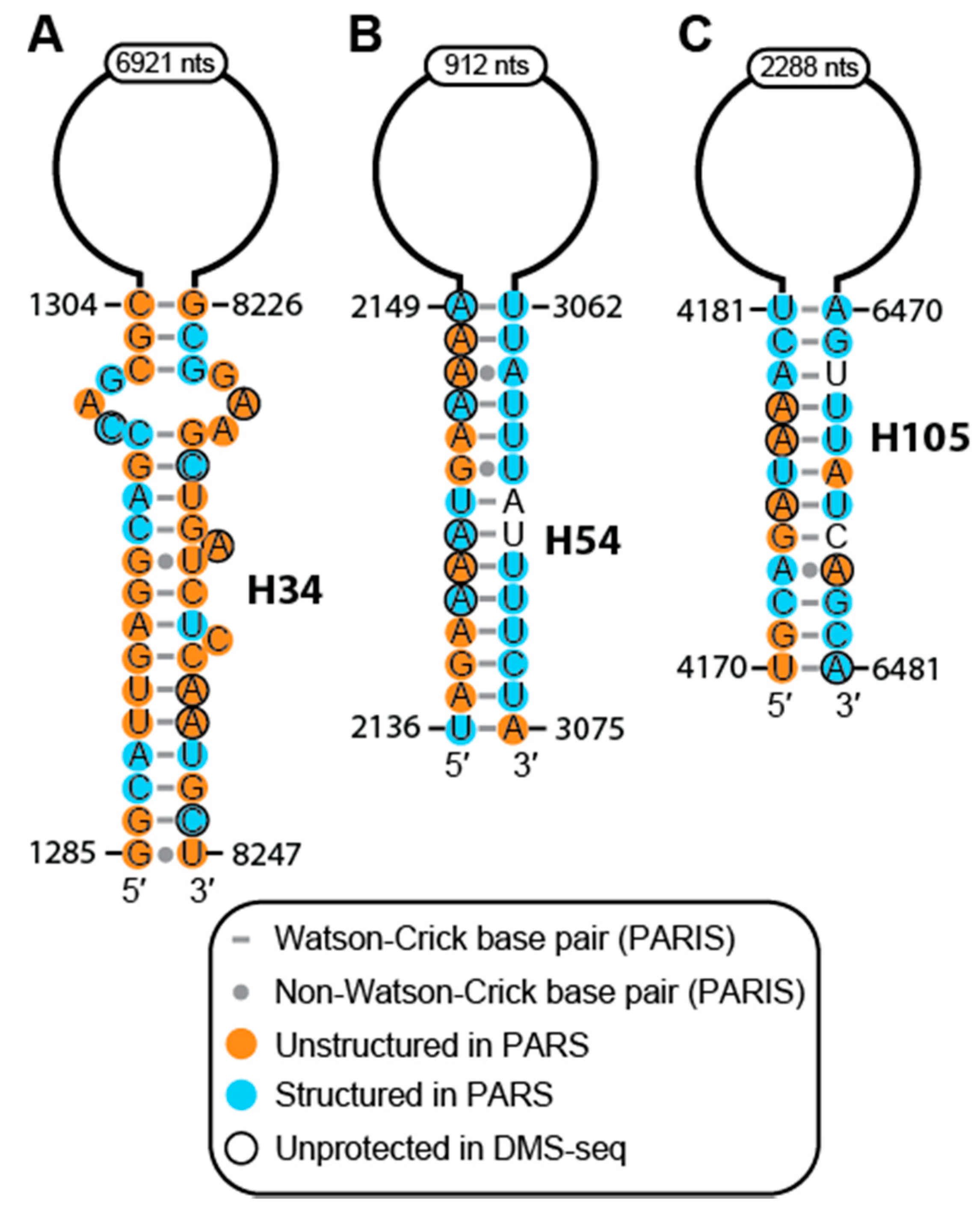
2.2. Conservation and Covariation Analyses Identified Evolutionarily Conserved Features in MALAT1 Homologs
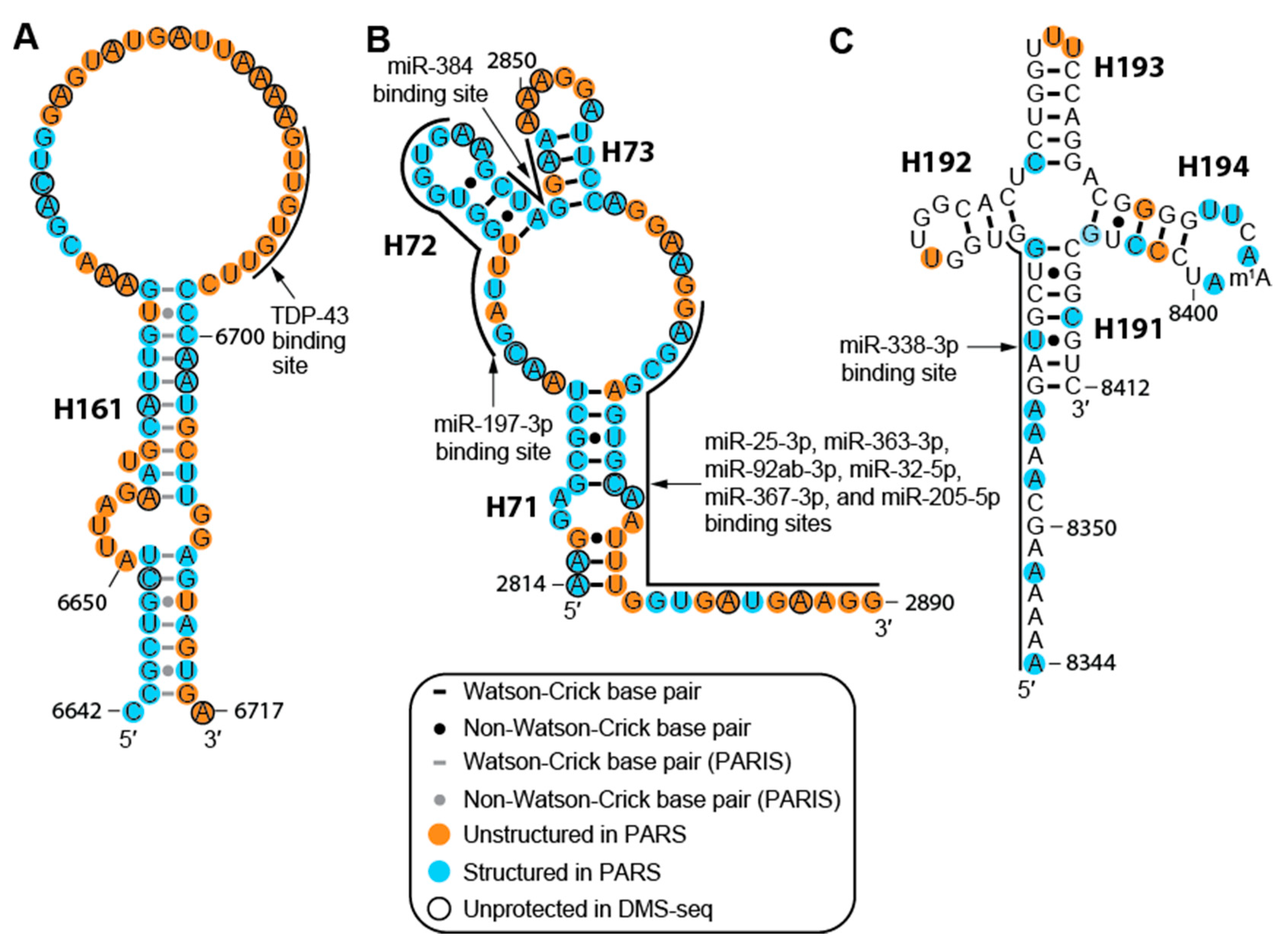
2.3. The MALAT1 Structural Model is Consistent with Known Protein-Binding Motifs
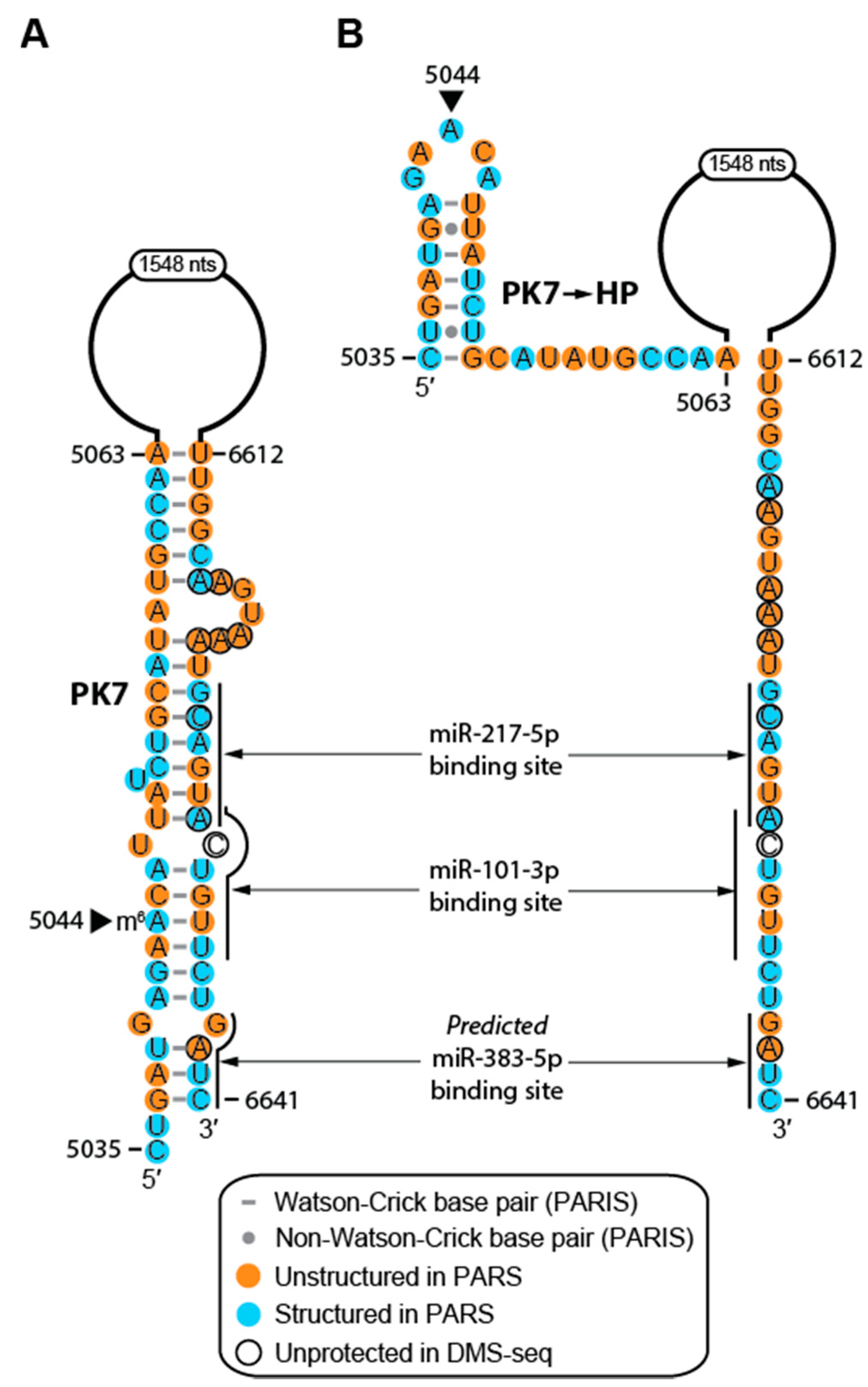
2.4. Structure of MALAT1 May Regulate Binding-site Accessibility for Diverse Classes of RNAs
2.5. RNA Modifications Alter the Structure and RNA-interacting Partners of MALAT1
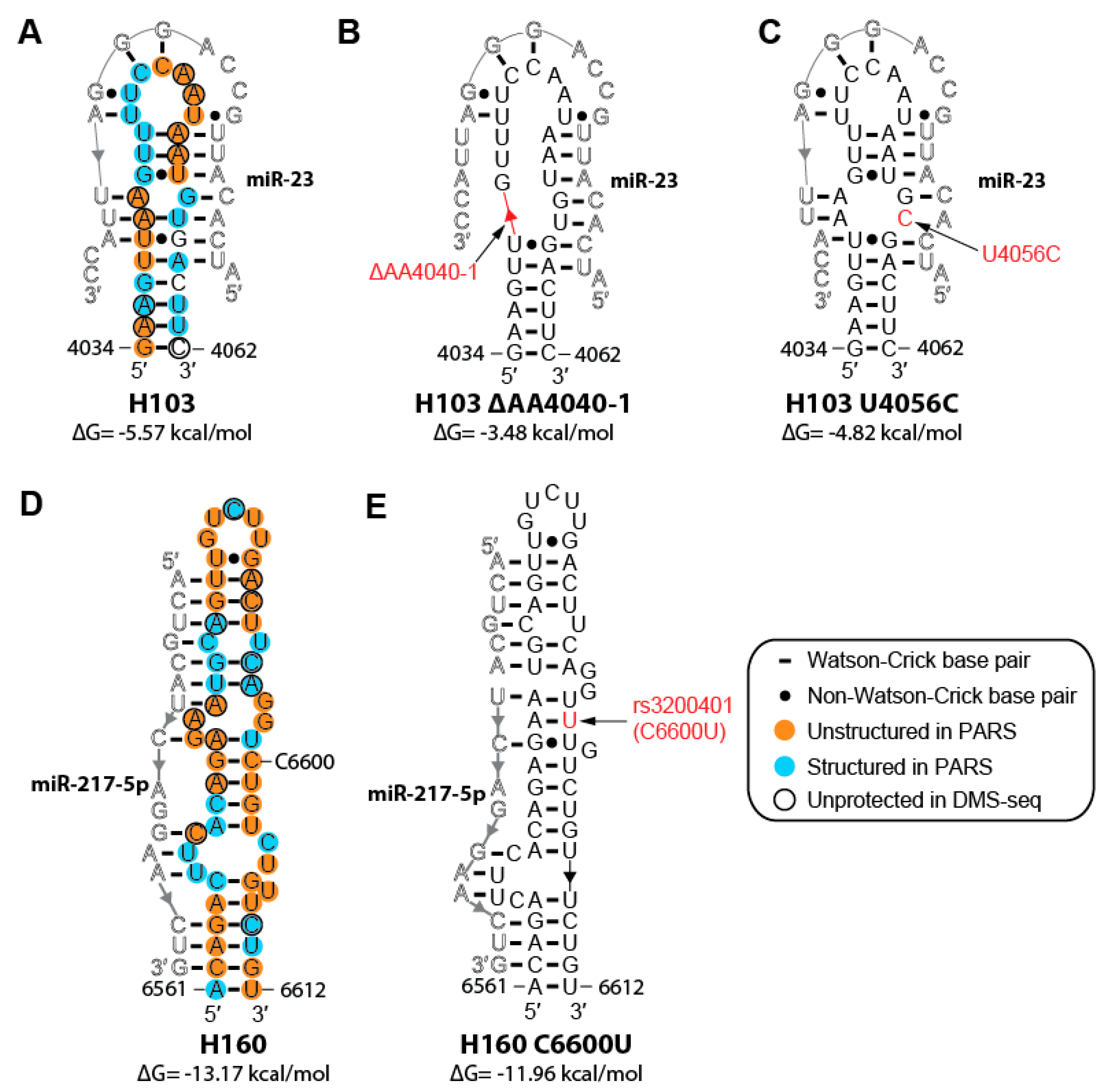
2.6. Cancer-associated Mutations and SNPs May Affect the Structure of MALAT1
3. Discussion
4. Materials and Methods
4.1. Datasets
4.2. Secondary Structure Modeling of MALAT1
4.3. Comparing Structural Assignments of Nucleotides
4.4. Conservation and Covariation Analyses of MALAT1 Structural Features
Supplementary Materials
Data Availability Statement
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Uszczynska-Ratajczak, B.; Lagarde, J.; Frankish, A.; Guigó, R.; Johnson, R. Towards a complete map of the human long non-coding RNA transcriptome. Nat. Rev. Genet. 2018, 19, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Zampetaki, A.; Albrecht, A.; Steinhofel, K. Long non-coding RNA structure and function: Is there a link? Front. Physiol. 2018, 9, 1201. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.N.; Ensminger, A.W.; Clemson, C.M.; Lynch, C.R.; Lawrence, J.B.; Chess, A. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics 2007, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Stadler, P.F. Evolution of the Long Non-Coding RNAs MALAT1 and MENβ/ε. In Proceedings of the Brazilian Symposium on Bioinformatics, Rio de Janeiro, Brasil, 31 August–3 September 2010; pp. 1–12. [Google Scholar]
- Gutschner, T.; Hammerle, M.; Eissmann, M.; Hsu, J.; Kim, Y.; Hung, G.; Revenko, A.; Arun, G.; Stentrup, M.; Gross, M.; et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013, 73, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Arun, G.; Diermeier, S.; Akerman, M.; Chang, K.-C.; Wilkinson, J.E.; Hearn, S.; Kim, Y.; MacLeod, A.R.; Krainer, A.R.; Norton, L.; et al. Differentiation of mammary tumors and reduction in metastasis upon Malat1 lncRNA loss. Genes Dev. 2016, 30, 34–51. [Google Scholar] [CrossRef]
- Amodio, N.; Raimondi, L.; Juli, G.; Stamato, M.A.; Caracciolo, D.; Tagliaferri, P.; Tassone, P. MALAT1: A druggable long non-coding RNA for targeted anti-cancer approaches. J. Hematol. Oncol. 2018, 11, 63. [Google Scholar] [CrossRef]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef]
- Mukherjee, N.; Corcoran, D.L.; Nusbaum, J.D.; Reid, D.W.; Georgiev, S.; Hafner, M.; Ascano, M.; Tuschl, T.; Ohler, U.; Keene, J.D. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol. Cell 2011, 43, 327–339. [Google Scholar] [CrossRef]
- Guo, F.; Jiao, F.; Song, Z.; Li, S.; Liu, B.; Yang, H.; Zhou, Q.; Li, Z. Regulation of MALAT1 expression by TDP43 controls the migration and invasion of non-small cell lung cancer cells in vitro. Biochem. Biophys. Res. Commun. 2015, 465, 293–298. [Google Scholar] [CrossRef]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N6-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef]
- Liu, N.; Zhou, K.I.; Parisien, M.; Dai, Q.; Diatchenko, L.; Pan, T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucl. Acids Res. 2017, 45, 6051–6063. [Google Scholar] [CrossRef] [PubMed]
- Engreitz, J.M.; Sirokman, K.; McDonel, P.; Shishkin, A.A.; Surka, C.; Russell, P.; Grossman, S.R.; Chow, A.Y.; Guttman, M.; Lander, E.S. RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent pre-mRNAs and chromatin sites. Cell 2014, 159, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Leucci, E.; Patella, F.; Waage, J.; Holmstrøm, K.; Lindow, M.; Porse, B.; Kauppinen, S.; Lund, A.H. MicroRNA-9 targets the long non-coding RNA MALAT1 for degradation in the nucleus. Sci. Rep. 2013, 3, 2535. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-H.; Liu, S.; Zhou, H.; Qu, L.-H.; Yang, J.-H. StarBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucl. Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Yang, H.; Zhang, J.; Peng, X.; Lu, Z.; Tong, W.; Chen, J. The lncRNA MALAT1 acts as a competing endogenous RNA to regulate KRAS expression by sponging miR-217 in pancreatic ductal adenocarcinoma. Sci. Rep. 2017, 7, 5186. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Bulkley, D.; Wang, J.; Valenstein, M.L.; Yario, T.A.; Steitz, T.A.; Steitz, J.A. Structural insights into the stabilization of MALAT1 noncoding RNA by a bipartite triple helix. Nat. Struct. Mol. Biol. 2014, 21, 633–640. [Google Scholar] [CrossRef]
- Wilusz, J.E.; Freier, S.M.; Spector, D.L. 3′-end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell 2008, 135, 919–932. [Google Scholar] [CrossRef]
- Smith, M.A.; Gesell, T.; Stadler, P.F.; Mattick, J.S. Widespread purifying selection on RNA structure in mammals. Nucl. Acids Res. 2013, 41, 8220–8236. [Google Scholar] [CrossRef]
- Zhang, B.; Mao, Y.S.; Diermeier, S.D.; Novikova, I.V.; Nawrocki, E.P.; Jones, T.A.; Lazar, Z.; Tung, C.-S.; Luo, W.; Eddy, S.R.; et al. Identification and characterization of a class of MALAT1-like genomic loci. Cell Rep. 2017, 19, 1723–1738. [Google Scholar] [CrossRef]
- Wan, Y.; Qu, K.; Zhang, Q.C.; Flynn, R.A.; Manor, O.; Ouyang, Z.; Zhang, J.; Spitale, R.C.; Snyder, M.P.; Segal, E.; et al. Landscape and variation of RNA secondary structure across the human transcriptome. Nature 2014, 505, 706–709. [Google Scholar] [CrossRef]
- Rouskin, S.; Zubradt, M.; Washietl, S.; Kellis, M.; Weissman, J.S. Genome-Wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 2014, 505, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Somarowthu, S.; Legiewicz, M.; Chillón, I.; Marcia, M.; Liu, F.; Pyle, A.M. HOTAIR forms an intricate and modular secondary structure. Mol. Cell 2015, 58, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.; Moss, W.N.; Rutenberg-Schoenberg, M.; Simon, M.D. Probing Xist RNA structure in cells using targeted structure-seq. PLoS Genet. 2015, 11, e1005668. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Schmidt, B.F.; Bruchez, M.P.; McManus, C.J. Structural analyses of NEAT1 lncRNAs suggest long-range RNA interactions that may contribute to paraspeckle architecture. Nucl. Acids Res. 2018, 46, 3742–3752. [Google Scholar] [CrossRef]
- Lu, Z.; Zhang, Q.C.; Lee, B.; Flynn, R.A.; Smith, M.A.; Robinson, J.T.; Davidovich, C.; Gooding, A.R.; Goodrich, K.J.; Mattick, J.S.; et al. RNA duplex map in living cells reveals higher-order transcriptome structure. Cell 2016, 165, 1267–1279. [Google Scholar] [CrossRef]
- Lu, Z.; Gong, J.; Zhang, Q.C. PARIS: Psoralen analysis of RNA interactions and structures with high throughput and resolution. Methods Mol. Biol. 2018, 1649, 59–84. [Google Scholar]
- Rivas, E.; Clements, J.; Eddy, S.R. A statistical test for conserved RNA structure shows lack of evidence for structure in lncRNAs. Nat. Methods 2017, 14, 45–48. [Google Scholar] [CrossRef]
- Turner, D.H.; Mathews, D.H. NNDB: The nearest neighbor parameter database for predicting stability of nucleic acid secondary structure. Nucl. Acids Res. 2010, 38, D280–D282. [Google Scholar] [CrossRef]
- Mortimer, S.A.; Trapnell, C.; Aviran, S.; Pachter, L.; Lucks, J.B. SHAPE-Seq: High-throughput RNA structure analysis. In Current Protocols in Chemical Biology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Aw, J.G.A.; Shen, Y.; Wilm, A.; Sun, M.; Lim, X.N.; Boon, K.-L.; Tapsin, S.; Chan, Y.-S.; Tan, C.-P.; Sim, A.Y.L.; et al. In vivo mapping of eukaryotic RNA interactomes reveals principles of higher-order organization and regulation. Mol. Cell 2016, 62, 603–617. [Google Scholar] [CrossRef]
- Lorenz, R.; Bernhart, S.H.; Höner Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef]
- Li, F.; Ryvkin, P.; Childress, D.M.; Valladares, O.; Gregory, B.D.; Wang, L.-S. SAVoR: A server for sequencing annotation and visualization of RNA structures. Nucl. Acids Res. 2012, 40, W59–W64. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Darty, K.; Denise, A.; Ponty, Y. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics 2009, 25, 1974–1975. [Google Scholar] [CrossRef] [PubMed]
- Tavares, R.C.A.; Pyle, A.M.; Somarowthu, S. Phylogenetic analysis with improved parameters reveals conservation in lncRNA structures. J. Mol. Biol. 2019, 431, 1592–1603. [Google Scholar] [CrossRef] [PubMed]
- Novikova, I.V.; Hennelly, S.P.; Sanbonmatsu, K.Y. Structural architecture of the human long non-coding RNA, steroid receptor RNA activator. Nucl. Acids Res. 2012, 40, 5034–5051. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Shkumatava, A.; Jan, C.H.; Sive, H.; Bartel, D.P. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 2011, 147, 1537–1550. [Google Scholar] [CrossRef] [PubMed]
- West, J.A.; Davis, C.P.; Sunwoo, H.; Simon, M.D.; Sadreyev, R.I.; Wang, P.I.; Tolstorukov, M.Y.; Kingston, R.E. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol. Cell 2014, 55, 791–802. [Google Scholar] [CrossRef]
- Chen, R.; Liu, Y.; Zhuang, H.; Yang, B.; Hei, K.; Xiao, M.; Hou, C.; Gao, H.; Zhang, X.; Jia, C.; et al. Quantitative proteomics reveals that long non-coding RNA MALAT1 interacts with DBC1 to regulate p53 acetylation. Nucl. Acids Res. 2017, 45, 9947–9959. [Google Scholar] [CrossRef]
- Spiniello, M.; Knoener, R.A.; Steinbrink, M.I.; Yang, B.; Cesnik, A.J.; Buxton, K.E.; Scalf, M.; Jarrard, D.F.; Smith, L.M. HyPR-MS for multiplexed discovery of MALAT1, NEAT1, and NORAD lncRNA protein interactomes. J. Proteome Res. 2018, 17, 3022–3038. [Google Scholar] [CrossRef]
- Brown, J.A.; Kinzig, C.G.; DeGregorio, S.J.; Steitz, J.A. Methyltransferase-like protein 16 binds the 3′-terminal triple helix of MALAT1 long noncoding RNA. Proc. Natl. Acad. Sci. 2016, 113, 14013–14018. [Google Scholar] [CrossRef]
- Linder, B.; Grozhik, A.V.; Olarerin-George, A.O.; Meydan, C.; Mason, C.E.; Jaffrey, S.R. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 2015, 12, 767–772. [Google Scholar] [CrossRef]
- Liu, N.; Parisien, M.; Dai, Q.; Zheng, G.; He, C.; Pan, T. Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA 2013, 19, 1848–1856. [Google Scholar] [CrossRef] [PubMed]
- Hirata, H.; Hinoda, Y.; Shahryari, V.; Deng, G.; Nakajima, K.; Tabatabai, Z.L.; Ishii, N.; Dahiya, R. Long noncoding RNA MALAT1 promotes aggressive renal cell carcinoma through Ezh2 and interacts with miR-205. Cancer Res. 2015, 75, 1322–1331. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-H.; Zhang, W.-J.; Wu, X.-C.; Weng, M.-Z.; Zhang, M.-D.; Cai, Q.; Zhou, D.; Wang, J.-D.; Quan, Z.-W. The lncRNA MALAT1 functions as a competing endogenous RNA to regulate MCL-1 expression by sponging miR-363-3p in gallbladder cancer. J. Cell. Mol. Med. 2016, 20, 2299–2308. [Google Scholar] [CrossRef] [PubMed]
- Sárközy, M.; Kahán, Z.; Csont, T. A myriad of roles of miR-25 in health and disease. Oncotarget 2018, 9, 21580–21612. [Google Scholar] [CrossRef]
- Kong, X.; Wang, J.; Cao, Y.; Zhang, H.; Lu, X.; Wang, Y.; Bo, C.; Wang, T.; Li, S.; Tian, K.; et al. The long noncoding RNA MALAT-1 functions as a competing endogenous RNA to regulate MSL2 expression by sponging miR-338-3p in myasthenia gravis. J. Cell. Biochem. 2019, 120, 5542–5550. [Google Scholar] [CrossRef]
- Gupta, S.; Stamatoyannopoulos, J.A.; Bailey, T.L.; Noble, W. Quantifying similarity between motifs. Genome Biol. 2007, 8, R24. [Google Scholar] [CrossRef]
- Hua, W.-F.; Zhong, Q.; Xia, T.-L.; Chen, Q.; Zhang, M.-Y.; Zhou, A.-J.; Tu, Z.-W.; Qu, C.; Li, M.-Z.; Xia, Y.-F.; et al. RBM24 suppresses cancer progression by upregulating miR-25 to target MALAT1 in nasopharyngeal carcinoma. Cell Death Dis. 2016, 7, e2352. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Saini, H.K.; van Dongen, S.; Enright, A.J. miRBase: Tools for microRNA genomics. Nucl. Acids Res. 2007, 36, D154–D158. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From microRNA sequences to function. Nucl. Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Chu, P.; Liang, A.; Jiang, A.; Zong, L. MiR-205 regulates the proliferation and invasion of ovarian cancer cells via suppressing PTEN/SMAD4 expression. Oncol. Lett. 2018, 15, 7571–7578. [Google Scholar] [CrossRef]
- Li, Q.; Pan, X.; Wang, X.; Jiao, X.; Zheng, J.; Li, Z.; Huo, Y. Long noncoding RNA MALAT1 promotes cell proliferation through suppressing miR-205 and promoting SMAD4 expression in osteosarcoma. Oncotarget 2017, 8, 106648–106660. [Google Scholar] [CrossRef] [PubMed]
- Jacob, R.; Zander, S.; Gutschner, T. The dark side of the epitranscriptome: Chemical modifications in long non-coding RNAs. Int. J. Mol. Sci. 2017, 18, 2387. [Google Scholar] [CrossRef] [PubMed]
- Safra, M.; Sas-Chen, A.; Nir, R.; Winkler, R.; Nachshon, A.; Bar-Yaacov, D.; Erlacher, M.; Rossmanith, W.; Stern-Ginossar, N.; Schwartz, S. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature 2017, 551, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Squires, J.E.; Patel, H.R.; Nousch, M.; Sibbritt, T.; Humphreys, D.T.; Parker, B.J.; Suter, C.M.; Preiss, T. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucl. Acids Res. 2012, 40, 5023–5033. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-S.; Liu, C.; Ma, H.; Dai, Q.; Sun, H.-L.; Luo, G.; Zhang, Z.; Zhang, L.; Hu, L.; Dong, X.; et al. Transcriptome-wide mapping of internal N7-methylguanosine methylome in mammalian mRNA. Mol. Cell 2019. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Moshitch-Moshkovitz, S.; Han, D.; Kol, N.; Amariglio, N.; Rechavi, G.; Dominissini, D.; He, C. Nm-seq maps 2′-O-methylation sites in human mRNA with base precision. Nat. Methods 2017, 14, 695–698. [Google Scholar] [CrossRef]
- Zheng, Y.; Nie, P.; Peng, D.; He, Z.; Liu, M.; Xie, Y.; Miao, Y.; Zuo, Z.; Ren, J. m6AVar: A database of functional variants involved in m6A modification. Nucl. Acids Res. 2018, 46, D139–D145. [Google Scholar] [CrossRef]
- Liu, B.; Merriman, D.K.; Choi, S.H.; Schumacher, M.A.; Plangger, R.; Kreutz, C.; Horner, S.M.; Meyer, K.D.; Al-Hashimi, H.M. A potentially abundant junctional RNA motif stabilized by m6A and Mg2. Nat. Commun. 2018, 9, 2761. [Google Scholar] [CrossRef]
- Wang, X.; Li, M.; Wang, Z.; Han, S.; Tang, X.; Ge, Y.; Zhou, L.; Zhou, C.; Yuan, Q.; Yang, M. Silencing of long noncoding RNA MALAT1 by miR-101 and miR-217 inhibits proliferation, migration, and invasion of esophageal squamous cell carcinoma cells. J. Biol. Chem. 2015, 290, 3925–3935. [Google Scholar] [CrossRef]
- Wang, H.; Wang, L.; Zhang, G.; Lu, C.; Chu, H.; Yang, R.; Zhao, G. MALAT1/miR-101-3p/MCL1 axis mediates cisplatin resistance in lung cancer. Oncotarget 2018, 9, 7501–7512. [Google Scholar] [CrossRef]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a shared vision for cancer genomic data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Yi Ren, H.; Ying Cong, Y.; Sunwu, Y.; Keqin, L.; Xiaochun, T.; Senrui, C.; Ende, C.; Xi Zhou, L.; Yanfan, C. Long noncoding RNA MALAT1 regulates autophagy associated chemoresistance via miR-23b-3p sequestration in gastric cancer. Mol. Cancer 2017, 16, 174. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Wu, X.; Xia, M.; Wu, F.; Ding, J.; Jiao, Y.; Zhan, Q.; An, F. Upregulated exosomic miR-23b-3p plays regulatory roles in the progression of pancreatic cancer. Oncol. Rep. 2017, 38, 2182–2188. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, K.; Han, K.; Tang, H.; Yin, X.; Zhang, J.; Zhang, X.; Zhang, L. Up-Regulation of plasma miR-23b is associated with poor prognosis of gastric cancer. Med. Sci. Monit. 2016, 22, 356–361. [Google Scholar] [CrossRef]
- Roufayel, R.; Kadry, S. Expression of miR-23a by apoptotic regulators in human cancer: A review. Cancer Biol. Ther. 2017, 18, 269–276. [Google Scholar] [CrossRef]
- Wang, J.-Z.; Xiang, J.-J.; Wu, L.-G.; Bai, Y.-S.; Chen, Z.-W.; Yin, X.-Q.; Wang, Q.; Guo, W.-H.; Peng, Y.; Guo, H.; et al. A genetic variant in long non-coding RNA MALAT1 associated with survival outcome among patients with advanced lung adenocarcinoma: A survival cohort analysis. BMC Cancer 2017, 17, 167. [Google Scholar] [CrossRef]
- Andrews, R.J.; Baber, L.; Moss, W.N. RNAStructuromeDB: A genome-wide database for RNA structural inference. Sci. Rep. 2017, 7, 17269. [Google Scholar] [CrossRef]
- Ratti, A.; Buratti, E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J. Neurochem. 2016, 138, 95–111. [Google Scholar] [CrossRef]
- Lai, W.-J.C.; Kayedkhordeh, M.; Cornell, E.V.; Farah, E.; Bellaousov, S.; Rietmeijer, R.; Salsi, E.; Mathews, D.H.; Ermolenko, D.N. mRNAs and lncRNAs intrinsically form secondary structures with short end-to-end distances. Nat. Commun. 2018, 9, 4328. [Google Scholar] [CrossRef]
- Si, Y.; Yang, Z.; Ge, Q.; Yu, L.; Yao, M.; Sun, X.; Ren, Z.; Ding, C. Long non-coding RNA MALAT1 activated autophagy, hence promoting cell proliferation and inhibiting apoptosis by sponging miR-101 in colorectal cancer. Cell. Mol. Biol. Lett. 2019, 24, 50. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, A.D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucl. Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, Z.; Breaker, R.R. R2R—Software to speed the depiction of aesthetic consensus RNA secondary structures. BMC Bioinf. 2011, 12, 3. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Goodarzi, H.; Nguyen, H.C.B.; Zhang, S.; Dill, B.D.; Molina, H.; Tavazoie, S.F. Modulated expression of specific tRNAs drives gene expression and cancer progression. Cell 2016, 165, 1416–1427. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| m6A | CD8T | GM | HEK 293T | hESC | Neuro | A549 | AML | H1299 | HepG2 | HeLa | PA-HeLa |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1763 | - | - | + | - | - | - | - | - | - | - | - |
| 2414 | - | - | + | - | - | - | - | - | - | - | - |
| 2515 | + | - | + | + | + | + | + | + | + | + | - |
| 2577 | + | + | + | + | + | + | + | + | + | + | - |
| 2611 | + | + | + | + | + | + | + | + | + | + | + |
| 2720 | + | - | + | - | - | + | - | - | - | + | + |
| 3752 | - | - | + | - | - | - | - | - | - | - | - |
| 4457 | - | - | + | - | - | - | - | - | - | - | + |
| 5044 | + | - | + | - | + | + | - | - | - | - | - |
| 6924 | - | - | + | - | - | - | - | - | - | - | - |
| 8181 | + | - | + | - | - | - | - | - | - | - | - |
| 8290 | - | - | + | - | - | - | - | - | - | - | - |
| Normal | Cancer | ||||||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCown, P.J.; Wang, M.C.; Jaeger, L.; Brown, J.A. Secondary Structural Model of Human MALAT1 Reveals Multiple Structure–Function Relationships. Int. J. Mol. Sci. 2019, 20, 5610. https://doi.org/10.3390/ijms20225610
McCown PJ, Wang MC, Jaeger L, Brown JA. Secondary Structural Model of Human MALAT1 Reveals Multiple Structure–Function Relationships. International Journal of Molecular Sciences. 2019; 20(22):5610. https://doi.org/10.3390/ijms20225610
Chicago/Turabian StyleMcCown, Phillip J., Matthew C. Wang, Luc Jaeger, and Jessica A. Brown. 2019. "Secondary Structural Model of Human MALAT1 Reveals Multiple Structure–Function Relationships" International Journal of Molecular Sciences 20, no. 22: 5610. https://doi.org/10.3390/ijms20225610
APA StyleMcCown, P. J., Wang, M. C., Jaeger, L., & Brown, J. A. (2019). Secondary Structural Model of Human MALAT1 Reveals Multiple Structure–Function Relationships. International Journal of Molecular Sciences, 20(22), 5610. https://doi.org/10.3390/ijms20225610