Distinct Subcellular Compartments of Dendritic Cells Used for Cross-Presentation

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. DC Subsets
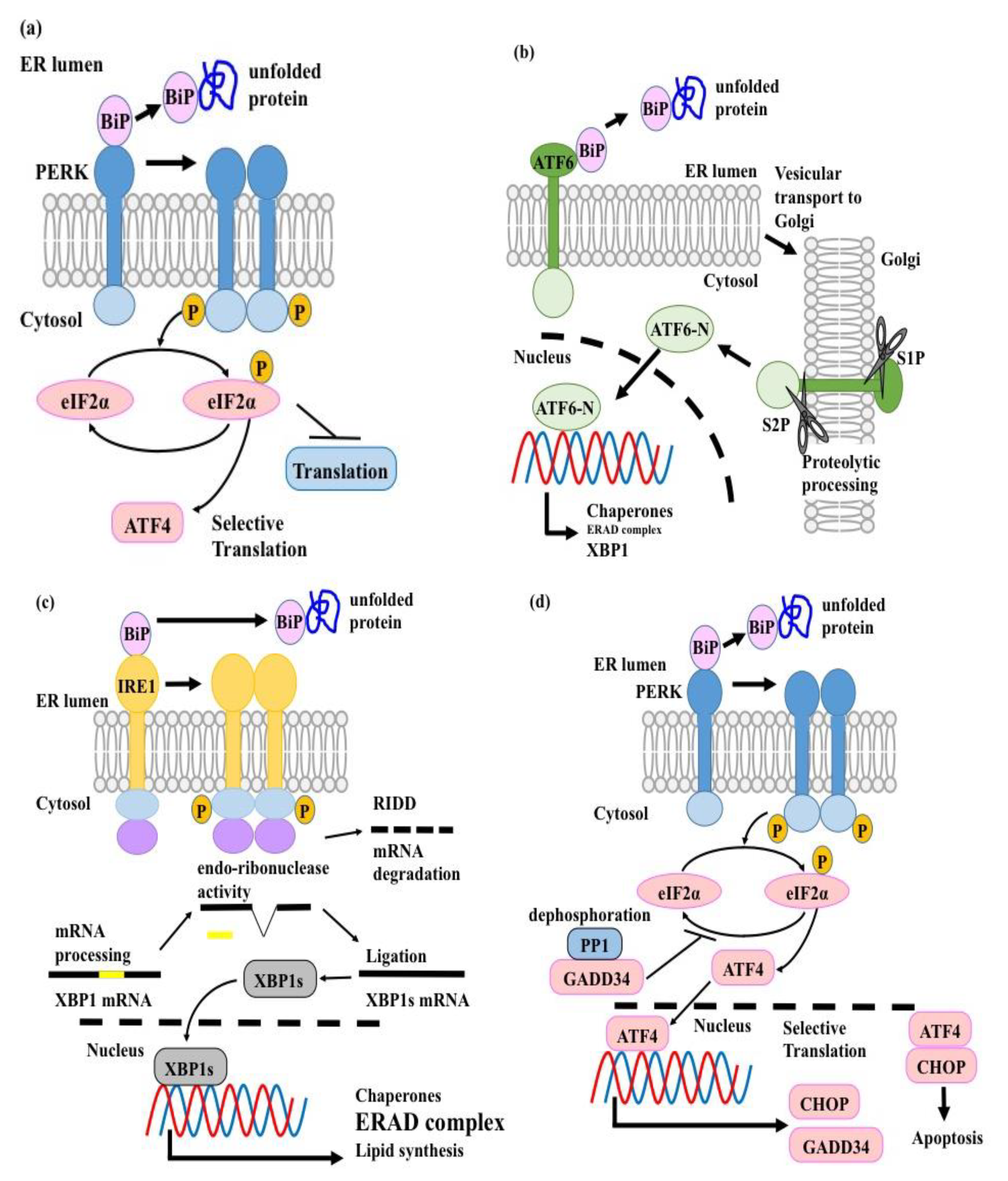
3. The UPR
4. Processing of Exogenous Proteins
5. Internalization and Transport of Exogenous Proteins
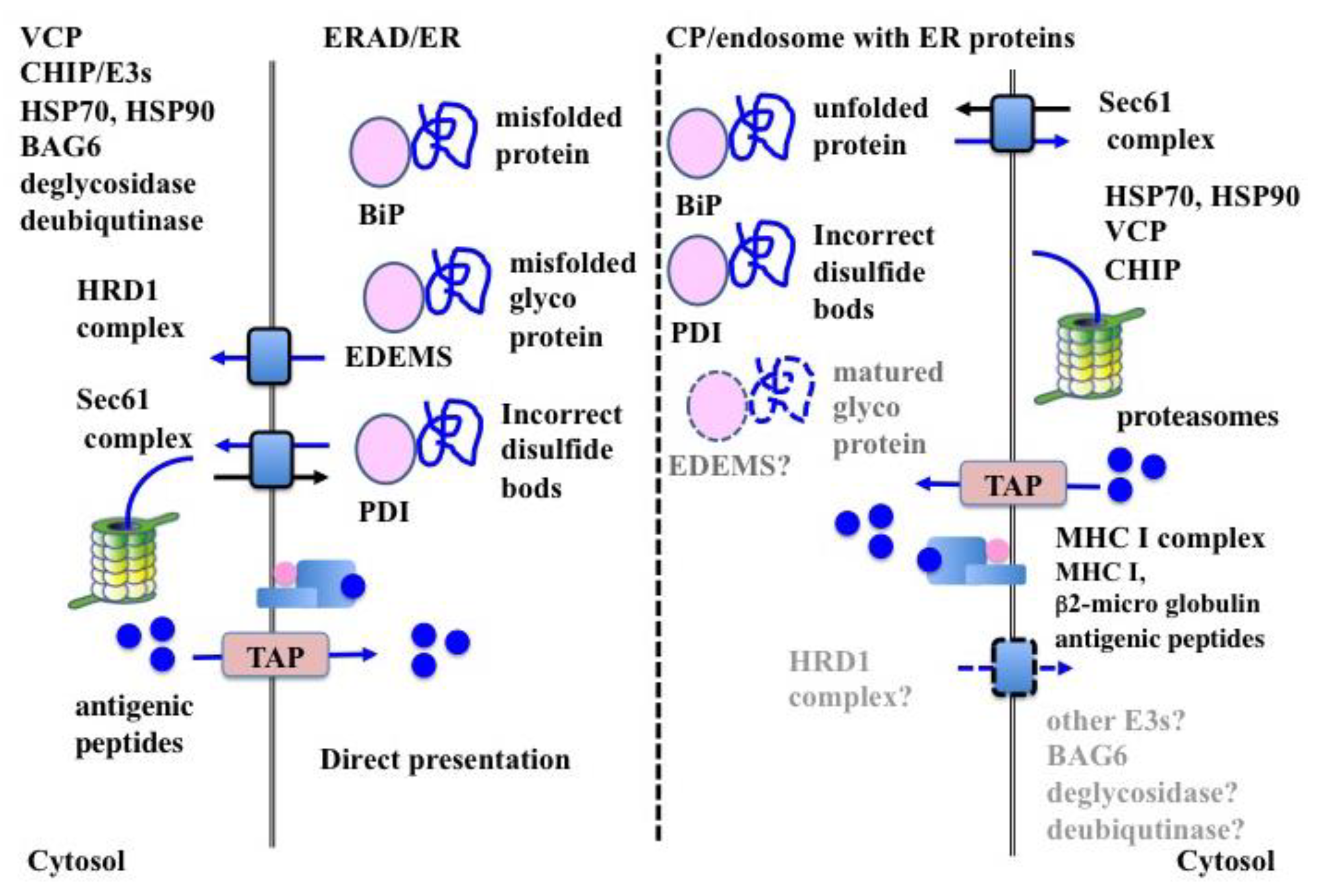
6. ERAD-Dependent Processing in CP
6.1. Substrate Recognition
6.1.1. BiP
6.1.2. Mannose-Specific Lectins
6.1.3. PDIs
6.2. Retro-Transport of Substrates
6.2.1. HRD1 Complex in ERAD
6.2.2. SEC61 Complex in ERAD
6.2.3. Translocon in CP
6.3. ERAD-Related Molecules
7. The UPR Induces Inflammation
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| APC | antigen-presenting cells |
| BMDC | bone-marrow-derived dendritic cell |
| cDC | conventional or classical DC |
| CP | cross-presentation |
| DC | dendritic cell |
| ERAD | endoplasmic reticulum-associated degradation |
| EDEMs | enhancing α-mannosidase-like proteins |
| IRAP | insulin-responsive aminopeptidase |
| MDSC | myeloid-derived suppressor ell |
| MHC I | major histocompatibility class I |
| MHC II | major histocompatibility class II |
| moDC | monocyte-derived DC |
| Mφ | macrophage |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| pDC | plasmacytoid DC |
| PDI | protein disulfide isomerase |
| PLC | peptide-loading complex |
| PRRs | pattern recognition receptors |
| RIDD | regulated IRE1-dependent decay |
| ROS | reactive oxygen species |
| TAP | transporter associated with antigen processing |
| UPS | ubiquitin-proteasome system |
References
- Steinman, R.M.; Cohn, Z.A. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J. Exp. Med. 1973, 137, 1142–1162. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2019. [Google Scholar] [CrossRef]
- Huang, A.Y.; Golumbek, P.; Ahmadzadeh, M.; Jaffee, E.; Pardoll, D.; Levitsky, H. Role of bone marrow-derived cells in presenting MHC class I-restricted tumor antigens. Science 1994, 264, 961–965. [Google Scholar] [CrossRef]
- Kurts, C.; Heath, W.R.; Carbone, F.R.; Allison, J.; Miller, J.F.; Kosaka, H. Constitutive class I-restricted exogenous presentation of self antigens in vivo. J. Exp. Med. 1996, 184, 923–930. [Google Scholar] [CrossRef]
- Kurts, C.; Kosaka, H.; Carbone, F.R.; Miller, J.F.; Heath, W.R. Class I-restricted cross-presentation of exogenous self-antigens leads to deletion of autoreactive CD8(+) T cells. J. Exp. Med. 1997, 186, 239–245. [Google Scholar] [CrossRef]
- Sigal, L.J.; Crotty, S.; Andino, R.; Rock, K.L. Cytotoxic T-cell immunity to virus-infected non-haematopoietic cells requires presentation of exogenous antigen. Nature 1999, 398, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Bonifaz, L.; Bonnyay, D.; Mahnke, K.; Rivera, M.; Nussenzweig, M.C.; Steinman, R.M. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J. Exp. Med. 2002, 196, 1627–1638. [Google Scholar] [CrossRef] [PubMed]
- Den Haan, J.M.; Bevan, M.J. Antigen presentation to CD8+ T cells: Cross-priming in infectious diseases. Curr. Opin. Immunol. 2001, 13, 437–441. [Google Scholar] [CrossRef]
- Heath, W.R.; Carbone, F.R. Cross-presentation in viral immunity and self-tolerance. Nat. Rev. Immunol. 2001, 1, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Nedjic, J.; Aichinger, M.; Emmerich, J.; Mizushima, N.; Klein, L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature 2008, 455, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Hampton, R.Y. ER-associated degradation in protein quality control and cellular regulation. Curr. Opin. Cell Biol. 2002, 14, 476–482. [Google Scholar] [CrossRef]
- Tsai, B.; Ye, Y.; Rapoport, T.A. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat. Rev. Mol. Cell Biol. 2002, 3, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Wiertz, E.J.; Tortorella, D.; Bogyo, M.; Yu, J.; Mothes, W.; Jones, T.R.; Rapoport, T.A.; Ploegh, H.L. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature 1996, 384, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Zehner, M.; Marschall, A.L.; Bos, E.; Schloetel, J.G.; Kreer, C.; Fehrenschild, D.; Limmer, A.; Ossendorp, F.; Lang, T.; Koster, A.J.; et al. The translocon protein Sec61 mediates antigen transport from endosomes in the cytosol for cross-presentation to CD8(+) T cells. Immunity 2015, 42, 850–863. [Google Scholar] [CrossRef] [PubMed]
- Imai, J.; Otani, M.; Sakai, T.; Hatta, S. Purification of the subcellular compartment in which exogenous antigens undergo endoplasmic reticulum-associated degradation from dendritic cells. Heliyon 2016, 2, e00151. [Google Scholar] [CrossRef]
- Osorio, F.; Tavernier, S.J.; Hoffmann, E.; Saeys, Y.; Martens, L.; Vetters, J.; Delrue, I.; De Rycke, R.; Parthoens, E.; Pouliot, P.; et al. The unfolded-protein-response sensor IRE-1alpha regulates the function of CD8alpha+ dendritic cells. Nat. Immunol. 2014, 15, 248–257. [Google Scholar] [CrossRef]
- Tavernier, S.J.; Osorio, F.; Vandersarren, L.; Vetters, J.; Vanlangenakker, N.; Van Isterdael, G.; Vergote, K.; De Rycke, R.; Parthoens, E.; van de Laar, L.; et al. Regulated IRE1-dependent mRNA decay sets the threshold for dendritic cell survival. Nat. Cell Biol. 2017, 19, 698–710. [Google Scholar] [CrossRef]
- Bourque, J.; Hawiger, D. Immunomodulatory Bonds of the Partnership between Dendritic Cells and T Cells. Crit. Rev. Immunol. 2018, 38, 379–401. [Google Scholar] [CrossRef]
- Reverendo, M.; Mendes, A.; Arguello, R.J.; Gatti, E.; Pierre, P. At the crossway of ER-stress and proinflammatory responses. FEBS J. 2019, 286, 297–310. [Google Scholar] [CrossRef]
- So, J.S. Roles of Endoplasmic Reticulum Stress in Immune Responses. Mol. Cells 2018, 41, 705–716. [Google Scholar] [CrossRef]
- Smith, J.A. Regulation of Cytokine Production by the Unfolded Protein Response; Implications for Infection and Autoimmunity. Front. Immunol. 2018, 9, 422. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Ruiz, J.R.; Silberman, P.C.; Rutkowski, M.R.; Chopra, S.; Perales-Puchalt, A.; Song, M.; Zhang, S.; Bettigole, S.E.; Gupta, D.; Holcomb, K.; et al. ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 2015, 161, 1527–1538. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.M.; Yao, F.H.; Yao, Y.M.; Dong, N.; Yu, Y.; Sheng, Z.Y. Endoplasmic reticulum stress and its regulator XBP-1 contributes to dendritic cell maturation and activation induced by high mobility group box-1 protein. Int. J. Biochem. Cell Biol. 2012, 44, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef]
- Segura, E.; Albiston, A.L.; Wicks, I.P.; Chai, S.Y.; Villadangos, J.A. Different cross-presentation pathways in steady-state and inflammatory dendritic cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20377–20381. [Google Scholar] [CrossRef]
- Mashayekhi, M.; Sandau, M.M.; Dunay, I.R.; Frickel, E.M.; Khan, A.; Goldszmid, R.S.; Sher, A.; Ploegh, H.L.; Murphy, T.L.; Sibley, L.D.; et al. CD8alpha(+) dendritic cells are the critical source of interleukin-12 that controls acute infection by Toxoplasma gondii tachyzoites. Immunity 2011, 35, 249–259. [Google Scholar] [CrossRef]
- Mattei, F.; Schiavoni, G.; Belardelli, F.; Tough, D.F. IL-15 is expressed by dendritic cells in response to type I IFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J. Immunol. 2001, 167, 1179–1187. [Google Scholar] [CrossRef]
- Keller, A.M.; Schildknecht, A.; Xiao, Y.; van den Broek, M.; Borst, J. Expression of costimulatory ligand CD70 on steady-state dendritic cells breaks CD8+ T cell tolerance and permits effective immunity. Immunity 2008, 29, 934–946. [Google Scholar] [CrossRef]
- Kurche, J.S.; Burchill, M.A.; Sanchez, P.J.; Haluszczak, C.; Kedl, R.M. Comparison of OX40 ligand and CD70 in the promotion of CD4+ T cell responses. J. Immunol. 2010, 185, 2106–2115. [Google Scholar] [CrossRef]
- Soares, H.; Waechter, H.; Glaichenhaus, N.; Mougneau, E.; Yagita, H.; Mizenina, O.; Dudziak, D.; Nussenzweig, M.C.; Steinman, R.M. A subset of dendritic cells induces CD4+ T cells to produce IFN-gamma by an IL-12-independent but CD70-dependent mechanism in vivo. J. Exp. Med. 2007, 204, 1095–1106. [Google Scholar] [CrossRef]
- Flynn, R.; Hutchinson, T.; Murphy, K.M.; Ware, C.F.; Croft, M.; Salek-Ardakani, S. CD8 T cell memory to a viral pathogen requires trans cosignaling between HVEM and BTLA. PLoS ONE 2013, 8, e77991. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, M.W.; Huang, Y.; Wang-Zhu, Y.; Ware, C.F.; Cheroutre, H.; Kronenberg, M. BTLA interaction with HVEM expressed on CD8(+) T cells promotes survival and memory generation in response to a bacterial infection. PLoS ONE 2013, 8, e77992. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, C.; Damuzzo, V.; Gotot, J.; Kroczek, R.A.; Yagita, H.; Murphy, K.M.; Knolle, P.A.; Ludwig-Portugall, I.; Kurts, C. Batf3-dependent dendritic cells in the renal lymph node induce tolerance against circulating antigens. J. Am. Soc. Nephrol. JASN 2013, 24, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.; Bourque, J.; Kuehm, L.; Opejin, A.; Teague, R.M.; Gross, C.; Hawiger, D. Immunomodulatory Functions of BTLA and HVEM Govern Induction of Extrathymic Regulatory T Cells and Tolerance by Dendritic Cells. Immunity 2016, 45, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Boucard-Jourdin, M.; Kugler, D.; Endale Ahanda, M.L.; This, S.; De Calisto, J.; Zhang, A.; Mora, J.R.; Stuart, L.M.; Savill, J.; Lacy-Hulbert, A.; et al. beta8 Integrin Expression and Activation of TGF-beta by Intestinal Dendritic Cells Are Determined by Both Tissue Microenvironment and Cell Lineage. J. Immunol. 2016, 197, 1968–1978. [Google Scholar] [CrossRef] [PubMed]
- Luda, K.M.; Joeris, T.; Persson, E.K.; Rivollier, A.; Demiri, M.; Sitnik, K.M.; Pool, L.; Holm, J.B.; Melo-Gonzalez, F.; Richter, L.; et al. IRF8 Transcription-Factor-Dependent Classical Dendritic Cells Are Essential for Intestinal T Cell Homeostasis. Immunity 2016, 44, 860–874. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, A.O.; Guermonprez, P.; Dudziak, D.; Nussenzweig, M.C. Route of antigen uptake differentially impacts presentation by dendritic cells and activated monocytes. J. Immunol. 2010, 185, 3426–3435. [Google Scholar] [CrossRef]
- Theisen, D.J.; Davidson, J.T.; Briseno, C.G.; Gargaro, M.; Lauron, E.J.; Wang, Q.; Desai, P.; Durai, V.; Bagadia, P.; Brickner, J.R.; et al. WDFY4 is required for cross-presentation in response to viral and tumor antigens. Science 2018, 362, 694–699. [Google Scholar] [CrossRef]
- Weimershaus, M.; Maschalidi, S.; Sepulveda, F.; Manoury, B.; van Endert, P.; Saveanu, L. Conventional dendritic cells require IRAP-Rab14 endosomes for efficient cross-presentation. J. Immunol. 2012, 188, 1840–1846. [Google Scholar] [CrossRef]
- Lancaster, J.N.; Thyagarajan, H.M.; Srinivasan, J.; Li, Y.; Hu, Z.; Ehrlich, L.I.R. Live-cell imaging reveals the relative contributions of antigen-presenting cell subsets to thymic central tolerance. Nat. Commun. 2019, 10, 2220. [Google Scholar] [CrossRef]
- Iwakoshi, N.N.; Pypaert, M.; Glimcher, L.H. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J. Exp. Med. 2007, 204, 2267–2275. [Google Scholar] [CrossRef] [PubMed]
- Di Pucchio, T.; Chatterjee, B.; Smed-Sorensen, A.; Clayton, S.; Palazzo, A.; Montes, M.; Xue, Y.; Mellman, I.; Banchereau, J.; Connolly, J.E. Direct proteasome-independent cross-presentation of viral antigen by plasmacytoid dendritic cells on major histocompatibility complex class I. Nat. Immunol. 2008, 9, 551–557. [Google Scholar] [CrossRef] [PubMed]
- GeurtsvanKessel, C.H.; Willart, M.A.; van Rijt, L.S.; Muskens, F.; Kool, M.; Baas, C.; Thielemans, K.; Bennett, C.; Clausen, B.E.; Hoogsteden, H.C.; et al. Clearance of influenza virus from the lung depends on migratory langerin+CD11b- but not plasmacytoid dendritic cells. J. Exp. Med. 2008, 205, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Mouries, J.; Moron, G.; Schlecht, G.; Escriou, N.; Dadaglio, G.; Leclerc, C. Plasmacytoid dendritic cells efficiently cross-prime naive T cells in vivo after TLR activation. Blood 2008, 112, 3713–3722. [Google Scholar] [CrossRef]
- Leon, B.; Martinez del Hoyo, G.; Parrillas, V.; Vargas, H.H.; Sanchez-Mateos, P.; Longo, N.; Lopez-Bravo, M.; Ardavin, C. Dendritic cell differentiation potential of mouse monocytes: Monocytes represent immediate precursors of CD8- and CD8+ splenic dendritic cells. Blood 2004, 103, 2668–2676. [Google Scholar] [CrossRef]
- Auffray, C.; Fogg, D.K.; Narni-Mancinelli, E.; Senechal, B.; Trouillet, C.; Saederup, N.; Leemput, J.; Bigot, K.; Campisi, L.; Abitbol, M.; et al. CX3CR1+ CD115+ CD135+ common macrophage/DC precursors and the role of CX3CR1 in their response to inflammation. J. Exp. Med. 2009, 206, 595–606. [Google Scholar] [CrossRef]
- Segura, E.; Touzot, M.; Bohineust, A.; Cappuccio, A.; Chiocchia, G.; Hosmalin, A.; Dalod, M.; Soumelis, V.; Amigorena, S. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity 2013, 38, 336–348. [Google Scholar] [CrossRef]
- Shortman, K.; Liu, Y.J. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2002, 2, 151–161. [Google Scholar] [CrossRef]
- Aldridge, J.R., Jr.; Moseley, C.E.; Boltz, D.A.; Negovetich, N.J.; Reynolds, C.; Franks, J.; Brown, S.A.; Doherty, P.C.; Webster, R.G.; Thomas, P.G. TNF/iNOS-producing dendritic cells are the necessary evil of lethal influenza virus infection. Proc. Natl. Acad. Sci. USA 2009, 106, 5306–5311. [Google Scholar] [CrossRef]
- Ji, Q.; Castelli, L.; Goverman, J.M. MHC class I-restricted myelin epitopes are cross-presented by Tip-DCs that promote determinant spreading to CD8(+) T cells. Nat. Immunol. 2013, 14, 254–261. [Google Scholar] [CrossRef]
- Leon, B.; Lopez-Bravo, M.; Ardavin, C. Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against Leishmania. Immunity 2007, 26, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Cheong, C.; Matos, I.; Choi, J.H.; Dandamudi, D.B.; Shrestha, E.; Longhi, M.P.; Jeffrey, K.L.; Anthony, R.M.; Kluger, C.; Nchinda, G.; et al. Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209(+) dendritic cells for immune T cell areas. Cell 2010, 143, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Soullie, T.; van Nimwegen, M.; Willart, M.A.; Muskens, F.; Jung, S.; Hoogsteden, H.C.; Hammad, H.; Lambrecht, B.N. Alum adjuvant boosts adaptive immunity by inducing uric acid and activating inflammatory dendritic cells. J. Exp. Med. 2008, 205, 869–882. [Google Scholar] [CrossRef] [PubMed]
- Markov, O.V.; Mironova, N.L.; Vlasov, V.V.; Zenkova, M.A. Molecular and Cellular Mechanisms of Antitumor Immune Response Activation by Dendritic Cells. Acta Nat. 2016, 8, 17–30. [Google Scholar] [CrossRef]
- Medel, B.; Costoya, C.; Fernandez, D.; Pereda, C.; Lladser, A.; Sauma, D.; Pacheco, R.; Iwawaki, T.; Salazar-Onfray, F.; Osorio, F. IRE1alpha Activation in Bone Marrow-Derived Dendritic Cells Modulates Innate Recognition of Melanoma Cells and Favors CD8(+) T Cell Priming. Front. Immunol. 2018, 9, 3050. [Google Scholar] [CrossRef]
- Plantinga, M.; Guilliams, M.; Vanheerswynghels, M.; Deswarte, K.; Branco-Madeira, F.; Toussaint, W.; Vanhoutte, L.; Neyt, K.; Killeen, N.; Malissen, B.; et al. Conventional and monocyte-derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity 2013, 38, 322–335. [Google Scholar] [CrossRef]
- Briseno, C.G.; Haldar, M.; Kretzer, N.M.; Wu, X.; Theisen, D.J.; Kc, W.; Durai, V.; Grajales-Reyes, G.E.; Iwata, A.; Bagadia, P.; et al. Distinct Transcriptional Programs Control Cross-Priming in Classical and Monocyte-Derived Dendritic Cells. Cell Rep. 2016, 15, 2462–2474. [Google Scholar] [CrossRef]
- Wong, K.L.; Tai, J.J.; Wong, W.C.; Han, H.; Sem, X.; Yeap, W.H.; Kourilsky, P.; Wong, S.C. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood 2011, 118, e16–e31. [Google Scholar] [CrossRef]
- Warger, T.; Osterloh, P.; Rechtsteiner, G.; Fassbender, M.; Heib, V.; Schmid, B.; Schmitt, E.; Schild, H.; Radsak, M.P. Synergistic activation of dendritic cells by combined Toll-like receptor ligation induces superior CTL responses in vivo. Blood 2006, 108, 544–550. [Google Scholar] [CrossRef]
- Walter, P.; Blobel, G. Translocation of proteins across the endoplasmic reticulum. II. Signal recognition protein (SRP) mediates the selective binding to microsomal membranes of in-vitro-assembled polysomes synthesizing secretory protein. J. Cell Biol. 1981, 91, 551–556. [Google Scholar] [CrossRef]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 2015, 10, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Stevens, F.J.; Argon, Y. Protein folding in the ER. Semin. Cell Dev. Biol. 1999, 10, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.T.; Hegde, R.S. Regulation of basal cellular physiology by the homeostatic unfolded protein response. J. Cell Biol. 2010, 189, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Tirasophon, W.; Welihinda, A.A.; Kaufman, R.J. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998, 12, 1812–1824. [Google Scholar] [CrossRef]
- Yoshida, H.; Haze, K.; Yanagi, H.; Yura, T.; Mori, K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 1998, 273, 33741–33749. [Google Scholar] [CrossRef]
- Korennykh, A.V.; Egea, P.F.; Korostelev, A.A.; Finer-Moore, J.; Zhang, C.; Shokat, K.M.; Stroud, R.M.; Walter, P. The unfolded protein response signals through high-order assembly of Ire1. Nature 2009, 457, 687–693. [Google Scholar] [CrossRef]
- Shaffer, A.L.; Shapiro-Shelef, M.; Iwakoshi, N.N.; Lee, A.H.; Qian, S.B.; Zhao, H.; Yu, X.; Yang, L.; Tan, B.K.; Rosenwald, A.; et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 2004, 21, 81–93. [Google Scholar] [CrossRef]
- Shoulders, M.D.; Ryno, L.M.; Genereux, J.C.; Moresco, J.J.; Tu, P.G.; Wu, C.; Yates, J.R., 3rd; Su, A.I.; Kelly, J.W.; Wiseman, R.L. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 2013, 3, 1279–1292. [Google Scholar] [CrossRef] [PubMed]
- Sriburi, R.; Jackowski, S.; Mori, K.; Brewer, J.W. XBP1: A link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 2004, 167, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef]
- Song, B.; Scheuner, D.; Ron, D.; Pennathur, S.; Kaufman, R.J. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Investig. 2008, 118, 3378–3389. [Google Scholar] [CrossRef]
- Reimold, A.M.; Iwakoshi, N.N.; Manis, J.; Vallabhajosyula, P.; Szomolanyi-Tsuda, E.; Gravallese, E.M.; Friend, D.; Grusby, M.J.; Alt, F.; Glimcher, L.H. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001, 412, 300–307. [Google Scholar] [CrossRef]
- Embgenbroich, M.; Burgdorf, S. Current Concepts of Antigen Cross-Presentation. Front. Immunol. 2018, 9, 1643. [Google Scholar] [CrossRef] [PubMed]
- Gros, M.; Amigorena, S. Regulation of Antigen Export to the Cytosol During Cross-Presentation. Front. Immunol. 2019, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.Y.; Bruce, A.T.; Pardoll, D.M.; Levitsky, H.I. In vivo cross-priming of MHC class I-restricted antigens requires the TAP transporter. Immunity 1996, 4, 349–355. [Google Scholar] [CrossRef]
- Kloetzel, P.M. Antigen processing by the proteasome. Nat. Rev. Mol. Cell Biol. 2001, 2, 179–187. [Google Scholar] [CrossRef]
- Rodriguez, A.; Regnault, A.; Kleijmeer, M.; Ricciardi-Castagnoli, P.; Amigorena, S. Selective transport of internalized antigens to the cytosol for MHC class I presentation in dendritic cells. Nat. Cell Biol. 1999, 1, 362–368. [Google Scholar] [CrossRef]
- Shen, L.; Sigal, L.J.; Boes, M.; Rock, K.L. Important role of cathepsin S in generating peptides for TAP-independent MHC class I crosspresentation in vivo. Immunity 2004, 21, 155–165. [Google Scholar] [CrossRef]
- Belizaire, R.; Unanue, E.R. Targeting proteins to distinct subcellular compartments reveals unique requirements for MHC class I and II presentation. Proc. Natl. Acad. Sci. USA 2009, 106, 17463–17468. [Google Scholar] [CrossRef]
- Dudziak, D.; Kamphorst, A.O.; Heidkamp, G.F.; Buchholz, V.R.; Trumpfheller, C.; Yamazaki, S.; Cheong, C.; Liu, K.; Lee, H.W.; Park, C.G.; et al. Differential antigen processing by dendritic cell subsets in vivo. Science 2007, 315, 107–111. [Google Scholar] [CrossRef]
- Belz, G.T.; Behrens, G.M.; Smith, C.M.; Miller, J.F.; Jones, C.; Lejon, K.; Fathman, C.G.; Mueller, S.N.; Shortman, K.; Carbone, F.R.; et al. The CD8alpha(+) dendritic cell is responsible for inducing peripheral self-tolerance to tissue-associated antigens. J. Exp. Med. 2002, 196, 1099–1104. [Google Scholar] [CrossRef]
- Palmowski, M.J.; Gileadi, U.; Salio, M.; Gallimore, A.; Millrain, M.; James, E.; Addey, C.; Scott, D.; Dyson, J.; Simpson, E.; et al. Role of immunoproteasomes in cross-presentation. J. Immunol. 2006, 177, 983–990. [Google Scholar] [CrossRef]
- Jia, Y.; Omri, A.; Krishnan, L.; McCluskie, M.J. Potential applications of nanoparticles in cancer immunotherapy. Hum. Vaccines Immunother. 2017, 13, 63–74. [Google Scholar] [CrossRef]
- Dasari, V.; Rehan, S.; Tey, S.K.; Smyth, M.J.; Smith, C.; Khanna, R. Autophagy and proteasome interconnect to coordinate cross-presentation through MHC class I pathway in B cells. Immunol. Cell Biol. 2016, 94, 964–974. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, D.; Graham, M.; Liu, X.; Cresswell, P. Proteasomal degradation within endocytic organelles mediates antigen cross-presentation. EMBO J. 2019, 38, e99266. [Google Scholar] [CrossRef] [PubMed]
- Dingjan, I.; Verboogen, D.R.; Paardekooper, L.M.; Revelo, N.H.; Sittig, S.P.; Visser, L.J.; Mollard, G.F.; Henriet, S.S.; Figdor, C.G.; Ter Beest, M.; et al. Lipid peroxidation causes endosomal antigen release for cross-presentation. Sci. Rep. 2016, 6, 22064. [Google Scholar] [CrossRef] [PubMed]
- Mant, A.; Chinnery, F.; Elliott, T.; Williams, A.P. The pathway of cross-presentation is influenced by the particle size of phagocytosed antigen. Immunology 2012, 136, 163–175. [Google Scholar] [CrossRef]
- Kretzer, N.M.; Theisen, D.J.; Tussiwand, R.; Briseno, C.G.; Grajales-Reyes, G.E.; Wu, X.; Durai, V.; Albring, J.; Bagadia, P.; Murphy, T.L.; et al. RAB43 facilitates cross-presentation of cell-associated antigens by CD8alpha+ dendritic cells. J. Exp. Med. 2016, 213, 2871–2883. [Google Scholar] [CrossRef]
- Nair-Gupta, P.; Baccarini, A.; Tung, N.; Seyffer, F.; Florey, O.; Huang, Y.; Banerjee, M.; Overholtzer, M.; Roche, P.A.; Tampe, R.; et al. TLR signals induce phagosomal MHC-I delivery from the endosomal recycling compartment to allow cross-presentation. Cell 2014, 158, 506–521. [Google Scholar] [CrossRef]
- Weimershaus, M.; Mauvais, F.X.; Saveanu, L.; Adiko, C.; Babdor, J.; Abramova, A.; Montealegre, S.; Lawand, M.; Evnouchidou, I.; Huber, K.J.; et al. Innate Immune Signals Induce Anterograde Endosome Transport Promoting MHC Class I Cross-Presentation. Cell Rep. 2018, 24, 3568–3581. [Google Scholar] [CrossRef]
- Menager, J.; Ebstein, F.; Oger, R.; Hulin, P.; Nedellec, S.; Duverger, E.; Lehmann, A.; Kloetzel, P.M.; Jotereau, F.; Guilloux, Y. Cross-presentation of synthetic long peptides by human dendritic cells: A process dependent on ERAD component p97/VCP but Not sec61 and/or Derlin-1. PLoS ONE 2014, 9, e89897. [Google Scholar] [CrossRef]
- Cohn, L.; Chatterjee, B.; Esselborn, F.; Smed-Sorensen, A.; Nakamura, N.; Chalouni, C.; Lee, B.C.; Vandlen, R.; Keler, T.; Lauer, P.; et al. Antigen delivery to early endosomes eliminates the superiority of human blood BDCA3+ dendritic cells at cross presentation. J. Exp. Med. 2013, 210, 1049–1063. [Google Scholar] [CrossRef]
- Delamarre, L.; Pack, M.; Chang, H.; Mellman, I.; Trombetta, E.S. Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science 2005, 307, 1630–1634. [Google Scholar] [CrossRef]
- Han, D.; Liu, J.; Chen, C.; Dong, L.; Liu, Y.; Chang, R.; Huang, X.; Liu, Y.; Wang, J.; Dougherty, U.; et al. Anti-tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature 2019, 566, 270–274. [Google Scholar] [CrossRef]
- Lennon-Dumenil, A.M.; Bakker, A.H.; Wolf-Bryant, P.; Ploegh, H.L.; Lagaudriere-Gesbert, C. A closer look at proteolysis and MHC-class-II-restricted antigen presentation. Curr. Opin. Immunol. 2002, 14, 15–21. [Google Scholar] [CrossRef]
- Pauwels, A.M.; Hartlova, A.; Peltier, J.; Driege, Y.; Baudelet, G.; Brodin, P.; Trost, M.; Beyaert, R.; Hoffmann, E. Spatiotemporal Changes of the Phagosomal Proteome in Dendritic Cells in Response to LPS Stimulation. Mol. Cell. Proteom. MCP 2019, 18, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Asano, J.; Tada, H.; Onai, N.; Sato, T.; Horie, Y.; Fujimoto, Y.; Fukase, K.; Suzuki, A.; Mak, T.W.; Ohteki, T. Nucleotide oligomerization binding domain-like receptor signaling enhances dendritic cell-mediated cross-priming in vivo. J. Immunol. 2010, 184, 736–745. [Google Scholar] [CrossRef]
- Corridoni, D.; Shiraishi, S.; Chapman, T.; Steevels, T.; Muraro, D.; Thezenas, M.L.; Prota, G.; Chen, J.L.; Gileadi, U.; Ternette, N.; et al. NOD2 and TLR2 Signal via TBK1 and PI31 to Direct Cross-Presentation and CD8 T Cell Responses. Front. Immunol. 2019, 10, 958. [Google Scholar] [CrossRef]
- Savina, A.; Peres, A.; Cebrian, I.; Carmo, N.; Moita, C.; Hacohen, N.; Moita, L.F.; Amigorena, S. The small GTPase Rac2 controls phagosomal alkalinization and antigen crosspresentation selectively in CD8(+) dendritic cells. Immunity 2009, 30, 544–555. [Google Scholar] [CrossRef]
- Claus, V.; Jahraus, A.; Tjelle, T.; Berg, T.; Kirschke, H.; Faulstich, H.; Griffiths, G. Lysosomal enzyme trafficking between phagosomes, endosomes, and lysosomes in J774 macrophages. Enrichment of cathepsin H in early endosomes. J. Biol. Chem. 1998, 273, 9842–9851. [Google Scholar] [CrossRef] [PubMed]
- Trombetta, E.S.; Ebersold, M.; Garrett, W.; Pypaert, M.; Mellman, I. Activation of lysosomal function during dendritic cell maturation. Science 2003, 299, 1400–1403. [Google Scholar] [CrossRef]
- Jancic, C.; Savina, A.; Wasmeier, C.; Tolmachova, T.; El-Benna, J.; Dang, P.M.; Pascolo, S.; Gougerot-Pocidalo, M.A.; Raposo, G.; Seabra, M.C.; et al. Rab27a regulates phagosomal pH and NADPH oxidase recruitment to dendritic cell phagosomes. Nat. Cell Biol. 2007, 9, 367–378. [Google Scholar] [CrossRef]
- Paardekooper, L.M.; Dingjan, I.; Linders, P.T.A.; Staal, A.H.J.; Cristescu, S.M.; Verberk, W.; van den Bogaart, G. Human Monocyte-Derived Dendritic Cells Produce Millimolar Concentrations of ROS in Phagosomes Per Second. Front. Immunol. 2019, 10, 1216. [Google Scholar] [CrossRef] [PubMed]
- Dingjan, I.; Paardekooper, L.M.; Verboogen, D.R.J.; von Mollard, G.F.; Ter Beest, M.; van den Bogaart, G. VAMP8-mediated NOX2 recruitment to endosomes is necessary for antigen release. Eur. J. Cell Biol. 2017, 96, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Matheoud, D.; Moradin, N.; Bellemare-Pelletier, A.; Shio, M.T.; Hong, W.J.; Olivier, M.; Gagnon, E.; Desjardins, M.; Descoteaux, A. Leishmania evades host immunity by inhibiting antigen cross-presentation through direct cleavage of the SNARE VAMP8. Cell Host Microbe 2013, 14, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Baptista, M.A.; Keszei, M.; Oliveira, M.; Sunahara, K.K.; Andersson, J.; Dahlberg, C.I.; Worth, A.J.; Lieden, A.; Kuo, I.C.; Wallin, R.P.; et al. Deletion of Wiskott-Aldrich syndrome protein triggers Rac2 activity and increased cross-presentation by dendritic cells. Nat. Commun. 2016, 7, 12175. [Google Scholar] [CrossRef]
- Ding, Y.; Guo, Z.; Liu, Y.; Li, X.; Zhang, Q.; Xu, X.; Gu, Y.; Zhang, Y.; Zhao, D.; Cao, X. The lectin Siglec-G inhibits dendritic cell cross-presentation by impairing MHC class I-peptide complex formation. Nat. Immunol. 2016, 17, 1167–1175. [Google Scholar] [CrossRef]
- Samie, M.; Cresswell, P. The transcription factor TFEB acts as a molecular switch that regulates exogenous antigen-presentation pathways. Nat. Immunol. 2015, 16, 729–736. [Google Scholar] [CrossRef]
- Accapezzato, D.; Visco, V.; Francavilla, V.; Molette, C.; Donato, T.; Paroli, M.; Mondelli, M.U.; Doria, M.; Torrisi, M.R.; Barnaba, V. Chloroquine enhances human CD8+ T cell responses against soluble antigens in vivo. J. Exp. Med. 2005, 202, 817–828. [Google Scholar] [CrossRef]
- Chatterjee, B.; Smed-Sorensen, A.; Cohn, L.; Chalouni, C.; Vandlen, R.; Lee, B.C.; Widger, J.; Keler, T.; Delamarre, L.; Mellman, I. Internalization and endosomal degradation of receptor-bound antigens regulate the efficiency of cross presentation by human dendritic cells. Blood 2012, 120, 2011–2020. [Google Scholar] [CrossRef]
- Liu, J.; Liu, X.; Han, Y.; Zhang, J.; Liu, D.; Ma, G.; Li, C.; Liu, L.; Kong, D. Nanovaccine Incorporated with Hydroxychloroquine Enhances Antigen Cross-Presentation and Promotes Antitumor Immune Responses. ACS Appl. Mater. Interfaces 2018, 10, 30983–30993. [Google Scholar] [CrossRef]
- Gagnon, E.; Duclos, S.; Rondeau, C.; Chevet, E.; Cameron, P.H.; Steele-Mortimer, O.; Paiement, J.; Bergeron, J.J.; Desjardins, M. Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell 2002, 110, 119–131. [Google Scholar] [CrossRef]
- Houde, M.; Bertholet, S.; Gagnon, E.; Brunet, S.; Goyette, G.; Laplante, A.; Princiotta, M.F.; Thibault, P.; Sacks, D.; Desjardins, M. Phagosomes are competent organelles for antigen cross-presentation. Nature 2003, 425, 402–406. [Google Scholar] [CrossRef]
- Guermonprez, P.; Saveanu, L.; Kleijmeer, M.; Davoust, J.; Van Endert, P.; Amigorena, S. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature 2003, 425, 397–402. [Google Scholar] [CrossRef]
- You, X.; Xu, D.D.; Zhang, D.; Chen, J.; Gao, F.G. PYR-41 and Thalidomide Impair Dendritic Cell Cross-Presentation by Inhibiting Myddosome Formation and Attenuating the Endosomal Recruitments of p97 and Sec61 via NF-kappaB Inactivation. J. Immunol. Res. 2018, 2018, 5070573. [Google Scholar] [CrossRef]
- Romisch, K. A Case for Sec61 Channel Involvement in ERAD. Trends Biochem. Sci. 2017, 42, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Ruggiano, A.; Foresti, O.; Carvalho, P. Quality control: ER-associated degradation: Protein quality control and beyond. J. Cell Biol. 2014, 204, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Molinari, M.; Calanca, V.; Galli, C.; Lucca, P.; Paganetti, P. Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science 2003, 299, 1397–1400. [Google Scholar] [CrossRef] [PubMed]
- Oda, Y.; Hosokawa, N.; Wada, I.; Nagata, K. EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science 2003, 299, 1394–1397. [Google Scholar] [CrossRef]
- Olivari, S.; Galli, C.; Alanen, H.; Ruddock, L.; Molinari, M. A novel stress-induced EDEM variant regulating endoplasmic reticulum-associated glycoprotein degradation. J. Biol. Chem. 2005, 280, 2424–2428. [Google Scholar] [CrossRef]
- Grubb, S.; Guo, L.; Fisher, E.A.; Brodsky, J.L. Protein disulfide isomerases contribute differentially to the endoplasmic reticulum-associated degradation of apolipoprotein B and other substrates. Mol. Biol. Cell 2012, 23, 520–532. [Google Scholar] [CrossRef]
- He, K.; Cunningham, C.N.; Manickam, N.; Liu, M.; Arvan, P.; Tsai, B. PDI reductase acts on Akita mutant proinsulin to initiate retrotranslocation along the Hrd1/Sel1L-p97 axis. Mol. Biol. Cell 2015, 26, 3413–3423. [Google Scholar] [CrossRef]
- Sato, Y.; Kojima, R.; Okumura, M.; Hagiwara, M.; Masui, S.; Maegawa, K.; Saiki, M.; Horibe, T.; Suzuki, M.; Inaba, K. Synergistic cooperation of PDI family members in peroxiredoxin 4-driven oxidative protein folding. Sci. Rep. 2013, 3, 2456. [Google Scholar] [CrossRef] [PubMed]
- Imai, J.; Hasegawa, H.; Maruya, M.; Koyasu, S.; Yahara, I. Exogenous antigens are processed through the endoplasmic reticulum-associated degradation (ERAD) in cross-presentation by dendritic cells. Int. Immunol. 2005, 17, 45–53. [Google Scholar] [CrossRef]
- Brodsky, J.L. Cleaning up: ER-associated degradation to the rescue. Cell 2012, 151, 1163–1167. [Google Scholar] [CrossRef]
- Rose, M.D.; Misra, L.M.; Vogel, J.P. KAR2, a karyogamy gene, is the yeast homolog of the mammalian BiP/GRP78 gene. Cell 1989, 57, 1211–1221. [Google Scholar] [CrossRef]
- Ushioda, R.; Hoseki, J.; Nagata, K. Glycosylation-independent ERAD pathway serves as a backup system under ER stress. Mol. Biol. Cell 2013, 24, 3155–3163. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Hirohashi, Y.; Kutomi, G.; Nakanishi, K.; Kamiguchi, K.; Torigoe, T.; Sato, N. Tumor-produced secreted form of binding of immunoglobulin protein elicits antigen-specific tumor immunity. J. Immunol. 2011, 186, 4325–4330. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Srivastava, P.K. Heat shock proteins: The fountainhead of innate and adaptive immune responses. Cell Stress Chaperones 2000, 5, 443–451. [Google Scholar] [CrossRef]
- Cormier, J.H.; Tamura, T.; Sunryd, J.C.; Hebert, D.N. EDEM1 recognition and delivery of misfolded proteins to the SEL1L-containing ERAD complex. Mol. Cell 2009, 34, 627–633. [Google Scholar] [CrossRef]
- Hosokawa, N.; Wada, I.; Hasegawa, K.; Yorihuzi, T.; Tremblay, L.O.; Herscovics, A.; Nagata, K. A novel ER alpha-mannosidase-like protein accelerates ER-associated degradation. EMBO Rep. 2001, 2, 415–422. [Google Scholar] [CrossRef]
- Helenius, A.; Aebi, M. Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 2004, 73, 1019–1049. [Google Scholar] [CrossRef]
- Yoo, Y.S.; Han, H.G.; Jeon, Y.J. Unfolded Protein Response of the Endoplasmic Reticulum in Tumor Progression and Immunogenicity. Oxidative Med. Cell. Longev. 2017, 2017, 2969271. [Google Scholar] [CrossRef]
- Bernasconi, R.; Pertel, T.; Luban, J.; Molinari, M. A dual task for the Xbp1-responsive OS-9 variants in the mammalian endoplasmic reticulum: Inhibiting secretion of misfolded protein conformers and enhancing their disposal. J. Biol. Chem. 2008, 283, 16446–16454. [Google Scholar] [CrossRef]
- Hosokawa, N.; Kamiya, Y.; Kamiya, D.; Kato, K.; Nagata, K. Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans. J. Biol. Chem. 2009, 284, 17061–17068. [Google Scholar] [CrossRef] [PubMed]
- Stanley, P. Golgi glycosylation. Cold Spring Harb. Perspect. Biol. 2011, 3, a005199. [Google Scholar] [CrossRef] [PubMed]
- Zehner, M.; Chasan, A.I.; Schuette, V.; Embgenbroich, M.; Quast, T.; Kolanus, W.; Burgdorf, S. Mannose receptor polyubiquitination regulates endosomal recruitment of p97 and cytosolic antigen translocation for cross-presentation. Proc. Natl. Acad. Sci. USA 2011, 108, 9933–9938. [Google Scholar] [CrossRef]
- Martinez-Pomares, L. The mannose receptor. J. Leukoc. Biol. 2012, 92, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Kreer, C.; Kuepper, J.M.; Zehner, M.; Quast, T.; Kolanus, W.; Schumak, B.; Burgdorf, S. N-glycosylation converts non-glycoproteins into mannose receptor ligands and reveals antigen-specific T cell responses in vivo. Oncotarget 2017, 8, 6857–6872. [Google Scholar] [CrossRef]
- Okumura, M.; Kadokura, H.; Inaba, K. Structures and functions of protein disulfide isomerase family members involved in proteostasis in the endoplasmic reticulum. Free Radic. Biol. Med. 2015, 83, 314–322. [Google Scholar] [CrossRef]
- Singh, R.; Cresswell, P. Defective cross-presentation of viral antigens in GILT-free mice. Science 2010, 328, 1394–1398. [Google Scholar] [CrossRef]
- Alcock, F.; Swanton, E. Mammalian OS-9 is upregulated in response to endoplasmic reticulum stress and facilitates ubiquitination of misfolded glycoproteins. J. Mol. Biol. 2009, 385, 1032–1042. [Google Scholar] [CrossRef]
- Carvalho, P.; Stanley, A.M.; Rapoport, T.A. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell 2010, 143, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Christianson, J.C.; Shaler, T.A.; Tyler, R.E.; Kopito, R.R. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 2008, 10, 272–282. [Google Scholar] [CrossRef]
- Katsanis, N.; Fisher, E.M. Identification, expression, and chromosomal localization of ubiquitin conjugating enzyme 7 (UBE2G2), a human homologue of the Saccharomyces cerevisiae ubc7 gene. Genomics 1998, 51, 128–131. [Google Scholar] [CrossRef]
- Klemm, E.J.; Spooner, E.; Ploegh, H.L. Dual role of ancient ubiquitous protein 1 (AUP1) in lipid droplet accumulation and endoplasmic reticulum (ER) protein quality control. J. Biol. Chem. 2011, 286, 37602–37614. [Google Scholar] [CrossRef] [PubMed]
- Lilley, B.N.; Ploegh, H.L. Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. USA 2005, 102, 14296–14301. [Google Scholar] [CrossRef] [PubMed]
- Mueller, B.; Klemm, E.J.; Spooner, E.; Claessen, J.H.; Ploegh, H.L. SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc. Natl. Acad. Sci. USA 2008, 105, 12325–12330. [Google Scholar] [CrossRef]
- Schulze, A.; Standera, S.; Buerger, E.; Kikkert, M.; van Voorden, S.; Wiertz, E.; Koning, F.; Kloetzel, P.M.; Seeger, M. The ubiquitin-domain protein HERP forms a complex with components of the endoplasmic reticulum associated degradation pathway. J. Mol. Biol. 2005, 354, 1021–1027. [Google Scholar] [CrossRef]
- Friedlander, R.; Jarosch, E.; Urban, J.; Volkwein, C.; Sommer, T. A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat. Cell Biol. 2000, 2, 379–384. [Google Scholar] [CrossRef]
- Stein, A.; Ruggiano, A.; Carvalho, P.; Rapoport, T.A. Key steps in ERAD of luminal ER proteins reconstituted with purified components. Cell 2014, 158, 1375–1388. [Google Scholar] [CrossRef]
- Neuber, O.; Jarosch, E.; Volkwein, C.; Walter, J.; Sommer, T. Ubx2 links the Cdc48 complex to ER-associated protein degradation. Nat. Cell Biol. 2005, 7, 993–998. [Google Scholar] [CrossRef]
- Schuberth, C.; Buchberger, A. Membrane-bound Ubx2 recruits Cdc48 to ubiquitin ligases and their substrates to ensure efficient ER-associated protein degradation. Nat. Cell Biol. 2005, 7, 999–1006. [Google Scholar] [CrossRef]
- Greenfield, J.J.; High, S. The Sec61 complex is located in both the ER and the ER-Golgi intermediate compartment. J. Cell Sci. 1999, 112 Pt 10, 1477–1486. [Google Scholar]
- Kalies, K.U.; Rapoport, T.A.; Hartmann, E. The beta subunit of the Sec61 complex facilitates cotranslational protein transport and interacts with the signal peptidase during translocation. J. Cell Biol. 1998, 141, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Braun, B.C.; Glickman, M.; Kraft, R.; Dahlmann, B.; Kloetzel, P.M.; Finley, D.; Schmidt, M. The base of the proteasome regulatory particle exhibits chaperone-like activity. Nat. Cell Biol. 1999, 1, 221–226. [Google Scholar] [CrossRef]
- Kaiser, M.L.; Romisch, K. Proteasome 19S RP binding to the Sec61 channel plays a key role in ERAD. PLoS ONE 2015, 10, e0117260. [Google Scholar] [CrossRef]
- Braunstein, I.; Zach, L.; Allan, S.; Kalies, K.U.; Stanhill, A. Proteasomal degradation of preemptive quality control (pQC) substrates is mediated by an AIRAPL-p97 complex. Mol. Biol. Cell 2015, 26, 3719–3727. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Ng, D.T. ERAD substrate recognition in budding yeast. Semin. Cell Dev. Biol. 2010, 21, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Grotzke, J.E.; Kozik, P.; Morel, J.D.; Impens, F.; Pietrosemoli, N.; Cresswell, P.; Amigorena, S.; Demangel, C. Sec61 blockade by mycolactone inhibits antigen cross-presentation independently of endosome-to-cytosol export. Proc. Natl. Acad. Sci. USA 2017, 114, E5910–E5919. [Google Scholar] [CrossRef]
- Ackerman, A.L.; Giodini, A.; Cresswell, P. A role for the endoplasmic reticulum protein retrotranslocation machinery during crosspresentation by dendritic cells. Immunity 2006, 25, 607–617. [Google Scholar] [CrossRef]
- Burgdorf, S.; Leister, P.; Scheidtmann, K.H. TSG101 interacts with apoptosis-antagonizing transcription factor and enhances androgen receptor-mediated transcription by promoting its monoubiquitination. J. Biol. Chem. 2004, 279, 17524–17534. [Google Scholar] [CrossRef] [PubMed]
- Giodini, A.; Cresswell, P. Hsp90-mediated cytosolic refolding of exogenous proteins internalized by dendritic cells. EMBO J. 2008, 27, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Kato, Y.; Kajiwara, C.; Mizukami, S.; Ishige, I.; Ichiyanagi, T.; Hikida, M.; Wang, J.Y.; Udono, H. Heat shock protein 90 (HSP90) contributes to cytosolic translocation of extracellular antigen for cross-presentation by dendritic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16363–16368. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, L.; Ye, Y. Regulation of retrotranslocation by p97-associated deubiquitinating enzyme ataxin-3. J. Cell Biol. 2006, 174, 963–971. [Google Scholar] [CrossRef]
- Zhang, K.; Shen, X.; Wu, J.; Sakaki, K.; Saunders, T.; Rutkowski, D.T.; Back, S.H.; Kaufman, R.J. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 2006, 124, 587–599. [Google Scholar] [CrossRef]
- Xue, X.; Piao, J.H.; Nakajima, A.; Sakon-Komazawa, S.; Kojima, Y.; Mori, K.; Yagita, H.; Okumura, K.; Harding, H.; Nakano, H. Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha. J. Biol. Chem. 2005, 280, 33917–33925. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Chen, X.; Lee, A.H.; Glimcher, L.H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 2010, 11, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Peters, L.R.; Raghavan, M. Endoplasmic reticulum calcium depletion impacts chaperone secretion, innate immunity, and phagocytic uptake of cells. J. Immunol. 2011, 187, 919–931. [Google Scholar] [CrossRef]
- Goodall, J.C.; Wu, C.; Zhang, Y.; McNeill, L.; Ellis, L.; Saudek, V.; Gaston, J.S. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc. Natl. Acad. Sci. USA 2010, 107, 17698–17703. [Google Scholar] [CrossRef]
- Hu, F.; Yu, X.; Wang, H.; Zuo, D.; Guo, C.; Yi, H.; Tirosh, B.; Subjeck, J.R.; Qiu, X.; Wang, X.Y. ER stress and its regulator X-box-binding protein-1 enhance polyIC-induced innate immune response in dendritic cells. Eur. J. Immunol. 2011, 41, 1086–1097. [Google Scholar] [CrossRef]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Et Biophys. Acta 2014, 1843, 2563–2582. [Google Scholar] [CrossRef]
- Kaser, A.; Blumberg, R.S. Paneth cells and inflammation dance together in Crohn’s disease. Cell Res. 2008, 18, 1160–1162. [Google Scholar] [CrossRef] [PubMed]
- Negroni, A.; Prete, E.; Vitali, R.; Cesi, V.; Aloi, M.; Civitelli, F.; Cucchiara, S.; Stronati, L. Endoplasmic reticulum stress and unfolded protein response are involved in paediatric inflammatory bowel disease. Dig. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2014, 46, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Retamal-Diaz, A.; Weiss, K.A.; Tognarelli, E.I.; Freire, M.; Bueno, S.M.; Herold, B.C.; Jacobs, W.R., Jr.; Gonzalez, P.A. US6 Gene Deletion in Herpes Simplex Virus Type 2 Enhances Dendritic Cell Function and T Cell Activation. Front. Immunol. 2017, 8, 1523. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, N.R.; Anufreichik, V.; Rodvold, J.J.; Chiu, K.T.; Sepulveda, H.; Zanetti, M. Cell-extrinsic effects of tumor ER stress imprint myeloid dendritic cells and impair CD8(+) T cell priming. PLoS ONE 2012, 7, e51845. [Google Scholar] [CrossRef]
- Martinon, F.; Glimcher, L.H. Regulation of innate immunity by signaling pathways emerging from the endoplasmic reticulum. Curr. Opin. Immunol. 2011, 23, 35–40. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Nakajima, S.; Kitamura, M. Bidirectional regulation of NF-kappaB by reactive oxygen species: A role of unfolded protein response. Free Radic. Biol. Med. 2013, 65, 162–174. [Google Scholar] [CrossRef]
- Tang, C.H.; Zundell, J.A.; Ranatunga, S.; Lin, C.; Nefedova, Y.; Del Valle, J.R.; Hu, C.C. Agonist-Mediated Activation of STING Induces Apoptosis in Malignant B Cells. Cancer Res. 2016, 76, 2137–2152. [Google Scholar] [CrossRef]
- Smith, J.A.; Turner, M.J.; DeLay, M.L.; Klenk, E.I.; Sowders, D.P.; Colbert, R.A. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-beta induction via X-box binding protein 1. Eur. J. Immunol. 2008, 38, 1194–1203. [Google Scholar] [CrossRef]
- Cho, J.A.; Lee, A.H.; Platzer, B.; Cross, B.C.S.; Gardner, B.M.; De Luca, H.; Luong, P.; Harding, H.P.; Glimcher, L.H.; Walter, P.; et al. The unfolded protein response element IRE1alpha senses bacterial proteins invading the ER to activate RIG-I and innate immune signaling. Cell Host Microbe 2013, 13, 558–569. [Google Scholar] [CrossRef]
- Lencer, W.I.; DeLuca, H.; Grey, M.J.; Cho, J.A. Innate immunity at mucosal surfaces: The IRE1-RIDD-RIG-I pathway. Trends Immunol. 2015, 36, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Mesman, A.W.; Zijlstra-Willems, E.M.; Kaptein, T.M.; de Swart, R.L.; Davis, M.E.; Ludlow, M.; Duprex, W.P.; Gack, M.U.; Gringhuis, S.I.; Geijtenbeek, T.B. Measles virus suppresses RIG-I-like receptor activation in dendritic cells via DC-SIGN-mediated inhibition of PP1 phosphatases. Cell Host Microbe 2014, 16, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Clavarino, G.; Claudio, N.; Dalet, A.; Terawaki, S.; Couderc, T.; Chasson, L.; Ceppi, M.; Schmidt, E.K.; Wenger, T.; Lecuit, M.; et al. Protein phosphatase 1 subunit Ppp1r15a/GADD34 regulates cytokine production in polyinosinic:polycytidylic acid-stimulated dendritic cells. Proc. Natl. Acad. Sci. USA 2012, 109, 3006–3011. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.W.; Cui, D.; Arellano, J.; Dorweiler, B.; Harding, H.; Fitzgerald, K.A.; Ron, D.; Tabas, I. Adaptive suppression of the ATF4-CHOP branch of the unfolded protein response by toll-like receptor signalling. Nat. Cell Biol. 2009, 11, 1473–1480. [Google Scholar] [CrossRef]
- Woo, C.W.; Kutzler, L.; Kimball, S.R.; Tabas, I. Toll-like receptor activation suppresses ER stress factor CHOP and translation inhibition through activation of eIF2B. Nat. Cell Biol. 2012, 14, 192–200. [Google Scholar] [CrossRef]
- Li, H.Y.; Liu, H.; Wang, C.H.; Zhang, J.Y.; Man, J.H.; Gao, Y.F.; Zhang, P.J.; Li, W.H.; Zhao, J.; Pan, X.; et al. Deactivation of the kinase IKK by CUEDC2 through recruitment of the phosphatase PP1. Nat. Immunol. 2008, 9, 533–541. [Google Scholar] [CrossRef]
- Heazlewood, C.K.; Cook, M.C.; Eri, R.; Price, G.R.; Tauro, S.B.; Taupin, D.; Thornton, D.J.; Png, C.W.; Crockford, T.L.; Cornall, R.J.; et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 2008, 5, e54. [Google Scholar] [CrossRef]
- Vattemi, G.; Engel, W.K.; McFerrin, J.; Askanas, V. Endoplasmic reticulum stress and unfolded protein response in inclusion body myositis muscle. Am. J. Pathol. 2004, 164, 1–7. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imai, J.; Otani, M.; Sakai, T. Distinct Subcellular Compartments of Dendritic Cells Used for Cross-Presentation. Int. J. Mol. Sci. 2019, 20, 5606. https://doi.org/10.3390/ijms20225606
Imai J, Otani M, Sakai T. Distinct Subcellular Compartments of Dendritic Cells Used for Cross-Presentation. International Journal of Molecular Sciences. 2019; 20(22):5606. https://doi.org/10.3390/ijms20225606
Chicago/Turabian StyleImai, Jun, Mayu Otani, and Takahiro Sakai. 2019. "Distinct Subcellular Compartments of Dendritic Cells Used for Cross-Presentation" International Journal of Molecular Sciences 20, no. 22: 5606. https://doi.org/10.3390/ijms20225606
APA StyleImai, J., Otani, M., & Sakai, T. (2019). Distinct Subcellular Compartments of Dendritic Cells Used for Cross-Presentation. International Journal of Molecular Sciences, 20(22), 5606. https://doi.org/10.3390/ijms20225606