The Impact of Hypoxia on the Host-Pathogen Interaction between Neutrophils and Staphylococcus aureus

Abstract
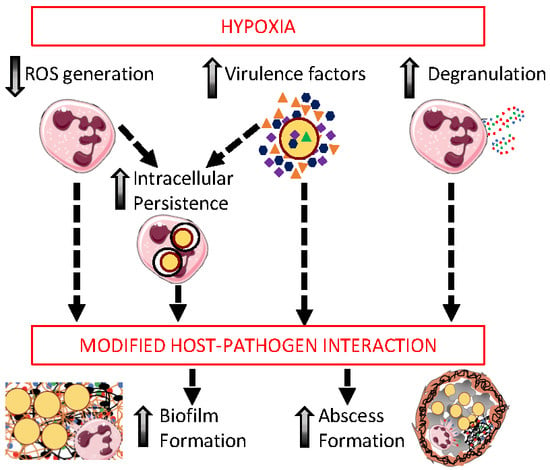
1. Introduction
2. Neutrophil Killing Mechanisms
3. Neutrophil Dysfunction and Staphylococcal Infection
4. Physiological and Pathological Hypoxia
5. Mammalian Oxygen Sensing and Response to Hypoxia
6. S. aureus Responses to Adverse Environmental Conditions
7. The Impact of Hypoxia on Neutrophil Bactericidal Functions
8. The Role of Neutrophils and Hypoxia in Shaping Staphylococcal Infections
9. Staphylococcal Abscess Formation
10. Staphylococcal Biofilms and Infection of Prosthetic Material
11. Conclusions
Funding
Disclosure Statement
Acknowledgments
Conflicts of Interest
References
- Thammavongsa, V.; Kim, H.K.; Missiakas, D.; Schneewind, O. Staphylococcal manipulation of host immune responses. Nat. Rev. Microbiol. 2015, 13, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.Y.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G., Jr. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [PubMed]
- Boldock, E.; Surewaard, B.G.J.; Shamarina, D.; Na, M.; Fei, Y.; Ali, A.; Williams, A.; Pollitt, E.J.G.; Szkuta, P.; Morris, P.; et al. Human skin commensals augment Staphylococcus aureus pathogenesis. Nat. Microbiol. 2018, 3, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.G.; Morris, A.J. Cannula-associated Staphylococcus aureus bacteraemia: Outcome in relation to treatment. Intern. Med. J. 2005, 35, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Galar, A.; Weil, A.A.; Dudzinski, D.M.; Muñoz, P.; Siedner, M.J. Methicillin-Resistant Staphylococcus aureus Prosthetic Valve Endocarditis: Pathophysiology, Epidemiology, Clinical Presentation, Diagnosis, and Management. Clin. Microbiol. Rev. 2019, 32. [Google Scholar] [CrossRef] [PubMed]
- Gardete, S.; Tomasz, A. Mechanisms of vancomycin resistance in Staphylococcus aureus. J. Clin. Investig. 2014, 124, 2836–2840. [Google Scholar] [CrossRef]
- Lu, T.; Porter, A.R.; Kennedy, A.D.; Kobayashi, S.D.; DeLeo, F.R. Phagocytosis and killing of Staphylococcus aureus by human neutrophils. J. Innate Immun. 2014, 6, 639–649. [Google Scholar] [CrossRef]
- Simmen, H.P.; Blaser, J. Analysis of pH and pO2 in abscesses, peritoneal fluid, and drainage fluid in the presence or absence of bacterial infection during and after abdominal surgery. Am. J. Surg. 1993, 166, 24–27. [Google Scholar] [CrossRef]
- Parker, L.C.; Whyte, M.K.; Dower, S.K.; Sabroe, I. The expression and roles of Toll-like receptors in the biology of the human neutrophil. J. Leukoc. Biol. 2005, 77, 886–892. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Berends, E.T.M.; Chan, R.; Schwab, E.; Roy, S.; Sen, C.K.; Torres, V.J.; Wozniak, D.J. Staphylococcus aureus biofilms release leukocidins to elicit extracellular trap formation and evade neutrophil-mediated killing. Proc. Natl. Acad. Sci. USA 2018, 115, 7416–7421. [Google Scholar] [CrossRef]
- Howard, M.W.; Strauss, R.G.; Johnston, R.B., Jr. Infections in patients with neutropenia. Am. J. Dis. Child. 1977, 131, 788–790. [Google Scholar] [CrossRef] [PubMed]
- Buvelot, H.; Posfay-Barbe, K.M.; Linder, P.; Schrenzel, J.; Krause, K.H. Staphylococcus aureus, phagocyte NADPH oxidase and chronic granulomatous disease. FEMS Microbiol. Rev. 2017, 41, 139–157. [Google Scholar] [CrossRef] [PubMed]
- Bortoletto, P.; Lyman, K.; Camacho, A.; Fricchione, M.; Khanolkar, A.; Katz, B.Z. Chronic Granulomatous Disease: A Large, Single-center US Experience. Pediatr. Infect. Dis. J. 2015, 34, 1110–1114. [Google Scholar] [CrossRef]
- Hill, H.R.; Ochs, H.D.; Quie, P.G.; Clark, R.A.; Pabst, H.F.; Klebanoff, S.J.; Wedgwood, R.J. Defect in neutrophil granulocyte chemotaxis in Job’s syndrome of recurrent “cold” staphylococcal abscesses. Lancet 1974, 2, 617–619. [Google Scholar] [CrossRef]
- Parry, M.F.; Root, R.K.; Metcalf, J.A.; Delaney, K.K.; Kaplow, L.S.; Richar, W.J. Myeloperoxidase deficiency: Prevalence and clinical significance. Ann. Intern. Med. 1981, 95, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Strauss, R.G.; Bove, K.E.; Jones, J.F.; Mauer, A.M.; Fulginiti, V.A. An anomaly of neutrophil morphology with impaired function. N. Engl. J. Med. 1974, 290, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Kyme, P.; Thoennissen, N.H.; Tseng, C.W.; Thoennissen, G.B.; Wolf, A.J.; Shimada, K.; Krug, U.O.; Lee, K.; Müller-Tidow, C.; Berdel, W.E.; et al. C/EBPε mediates nicotinamide-enhanced clearance of Staphylococcus aureus in mice. J. Clin. Investig. 2012, 122, 3316–3329. [Google Scholar] [CrossRef]
- Introne, W.; Boissy, R.E.; Gahl, W.A. Clinical, molecular, and cell biological aspects of Chediak-Higashi syndrome. Mol. Genet. Metab. 1999, 68, 283–303. [Google Scholar] [CrossRef]
- Bellinati-Pires, R.; Salgado, M.M.; Joazeiro, P.P.; Carneiro-Sampaio, M.M. Delayed phagocytosis and bacterial killing in Chediak-Higashi syndrome neutrophils detected by a fluorochrome assay. Ultrastructural aspects. Mem. Inst. Oswaldo Cruz. 1992, 87, 575–581. [Google Scholar] [CrossRef]
- Curnutte, J.T.; Whitten, D.M.; Babior, B.M. Defective superoxide production by granulocytes from patients with chronic granulomatous disease. N. Engl. J. Med. 1974, 290, 593–597. [Google Scholar] [CrossRef]
- Winkelstein, J.A.; Marino, M.C.; Johnston, R.B., Jr.; Boyle, J.; Curnutte, J.; Gallin, J.I.; Malech, H.L.; Holland, S.M.; Ochs, H.; Quie, P.; et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine 2000, 79, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Braun, R.D.; Lanzen, J.L.; Snyder, S.A.; Dewhirst, M.W. Comparison of tumor and normal tissue oxygen tension measurements using OxyLite or microelectrodes in rodents. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2533–H2544. [Google Scholar] [CrossRef]
- Campbell, E.L.; Bruyninckx, W.J.; Kelly, C.J.; Glover, L.E.; McNamee, E.N.; Bowers, B.E.; Bayless, A.J.; Scully, M.; Saeedi, B.J.; Golden-Mason, L.; et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity 2014, 40, 66–77. [Google Scholar] [CrossRef]
- Lone, A.G.; Atci, E.; Renslow, R.; Beyenal, H.; Noh, S.; Fransson, B.; Abu-Lail, N.; Park, J.J.; Gang, D.R.; Call, D.R. Staphylococcus aureus induces hypoxia and cellular damage in porcine dermal explants. Infect. Immun. 2015, 83, 2531–2541. [Google Scholar] [CrossRef]
- Werth, N.; Beerlage, C.; Rosenberger, C.; Yazdi, A.S.; Edelmann, M.; Amr, A.; Bernhardt, W.; Von Eiff, C.; Becker, K.; Schafer, A.; et al. Activation of hypoxia inducible Factor 1 is a general phenomenon in infections with human pathogens. PLoS ONE 2010, 5, e11576. [Google Scholar] [CrossRef] [PubMed]
- Koyasu, S.; Kobayashi, M.; Goto, Y.; Hiraoka, M.; Harada, H. Regulatory mechanisms of hypoxia-inducible factor 1 activity: Two decades of knowledge. Cancer Sci. 2018, 109, 560–571. [Google Scholar] [CrossRef] [PubMed]
- Watts, E.R.; Walmsley, S.R. Inflammation and Hypoxia: HIF and PHD Isoform Selectivity. Trends Mol. Med. 2019, 25, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 2002, 295, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Blouin, C.C.; Pagé, E.L.; Soucy, G.M.; Richard, D.E. Hypoxic gene activation by lipopolysaccharide in macrophages: Implication of hypoxia-inducible factor 1alpha. Blood 2004, 103, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Hoenderdos, K.; Lodge, K.M.; A Hirst, R.; Chen, C.; Palazzo, S.G.C.; Emerenciana, A.; Summers, C.; Angyal, A.; Porter, L.; Juss, J.K.; et al. Hypoxia upregulates neutrophil degranulation and potential for tissue injury. Thorax 2016, 71, 1030–1038. [Google Scholar] [CrossRef]
- Guerra, F.E.; Borgogna, T.R.; Patel, D.M.; Sward, E.W.; Voyich, J.M. Epic Immune Battles of History: Neutrophils vs. Staphylococcus aureus. Front. Cell Infect. Microbiol. 2017, 7, 286–305. [Google Scholar] [CrossRef] [PubMed]
- Horn, J.; Stelzner, K.; Rudel, T.; Fraunholz, M. Inside job: Staphylococcus aureus host-pathogen interactions. Int. J. Med. Microbiol. 2018, 308, 607–624. [Google Scholar] [CrossRef] [PubMed]
- Gresham, H.D.; Lowrance, J.H.; Caver, T.E.; Wilson, B.S.; Cheung, A.L.; Lindberg, F.P. Survival of Staphylococcus aureus inside neutrophils contributes to infection. J. Immunol. 2000, 164, 3713–3722. [Google Scholar] [CrossRef] [PubMed]
- Leliefeld, P.H.C.; Pillay, J.; Vrisekoop, N.; Heeres, M.; Tak, T.; Kox, M.; Rooijakkers, S.H.M.; Kuijpers, T.W.; Pickkers, P.; Leenen, L.P.H.; et al. Differential antibacterial control by neutrophil subsets. Blood Adv. 2018, 2, 1344–1355. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, D.; Harper, L.; Shopsin, B.; Torres, V.J. Staphylococcus aureus pathogenesis in diverse host environments. Pathog. Dis. 2017, 75. [Google Scholar] [CrossRef]
- Hall, J.W.; Yang, J.; Guo, H.; Ji, Y. The Staphylococcus aureus AirSR Two-Component System Mediates Reactive Oxygen Species Resistance via Transcriptional Regulation of Staphyloxanthin Production. Infect. Immun. 2017, 85. [Google Scholar] [CrossRef]
- Dunman, P.M.; Murphy, E.; Haney, S.; Palacios, D.; Tucker-Kellogg, G.; Wu, S.; Brown, E.L.; Zagursky, R.J.; Shlaes, D.; Projan, S.J. Transcription profiling-based identification of Staphylococcus aureus genes regulated by the agr and/or sarA loci. J. Bacteriol. 2001, 183, 7341–7353. [Google Scholar] [CrossRef]
- McGovern, N.N.; Cowburn, A.S.; Porter, L.; Walmsley, S.R.; Summers, C.; Thompson, A.A.R.; Anwar, S.; Willcocks, L.C.; Whyte, M.K.B.; Condliffe, A.M.; et al. Hypoxia selectively inhibits respiratory burst activity and killing of Staphylococcus aureus in human neutrophils. J. Immunol. 2011, 186, 453–463. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Datta, V.; Cramer, T.; Doedens, A.; Theodorakis, E.A.; Gallo, R.L.; Hurtado-Ziola, N.; Nizet, V.; Johnson, R.S. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J. Clin. Investig. 2005, 115, 1806–1815. [Google Scholar] [CrossRef]
- Zinkernagel, A.S.; Peyssonnaux, C.; Johnson, R.S.; Nizet, V. Pharmacologic augmentation of hypoxia-inducible factor-1alpha with mimosine boosts the bactericidal capacity of phagocytes. J. Infect. Dis. 2008, 197, 214–217. [Google Scholar] [CrossRef]
- Thompson, A.A.; Dickinson, R.S.; Murphy, F.; Thomson, J.P.; Marriott, H.M.; Tavares, A.; Willson, J.; Williams, L.; Lewis, A.; Mirchandani, A.; et al. Hypoxia determines survival outcomes of bacterial infection through HIF-1alpha dependent re-programming of leukocyte metabolism. Sci. Immunol. 2017, 2, eaal2861. [Google Scholar] [CrossRef] [PubMed]
- Fritzenwanger, M.; Jung, C.; Goebel, B.; Lauten, A.; Figulla, H.R. Impact of short-term systemic hypoxia on phagocytosis, cytokine production, and transcription factor activation in peripheral blood cells. Mediat. Inflamm. 2011, 2011, 429501. [Google Scholar] [CrossRef] [PubMed]
- Reeves, E.P.; Nagl, M.; Godovac-Zimmermann, J.; Segal, A.W. Reassessment of the microbicidal activity of reactive oxygen species and hypochlorous acid with reference to the phagocytic vacuole of the neutrophil granulocyte. J. Med. Microbiol. 2003, 52, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Babior, B.M.; Kipnes, R.S.; Curnutte, J.T. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J. Clin. Investig. 1973, 52, 741–744. [Google Scholar] [CrossRef]
- Eliasson, P.; Jonsson, J.I. The hematopoietic stem cell niche: Low in oxygen but a nice place to be. J. Cell Physiol. 2010, 222, 17–22. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Branitzki-Heinemann, K.; Möllerherm, H.; Völlger, L.; Husein, D.M.; de Buhr, N.; Blodkamp, S.; Reuner, F.; Brogden, G.; Naim, H.Y.; von Köckritz-Blickwede, M. Formation of Neutrophil Extracellular Traps under Low Oxygen Level. Front. Immunol. 2016, 7, 518. [Google Scholar] [CrossRef]
- Lerche, C.J.; Christophersen, L.J.; Kolpen, M.; Nielsen, P.R.; Trøstrup, H.; Thomsen, K.; Hyldegaard, O.; Bundgaard, H.; Jensen, P.Ø.; Høiby, N.; et al. Hyperbaric oxygen therapy augments tobramycin efficacy in experimental Staphylococcus aureus endocarditis. Int. J. Antimicrob. Agents 2017, 50, 406–412. [Google Scholar] [CrossRef]
- Jørgensen, N.P.; Hansen, K.; Andreasen, C.M.; Pedersen, M.; Fuursted, K.; Meyer, R.L.; Petersen, E. Hyperbaric Oxygen Therapy is Ineffective as an Adjuvant to Daptomycin with Rifampicin Treatment in a Murine Model of Staphylococcus aureus in Implant-Associated Osteomyelitis. Microorganisms 2017, 5, 21. [Google Scholar] [CrossRef]
- Sadiku, P.; Walmsley, S.R. Hypoxia and the regulation of myeloid cell metabolic imprinting: Consequences for the inflammatory response. EMBO Rep. 2019, 20, e47388. [Google Scholar] [CrossRef] [PubMed]
- Hannah, S.; Mecklenburgh, K.; Rahman, I.; Bellingan, G.J.; Greening, A.; Haslett, C.; Chilvers, E.R. Hypoxia prolongs neutrophil survival in vitro. FEBS Lett. 1995, 372, 233–237. [Google Scholar] [CrossRef]
- Wilde, A.D.; Snyder, D.J.; Putnam, N.E.; Valentino, M.D.; Hammer, N.D.; Lonergan, Z.R.; Hinger, S.A.; Aysanoa, E.E.; Blanchard, C.; Dunman, P.M.; et al. Bacterial Hypoxic Responses Revealed as Critical Determinants of the Host-Pathogen Outcome by TnSeq Analysis of Staphylococcus aureus Invasive Infection. PLoS Pathog. 2015, 11, e1005341. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, M.; Bastian, M.; Cramton, S.E.; Ziegler, K.; Pragman, A.A.; Bragonzi, A.; Memmi, G.; Wolz, C.; Schlievert, P.M.; Cheung, A.; et al. The staphylococcal respiratory response regulator SrrAB induces ica gene transcription and polysaccharide intercellular adhesin expression, protecting Staphylococcus aureus from neutrophil killing under anaerobic growth conditions. Mol. Microbiol. 2007, 65, 1276–1287. [Google Scholar] [CrossRef]
- Christmas, B.A.F.; Rolfe, M.D.; Rose, M.; Green, J. Staphylococcus aureus adaptation to aerobic low-redox-potential environments: Implications for an intracellular lifestyle. Microbiology 2019, 165, 779–791. [Google Scholar] [CrossRef]
- Kahl, B.C.; Becker, K.; Löffler, B. Clinical Significance and Pathogenesis of Staphylococcal Small Colony Variants in Persistent Infections. Clin. Microbiol. Rev. 2016, 29, 401–427. [Google Scholar] [CrossRef]
- Myles, I.A.; Anderson, E.D.; Earland, N.J.; Zarember, K.A.; Sastalla, I.; Williams, K.W.; Gough, P.; Moore, I.N.; Ganesan, S.; Fowler, C.J.; et al. TNF overproduction impairs epithelial staphylococcal response in hyper IgE syndrome. J. Clin. Investig. 2018, 128, 3595–3604. [Google Scholar] [CrossRef]
- Prajsnar, T.K.; Hamilton, R.; Garcia-Lara, J.; McVicker, G.; Williams, A.; Boots, M.; Foster, S.J.; Renshaw, S.A. A privileged intraphagocyte niche is responsible for disseminated infection of Staphylococcus aureus in a zebrafish model. Cell Microbiol. 2012, 14, 1600–1619. [Google Scholar] [CrossRef]
- Cheng, A.G.; Kim, H.K.; Burts, M.L.; Krausz, T.; Schneewind, O.; Missiakas, D.M. Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J. 2009, 23, 3393–3404. [Google Scholar] [CrossRef]
- Cheng, A.G.; McAdow, M.; Kim, H.K.; Bae, T.; Missiakas, D.M.; Schneewind, O. Contribution of coagulases towards Staphylococcus aureus disease and protective immunity. PLoS Pathog. 2010, 6, e1001036. [Google Scholar] [CrossRef]
- Beerlage, C.; Greb, J.; Kretschmer, D.; Assaggaf, M.; Trackman, P.C.; Hansmann, M.L.; Bonin, M.; Eble, J.A.; Peschel, A.; Brüne, B.; et al. Hypoxia-inducible factor 1-regulated lysyl oxidase is involved in Staphylococcus aureus abscess formation. Infect. Immun. 2013, 81, 2562–2573. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chan, P.F.; Foster, S.J. Role of SarA in virulence determinant production and environmental signal transduction in Staphylococcus aureus. J. Bacteriol. 1998, 180, 6232–6241. [Google Scholar] [PubMed]
- Wu, Y.; Klapper, I.; Stewart, P.S. Hypoxia arising from concerted oxygen consumption by neutrophils and microorganisms in biofilms. Pathog. Dis. 2018, 76. [Google Scholar] [CrossRef] [PubMed]
- Mashruwala, A.A.; Guchte, A.V.; Boyd, J.M. Impaired respiration elicits SrrAB-dependent programmed cell lysis and biofilm formation in Staphylococcus aureus. Elife 2017, 6, e23845. [Google Scholar] [CrossRef] [PubMed]
- Pabst, B.; Pitts, B.; Lauchnor, E.; Stewart, P.S. Gel-Entrapped Staphylococcus aureus Bacteria as Models of Biofilm Infection Exhibit Growth in Dense Aggregates, Oxygen Limitation, Antibiotic Tolerance, and Heterogeneous Gene Expression. Antimicrob. Agents Chemother. 2016, 60, 6294–6301. [Google Scholar] [CrossRef] [PubMed]
- Günther, F.; Wabnitz, G.H.; Stroh, P.; Prior, B.; Obst, U.; Samstag, Y.; Wagner, C.; Hänsch, G.M. Host defence against Staphylococcus aureus biofilms infection: Phagocytosis of biofilms by polymorphonuclear neutrophils (PMN). Mol. Immunol. 2009, 46, 1805–1813. [Google Scholar] [CrossRef]
- Gutierrez Jauregui, R.; Fleige, H.; Bubke, A.; Rohde, M.; Weiss, S.; Förster, R. IL-1β Promotes Staphylococcus aureus Biofilms on Implants in vivo. Front. Immunol. 2019, 10, 1082. [Google Scholar] [CrossRef]
- Josse, J.; Valour, F.; Maali, Y.; Diot, A.; Batailler, C.; Ferry, T.; Laurent, F. Interaction Between Staphylococcal Biofilm and Bone: How Does the Presence of Biofilm Promote Prosthesis Loosening? Front. Microbiol. 2019, 10, 1602. [Google Scholar] [CrossRef]




{kind=link}
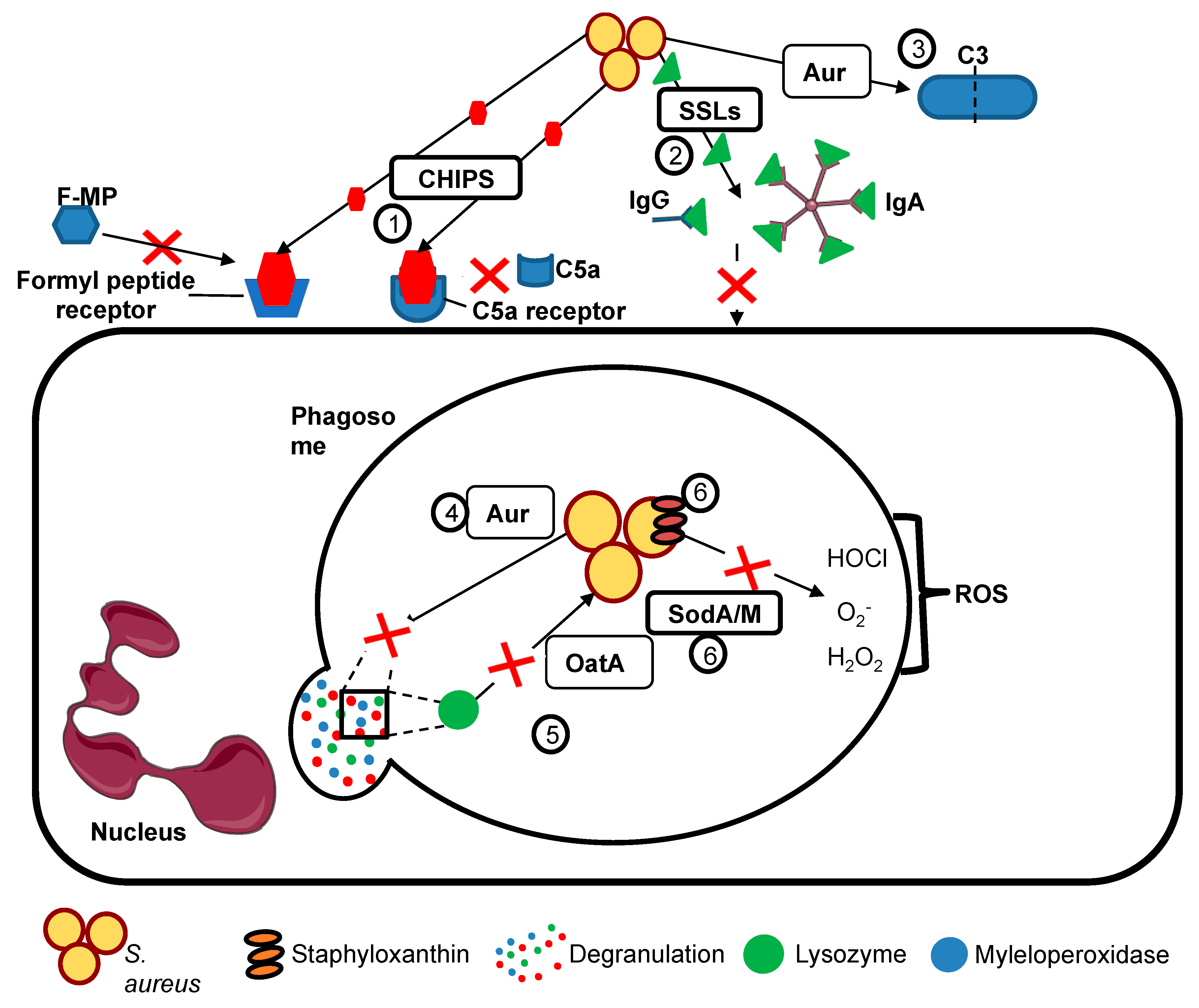{kind=link}
{kind=link}
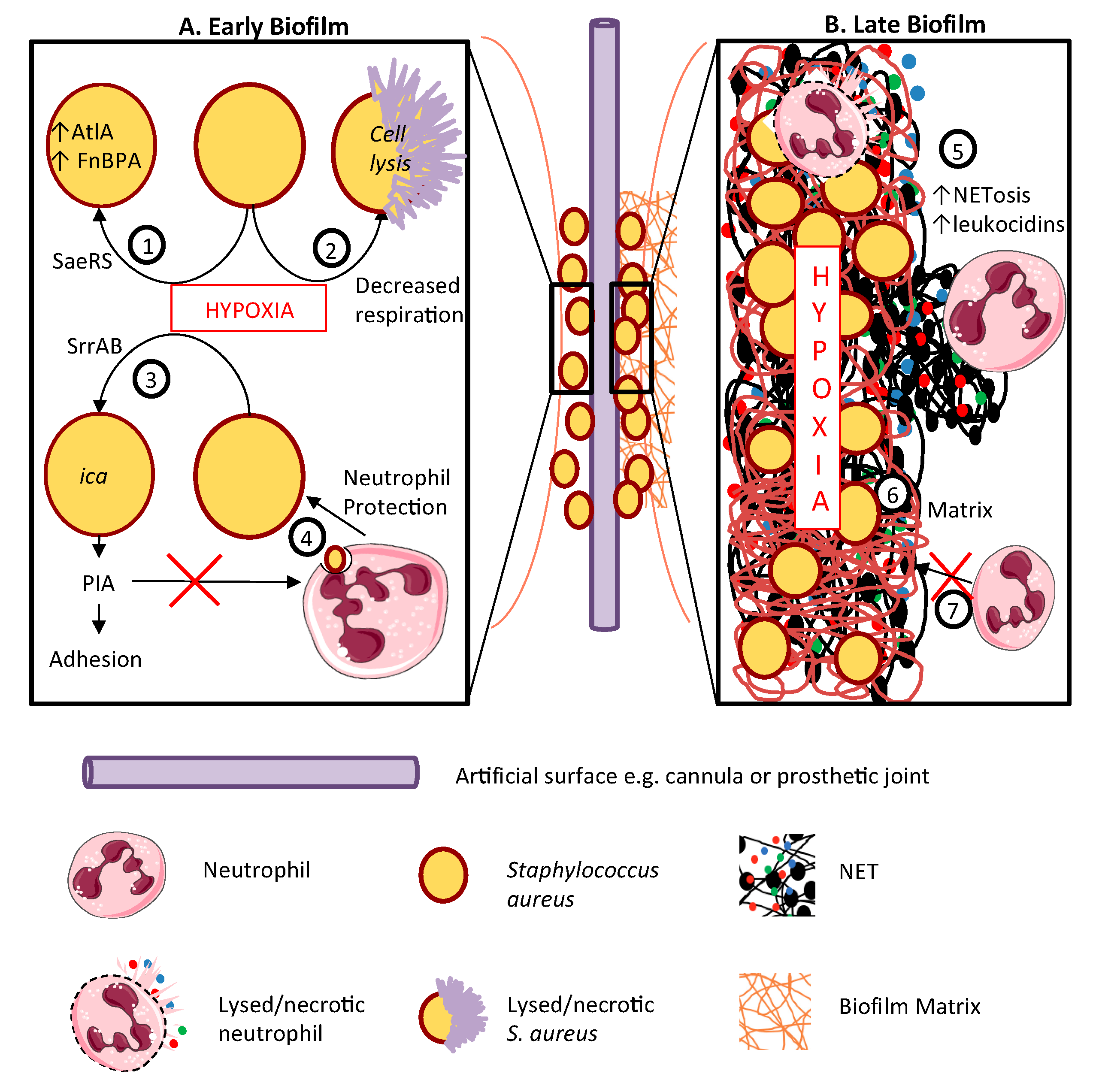{kind=link}
| Group | Virulence Factor | Mechanism |
|---|---|---|
| Prevention of phagocyte recognition by opsonisation and hence reduction of phagocytosis | Protein A (SpA) | Cross links Fab domain of IgM and binds Fcγ domain of immunoglobulin G. |
| Clumping factor A (ClfA) | Fibrinogen-binding surface protein causing platelet aggregation. Antiphagocytic effect with or without presence of fibrinogen. | |
| Staphylococcal complement inhibitor (SCIN) | Inhibits C3 complement convertase by preventing the C3b generation. | |
| Aureolysin | Anti-protease blocks C3 complement activity through cleaving C3 blocking C3a activation of neutrophils. Also cleaves granule-derived antimicrobial peptides. | |
| Induction of phagocyte damage and death | Panton-Valentine leukocidin (PVL) and other leukocidins such as gamma-haemolysin and LukED | Triggers apoptosis and necrosis of cells, initiated by pore formation. |
| Phenol-soluble modulins (PSMs) | Cause lysis of blood cells, assists in the structuring and dispersal of biofilms. | |
| Alpha-haemolysin (alpha toxin) | Forms pores in cells through its interaction with the ADAM10 receptor, resulting in cell lysis. | |
| Prevention of neutrophil chemotaxis and recruitment to sites of staphylococcal infection | Chemotaxsis inhibitory protein (CHIPs) | Blocks chemotaxis towards C5a and formylated peptides by binding to neutrophil C5a receptors formyl peptide receptors, preventing neutrophil recruitment to sites of staphylococcal infection. |
| Extracellular adherence protein (Eap) | Blocks complement activation and neutrophil adhesion to activated endothelium inhibiting neutrophil recruitment; suppresses NETosis. | |
| Staphylococcal superantigen like (SSLs) | A group of structurally similar antigens with functions including binding IgA, IgG, matrix metalloproteinases amd neutrophil adhesion molecules, which act together to inhibit neutrophil recruitment to staphylococcal infection. | |
| Evasion of phagocyte killing | OatA | Catalysing the O-acetylation of peptidoglycan in the Staphylococcal cell wall, rendering it insensitive to lysozyme (which is secreted by phagocytes and constitutively present in secretions such as tears). |
| SodA/M | Superoxide dismutases provide resistance to reactive oxygen species (ROS) produced by neutrophils including superoxide. | |
| Staphyloxanthin | A carotenoid which provides protection against oxidative stress and ROS | |
| Phenol-soluble modulins (PSMs) | Cause cell lysis, aid in biofilm development and stimulate inflammation. |
| Disease | Defect | PMN Dysfunction | Clinical Outcomes |
|---|---|---|---|
| Neutropenia | Decreased PMN numbers, either congenital (e.g., elastase deficiency) or acquired (most commonly drug-induced such as cancer chemotherapy). | Insufficient PMN numbers to respond to invading pathogens, life-threatening Gram-negative and Gram-positive infections. | Life-threatening infections during periods of neutropenia, susceptibility reduced when neutrophil count recovers. |
| Chronic granulomatous disease (CGD) | Mutations in NADPH oxidase components; reduced or absent ROS formation. | Reduced killing of certain pathogens e.g., Staphylococcus aureus, Aspergillus fumigatus, Gram- negative bacilli. | Life-threatening infections with Staphylococcus and Apergillus; aberrant healing (granulomas). |
| Hyper IgE Syndrome (formerly Job’s Syndrome) | Mutations in STAT3 (signal transducer and activator of transcription 3) or DOCK 8 (Dedicator of cytokinesis 8) or TYK2 leading to impaired T cell function and diminished neutrophil chemotaxis | Reduced killing of certain pathogens e.g., Staphylococcus aureus, Aspergillus fumigatus. | Staphylococcal and fungal skin infections, pulmonary and joint infections, ‘cold’ abscess formation (reduced cytokine release). |
| Myeloperoxidase deficiency | Decreased or lack of MPO/HOCl system required to generate the full range of ROS. | Increased chronic conditions mediated by adaptive immunity, decreased NET killing of microbes. | Susceptibility to chronic infections caused by Candida albicans, S. aureus. |
| SGD (Specific Granule Deficiency) | Absence of specific granules, bilobed neutrophils nuclei. Altered content of other granule populations. | Impaired chemotaxis, aberrant granule organisation, reduced respiratory burst, and deficient bactericidal activity (mainly to S. aureus). | Staphylococcal skin infections, aberrant skin lesion healing. |
| Chediak Higashi Syndrome | Mutations in lysosomal trafficking regulator (LYST) leading to failure of lysosomal trafficking in neutrophils and other cells | Giant granules, impaired phagocytosis and phagosomal maturation, oxidative burst and degranulation | Albinism, neurological defects, coagulopathy, recurrent skin (staphylococcal) infections and respiratory infection |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hajdamowicz, N.H.; Hull, R.C.; Foster, S.J.; Condliffe, A.M. The Impact of Hypoxia on the Host-Pathogen Interaction between Neutrophils and Staphylococcus aureus. Int. J. Mol. Sci. 2019, 20, 5561. https://doi.org/10.3390/ijms20225561
Hajdamowicz NH, Hull RC, Foster SJ, Condliffe AM. The Impact of Hypoxia on the Host-Pathogen Interaction between Neutrophils and Staphylococcus aureus. International Journal of Molecular Sciences. 2019; 20(22):5561. https://doi.org/10.3390/ijms20225561
Chicago/Turabian StyleHajdamowicz, Natalia H, Rebecca C Hull, Simon J Foster, and Alison M Condliffe. 2019. "The Impact of Hypoxia on the Host-Pathogen Interaction between Neutrophils and Staphylococcus aureus" International Journal of Molecular Sciences 20, no. 22: 5561. https://doi.org/10.3390/ijms20225561
APA StyleHajdamowicz, N. H., Hull, R. C., Foster, S. J., & Condliffe, A. M. (2019). The Impact of Hypoxia on the Host-Pathogen Interaction between Neutrophils and Staphylococcus aureus. International Journal of Molecular Sciences, 20(22), 5561. https://doi.org/10.3390/ijms20225561