Invariant NKT Cells and Rheumatic Disease: Focus on Primary Sjogren Syndrome


 , and
, and 
{kind=link}
Abstract
1. Primary Sjogren Syndrome
2. Invariant NKT Cells
3. iNKT and Their Controversial Role in Rheumatic Disease
3.1. iNKT in SLE
3.2. iNKT in RA
3.3. iNKT in SpA
3.4. iNKT in Systemic Sclerosis (SSc)
4. iNKT in Sjogren Syndrome
5. Conclusions and Future Perspectives
Conflicts of Interest
References
- Mariette, X.; Criswell, L.A. Primary Sjögren’s syndrome. N. Engl. J. Med. 2018, 378, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Rischmueller, M.; Tieu, J.; Lester, S. Primary Sjögren’s syndrome. Best Pract. Res. Clin. Rheumatol. 2016, 30, 189–220. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, S.J.; Brun, J.G.; Gøransson, L.G.; Småstuen, M.C.; Johannesen, T.B.; Haldorsen, K.; Harboe, E.; Jonsson, R.; Meyer, P.A.; Omdal, R.J. Risk of non-Hodgkin’s lymphoma in primary Sjögren’s syndrome: A population-based study. Arthritis Care Res. 2013, 65, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Carrasco, M.; Ramos-Casals, M.; Rosas, J.; Pallares, L.; Calvo-Alen, J.; Cervera, R.; Font, J.; Ingelmo, M.J.M. Primary Sjögren syndrome: Clinical and immunologic disease patterns in a cohort of 400 patients. Medicine 2002, 81, 270–280. [Google Scholar] [CrossRef]
- Tobon, G.J.; Saraux, A.; Gottenberg, J.E.; Quartuccio, L.; Fabris, M.; Seror, R.; Devauchelle-Pensec, V.; Morel, J.; Rist, S.; Mariette, X.; et al. Role of Fms-like tyrosine kinase 3 ligand as a potential biologic marker of lymphoma in primary Sjogren’s syndrome. Arthritis Rheum. 2013, 65, 3218–3227. [Google Scholar] [CrossRef]
- Kapsogeorgou, E.K.; Papageorgiou, A.; Protogerou, A.D.; Voulgarelis, M.; Tzioufas, A.G. Low miR200b-5p levels in minor salivary glands: A novel molecular marker predicting lymphoma development in patients with Sjogren’s syndrome. Ann. Rheum. Dis. 2018, 77, 1200–1207. [Google Scholar] [CrossRef]
- Patel, R.; Shahane, A. The epidemiology of Sjogren’s syndrome. Clin. Epidemiol. 2014, 6, 247–255. [Google Scholar]
- Voulgarelis, M.; Tzioufas, A.G. Pathogenetic mechanisms in the initiation and perpetuation of Sjogren’s syndrome. Nat. Rev. Rheum. 2010, 6, 529–537. [Google Scholar] [CrossRef]
- Bunya, V.Y.; Ying, G.S.; Maguire, M.G.; Kuklinski, E.; Lin, M.C.; Peskin, E.; Asbell, P.A. Prevalence of novel candidate sjogren syndrome autoantibodies in the dry eye assessment and management (DREAM) study. Cornea 2018, 37, 1425–1430. [Google Scholar] [CrossRef]
- Birnbaum, J.; Hoke, A.; Lalji, A.; Calabresi, P.; Bhargava, P.; Casciola-Rosen, L. Brief report: Anti-Calponin 3 autoantibodies: A newly identified specificity in patients with Sjogren’s syndrome. Arthritis Rheum. 2018, 70, 1610–1616. [Google Scholar] [CrossRef]
- Chen, W.; Cao, H.; Lin, J.; Olsen, N.; Zheng, S.G. Biomarkers for primary Sjogren’s syndrome. Genom. Proteom. Bioinf. 2015, 13, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Maria, N.I.; Brkic, Z.; Waris, M.; van Helden-Meeuwsen, C.G.; Heezen, K.; van de Merwe, J.P.; van Daele, P.L.; Dalm, V.A.; Drexhage, H.A.; Versnel, M.A. MxA as a clinically applicable biomarker for identifying systemic interferon type I in primary Sjogren’s syndrome. Ann. Rheum. Dis. 2014, 73, 1052–1059. [Google Scholar] [CrossRef] [PubMed]
- Hjelmervik, T.O.; Petersen, K.; Jonassen, I.; Jonsson, R.; Bolstad, A.I. Gene expression profiling of minor salivary glands clearly distinguishes primary Sjogren’s syndrome patients from healthy control subjects. Arthritis Rheum. 2005, 52, 1534–1544. [Google Scholar] [CrossRef] [PubMed]
- Nocturne, G.; Seror, R.; Fogel, O.; Belkhir, R.; Boudaoud, S.; Saraux, A.; Larroche, C.; Le Guern, V.; Gottenberg, J.E.; Mariette, X. CXCL13 and CCL11 serum levels and lymphoma and disease activity in primary Sjogren’s syndrome. Arthritis Rheumatol. 2015, 67, 3226–3233. [Google Scholar] [CrossRef]
- Versura, P.; Giannaccare, G.; Vukatana, G.; Mule, R.; Malavolta, N.; Campos, E.C. Predictive role of tear protein expression in the early diagnosis of Sjogren’s syndrome. Ann. Clin. Biochem 2018, 55, 561–570. [Google Scholar] [CrossRef]
- Jazzar, A.A.; Shirlaw, P.J.; Carpenter, G.H.; Challacombe, S.J.; Proctor, G.B. Salivary S100A8/A9 in Sjogren’s syndrome accompanied by lymphoma. J. Oral Pathol. Med. 2018, 47, 900–906. [Google Scholar] [CrossRef]
- Baldini, C.; Zabotti, A.; Filipovic, N.; Vukicevic, A.; Luciano, N.; Ferro, F.; Lorenzon, M.; De Vita, S. Imaging in primary Sjogren’s syndrome: The ‘obsolete and the new’. Clin. Exp. Rheumatol. 2018, 36, 215–221. [Google Scholar]
- Kiripolsky, J.; McCabe, L.G.; Kramer, J.M. Innate immunity in Sjogren’s syndrome. Clin. Immunol. 2017, 182, 4–13. [Google Scholar] [CrossRef]
- Bendelac, A.; Savage, P.B.; Teyton, L. The biology of NKT cells. Annu. Rev. Immunol. 2007, 25, 297–336. [Google Scholar] [CrossRef]
- Drennan, M.B.; Aspeslagh, S.; Elewaut, D. Invariant natural killer T cells in rheumatic disease: A joint dilemma. Nat. Rev. Rheumatol. 2010, 6, 90. [Google Scholar] [CrossRef]
- Bendelac, A. Positive selection of mouse NK1+ T cells by CD1-expressing cortical thymocytes. J. Exp. Med. 1995, 182, 2091–2096. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, D.I.; MacDonald, H.R.; Kronenberg, M.; Smyth, M.J.; Van Kaer, L. NKT cells: What’s in a name? Nat. Rev. Immunol. 2004, 4, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, J.L.; Naidenko, O.V.; Gapin, L.; Nakayama, T.; Taniguchi, M.; Wang, C.R.; Koezuka, Y.; Kronenberg, M. Tracking the response of natural killer T cells to a glycolipid antigen using CD1d tetramers. J. Exp. Med. 2000, 192, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Kojo, S.; Adachi, Y.; Keino, H.; Taniguchi, M.; Sumida, T.J.A. Dysfunction of T cell receptor AV24AJ18+, BV11+ double-negative regulatory natural killer T cells in autoimmune diseases. Arthritis Rheum. 2001, 44, 1127–1138. [Google Scholar] [CrossRef]
- Moody, D.B.; Reinhold, B.B.; Guy, M.R.; Beckman, E.M.; Frederique, D.E.; Furlong, S.T.; Ye, S.; Reinhold, V.N.; Sieling, P.A.; Modlin, R.L.J.S. Structural requirements for glycolipid antigen recognition by CD1b-restricted T cells. Science 1997, 278, 283–286. [Google Scholar] [CrossRef]
- Godfrey, D.I.; Kronenberg, M.J.T. Going both ways: Immune regulation via CD1d-dependent NKT cells. J. Clin. Invest. 2004, 114, 1379–1388. [Google Scholar] [CrossRef]
- Kronenberg, M.; Gapin, L.J. The unconventional lifestyle of NKT cells. Nat. Rev. Immunol. 2002, 2, 557. [Google Scholar] [CrossRef]
- Yang, J.Q.; Wen, X.; Kim, P.J.; Singh, R.R. Invariant NKT cells inhibit autoreactive B cells in a contact- and CD1d-dependent manner. J. Immunol. 2011, 186, 1512–1520. [Google Scholar] [CrossRef]
- Wu, D.; Xing, G.W.; Poles, M.A.; Horowitz, A.; Kinjo, Y.; Sullivan, B.; Bodmer-Narkevitch, V.; Plettenburg, O.; Kronenberg, M.; Tsuji, M.; et al. Bacterial glycolipids and analogs as antigens for CD1d-restricted NKT cells. Proc. Natl. Acad. Sci. USA 2005, 102, 1351–1356. [Google Scholar] [CrossRef]
- Brigl, M.; Bry, L.; Kent, S.C.; Gumperz, J.E.; Brenner, M.B. Mechanism of CD1d-restricted natural killer T cell activation during microbial infection. Nat. Immunol. 2003, 4, 1230–1237. [Google Scholar] [CrossRef]
- Mattner, J.; Debord, K.L.; Ismail, N.; Goff, R.D.; Cantu, C., 3rd; Zhou, D.; Saint-Mezard, P.; Wang, V.; Gao, Y.; Yin, N.; et al. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature 2005, 434, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Selmi, C.; Balkwill, D.L.; Invernizzi, P.; Ansari, A.A.; Coppel, R.L.; Podda, M.; Leung, P.S.; Kenny, T.P.; Van De Water, J.; Nantz, M.H.; et al. Patients with primary biliary cirrhosis react against a ubiquitous xenobiotic-metabolizing bacterium. Hepatology 2003, 38, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Torina, A.; Guggino, G.; La Manna, M.; Sireci, G.J.I. The janus face of NKT cell function in autoimmunity and infectious diseases. Int. J. Mol. Sci. 2018, 19, 440. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, A.; Wahren-Herlenius, M.J. Update on the immunobiology of Sjögren’s syndrome. Curr. Opin. Rheumatol. 2015, 27, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Kennell, A.S.; Larche, M.J.; Seifert, M.H.; Isenberg, D.A.; Salaman, M.R. Natural killer T cells in families of patients with systemic lupus erythematosus: Their possible role in regulation of IGG production. Arthritis Rheum. 2007, 56, 303–310. [Google Scholar] [CrossRef]
- Yang, J.-Q.; Singh, A.K.; Wilson, M.T.; Satoh, M.; Stanic, A.K.; Park, J.-J.; Hong, S.; Gadola, S.D.; Mizutani, A.; Kakumanu, S.R. Immunoregulatory role of CD1d in the hydrocarbon oil-induced model of lupus nephritis. J. Immunol. 2003, 171, 2142–2153. [Google Scholar] [CrossRef]
- Chan, O.T.; Paliwal, V.; McNiff, J.M.; Park, S.-H.; Bendelac, A.; Shlomchik, M.J. Deficiency in β2-microglobulin, but not CD1, accelerates spontaneous lupus skin disease while inhibiting nephritis in MRL-Faslpr mice: An example of disease regulation at the organ level. J. Immunol. 2001, 167, 2985–2990. [Google Scholar] [CrossRef]
- Zeng, D.; Liu, Y.; Sidobre, S.; Kronenberg, M.; Strober, S. Activation of natural killer T cells in NZB/W mice induces Th1-type immune responses exacerbating lupus. J. Clin. Invest. 2003, 112, 1211–1222. [Google Scholar] [CrossRef]
- Forestier, C.; Molano, A.; Im, J.S.; Dutronc, Y.; Diamond, B.; Davidson, A.; Illarionov, P.A.; Besra, G.S.; Porcelli, S.A. Expansion and hyperactivity of CD1d-restricted NKT cells during the progression of systemic lupus erythematosus in (New Zealand Black × New Zealand White) F1 mice. J. Immunol. 2005, 175, 763–770. [Google Scholar] [CrossRef]
- Tsukamoto, K.; Ohtsuji, M.; Shiroiwa, W.; Lin, Q.; Nakamura, K.; Tsurui, H.; Jiang, Y.; Sudo, K.; Nishimura, H.; Shirai, T. Aberrant genetic control of invariant TCR-bearing NKT cell function in New Zealand mouse strains: Possible involvement in systemic lupus erythematosus pathogenesis. J. Immunol. 2008, 180, 4530–4539. [Google Scholar] [CrossRef]
- Postól, E.; Meyer, A.; Cardillo, F.; De Alencar, R.; Pessina, D.; Nihei, J.; Mariano, M.; Mengel, J. Long-term administration of IgG2a anti-NK1. 1 monoclonal antibody ameliorates lupus-like disease in NZB/W mice in spite of an early worsening induced by an IgG2a-dependent BAFF/BLyS production. Immunology 2008, 125, 184–196. [Google Scholar]
- Singh, A.K.; Yang, J.Q.; Parekh, V.V.; Wei, J.; Wang, C.R.; Joyce, S.; Singh, R.R.; Van Kaer, L. The natural killer T cell ligand α-galactosylceramide prevents or promotes pristane-induced lupus in mice. Eur. J. Immunol. 2005, 35, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-Q.; Kim, P.J.; Singh, R.R. Brief treatment with iNKT cell ligand α-galactosylceramide confers a long-term protection against lupus. J. Clin. Immunol. 2012, 32, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Van Der Vliet, H.J.; Von Blomberg, B.M.E.; Nishi, N.; Reijm, M.; Voskuyl, A.E.; Van Bodegraven, A.A.; Polman, C.H.; Rustemeyer, T.; Lips, P.; Van Den Eertwegh, A. Circulating Vα24+ Vβ11+ NKT cell numbers are decreased in a wide variety of diseases that are characterized by autoreactive tissue damage. Clin. Immunol. 2001, 100, 144–148. [Google Scholar] [CrossRef]
- Gutowska-Owsiak, D.; Birchall, M.A.; Moots, R.J.; Christmas, S.E.; Pazmany, L. Proliferatory defect of invariant population and accumulation of non-invariant CD1d-restricted natural killer T cells in the joints of RA patients. Mod. Rheumatol. 2014, 24, 434–442. [Google Scholar] [CrossRef]
- Tang, X.; Zhang, B.; Jarrell, J.A.; Price, J.V.; Dai, H.; Utz, P.J.; Strober, S. Ly108 expression distinguishes subsets of invariant NKT cells that help autoantibody production and secrete IL-21 from those that secrete IL-17 in lupus prone NZB/W mice. J. Autoimmun. 2014, 50, 87–98. [Google Scholar] [CrossRef]
- Yoshiga, Y.; Goto, D.; Segawa, S.; Ohnishi, Y.; Matsumoto, I.; Ito, S.; Tsutsumi, A.; Taniguchi, M.; Sumida, T. [Corrigendum] Invariant NKT cells produce IL-17 through IL-23-dependent and-independent pathways with potential modulation of Th17 response in collagen-induced arthritis. Int. J. Mol. Med. 2013, 31, 998. [Google Scholar]
- Chiba, A.; Oki, S.; Miyamoto, K.; Hashimoto, H.; Yamamura, T.; Miyake, S.J.A. Suppression of collagen-induced arthritis by natural killer T cell activation with OCH, a sphingosine-truncated analog of α-galactosylceramide. Arthritis Rheum. 2004, 50, 305–313. [Google Scholar] [CrossRef]
- Takahashi, T.; Chiba, S.; Nieda, M.; Azuma, T.; Ishihara, S.; Shibata, Y.; Juji, T.; Hirai, H. Cutting edge: Analysis of human Vα24+ CD8+ NK T cells activated by α-galactosylceramide-pulsed monocyte-derived dendritic cells. J. Immunol. 2002, 168, 3140–3144. [Google Scholar] [CrossRef]
- Chiba, A.; Kaieda, S.; Oki, S.; Yamamura, T.; Miyake, S. Rheumatism, The involvement of Vα14 natural killer T cells in the pathogenesis of arthritis in murine models. Arthritis Rheum. 2005, 52, 1941–1948. [Google Scholar] [CrossRef]
- Kim, H.Y.; Kim, S.; Chung, D.H. FcγRIII engagement provides activating signals to NKT cells in antibody-induced joint inflammation. J. Clin. Invest. 2006, 116, 2484–2492. [Google Scholar] [PubMed]
- Li, X.; Shiratsuchi, T.; Chen, G.; Dellabona, P.; Casorati, G.; Franck, R.W.; Tsuji, M. Invariant TCR rather than CD1d shapes the preferential activities of C-glycoside analogues against human versus murine invariant NKT cells. J. Immunol. 2009, 183, 4415–4421. [Google Scholar] [CrossRef] [PubMed]
- Brossay, L.; Chioda, M.; Burdin, N.; Koezuka, Y.; Casorati, G.; Dellabona, P.; Kronenberg, M. CD1d-mediated recognition of an α-galactosylceramide by natural killer T cells is highly conserved through mammalian evolution. J. Exp. Med. 1998, 188, 1521–1528. [Google Scholar] [CrossRef]
- Grose, R.H.; Thompson, F.M.; Baxter, A.G.; Pellicci, D.G.; Cummins, A.G. Deficiency of invariant NK T cells in Crohn’s disease and ulcerative colitis. Dig. Dis. Sci. 2007, 52, 1415–1422. [Google Scholar] [CrossRef]
- Shibolet, O.; Kalish, Y.; Klein, A.; Alper, R.; Zolotarov, L.; Thalenfeld, B.; Engelhardt, D.; Rabbani, E.; Ilan, Y. Adoptive transfer of ex vivo immune-programmed NKT lymphocytes alleviates immune-mediated colitis. J. Leukoc. Biol. 2004, 75, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, F.; Rizzo, A.; Triolo, G. Subclinical gut inflammation in ankylosing spondylitis. Curr. Opin. Rheumatol. 2016, 28, 89–96. [Google Scholar] [CrossRef]
- Rizzo, A.; Ferrante, A.; Guggino, G.; Ciccia, F. Gut inflammation in spondyloarthritis. Best Pract Res. Clin. Rheumatol. 2017, 31, 863–876. [Google Scholar] [CrossRef]
- Jacques, P.; Venken, K.; Van Beneden, K.; Hammad, H.; Seeuws, S.; Drennan, M.B.; Deforce, D.; Verbruggen, G.; Apostolaki, M.; Kollias, G.; et al. Invariant natural killer T cells are natural regulators of murine spondylarthritis. Arthritis Rheum. 2010, 62, 988–999. [Google Scholar] [CrossRef]
- Wingender, G.; Stepniak, D.; Krebs, P.; Lin, L.; McBride, S.; Wei, B.; Braun, J.; Mazmanian, S.K.; Kronenberg, M. Intestinal microbes affect phenotypes and functions of invariant natural killer T cells in mice. Gastroenterology 2012, 143, 418–428. [Google Scholar] [CrossRef]
- Olszak, T.; An, D.; Zeissig, S.; Vera, M.P.; Richter, J.; Franke, A.; Glickman, J.N.; Siebert, R.; Baron, R.M.; Kasper, D.L.; et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science 2012, 336, 489–493. [Google Scholar] [CrossRef]
- Ferro, F.; Marcucci, E.; Orlandi, M.; Baldini, C.; Bartoloni-Bocci, E. One year in review 2017: Primary Sjögren’s syndrome. Clin. Exp. Rheumatol. 2017, 35, 179–191. [Google Scholar] [PubMed]
- Riccieri, V.; Parisi, G.; Spadaro, A.; Scrivo, R.; Barone, F.; Moretti, T.; Bernardini, G.; Strom, R.; Taccari, E.; Valesini, G. Reduced circulating natural killer T cells and gamma/delta T cells in patients with systemic sclerosis. J. Rheumatol. 2005, 32, 283–286. [Google Scholar] [PubMed]
- Mekinian, A.; Mahevas, T.; Mohty, M.; Jachiet, V.; Riviere, S.; Fain, O.; Gaugler, B. Mucosal-associated invariant cells are deficient in systemic sclerosis. Scand. J. Immunol. 2017, 86, 216–220. [Google Scholar] [CrossRef] [PubMed]
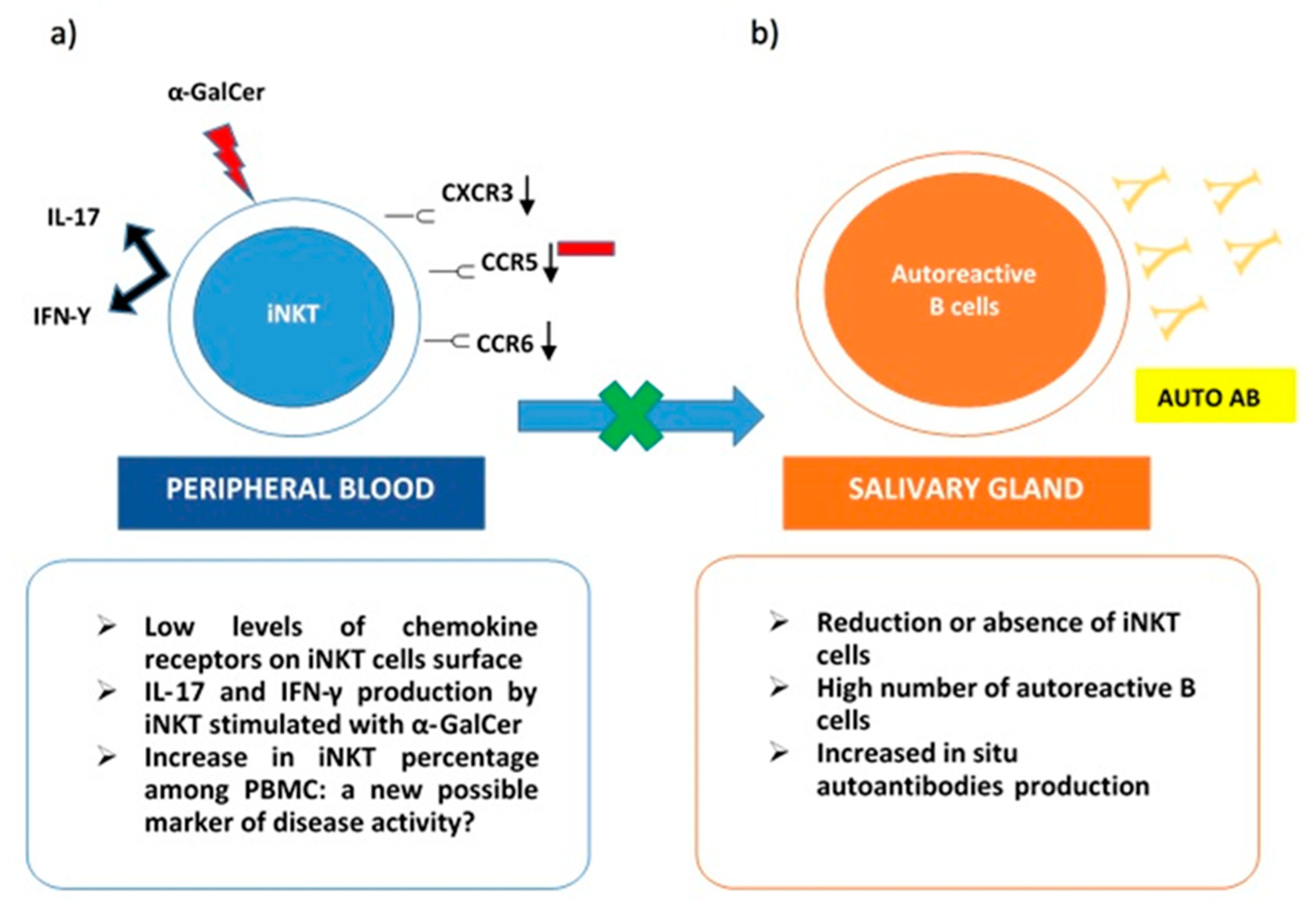
- Guggino, G.; Ciccia, F.; Raimondo, S.; Giardina, G.; Alessandro, R.; Dieli, F.; Sireci, G.; Triolo, G. Invariant NKT cells are expanded in peripheral blood but are undetectable in salivary glands of patients with primary Sjögren’s syndrome. Clin. Exp. Rheumatol. 2016, 34, 25–31. [Google Scholar]
- Awada, A.; Nicaise, C.; Ena, S.; Schandéné, L.; Rasschaert, J.; Popescu, I.; Gangji, V.; Soyfoo, M.S. Potential involvement of the IL-33–ST2 axis in the pathogenesis of primary Sjögren’s syndrome. Ann. Rheum. Dis. 2014, 73, 1259–1263. [Google Scholar] [CrossRef]
- Wermeling, F.; Lind, S.M.; Jordö, E.D.; Cardell, S.L.; Karlsson, M.C. Invariant NKT cells limit activation of autoreactive CD1d-positive B cells. J. Exp. Med. 2010, 207, 943–952. [Google Scholar] [CrossRef]
- Szodoray, P.; Papp, G.; Horvath, I.F.; Barath, S.; Sipka, S.; Nakken, B.; Zeher, M. Cells with regulatory function of the innate and adaptive immune system in primary Sjogren’s syndrome. Clin. Exp. Immunol. 2009, 157, 343–349. [Google Scholar] [CrossRef]
- Sudzius, G.; Mieliauskaite, D.; Siaurys, A.; Viliene, R.; Butrimiene, I.; Characiejus, D.; Dumalakiene, I. Distribution of peripheral lymphocyte populations in primary Sjogren’s syndrome Patients. J. Immunol. Res. 2015, 2015, 854706. [Google Scholar] [CrossRef] [PubMed]
- Davies, R.; Hammenfors, D.; Bergum, B.; Jakobsen, K.; Solheim, M.; Vogelsang, P.; Brun, J.G.; Bryceson, Y.; Jonsson, R.; Appel, S. Patients with primary Sjogren’s syndrome have alterations in absolute quantities of specific peripheral leucocyte populations. Scand. J. Immunol. 2017, 86, 491–502. [Google Scholar] [CrossRef]
- Jonsson, R.; Vogelsang, P.; Volchenkov, R.; Espinosa, A.; Wahren-Herlenius, M.; Appel, S. The complexity of Sjogren’s syndrome: Novel aspects on pathogenesis. Immunol. Lett. 2011, 141, 1–9. [Google Scholar] [CrossRef]
- Fogel, L.A.; Yokoyama, W.M.; French, A.R. Natural killer cells in human autoimmune disorders. Arthritis Res. Ther. 2013, 15, 216. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, Y.; Yoshida, M.; Takaya, M.; Uchiyama, M.; Shimizu, H.; Arimori, S.J.A. Circulating natural killer cells in Sjögren’s syndrome. Arthritis Rheum. 1985, 28, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Coquet, J.M.; Chakravarti, S.; Kyparissoudis, K.; McNab, F.W.; Pitt, L.A.; McKenzie, B.S.; Berzins, S.P.; Smyth, M.J.; Godfrey, D.I. Diverse cytokine production by NKT cell subsets and identification of an IL-17− producing CD4− NK1. 1− NKT cell population. Proc. Natl. Acad. Sci. USA 2008, 105, 11287–11292. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, R.; DelaRosa, O.; Peralbo, E.; Casado, J.; Pena, J.; Solana, R. Human NKT cells in health and disease. Inmunología 2003, 22, 359–370. [Google Scholar]
- Sag, D.; Özkan, M.; Kronenberg, M.; Wingender, G. Improved detection of cytokines produced by invariant NKT cells. Sci. Rep. 2017, 7, 16607. [Google Scholar] [CrossRef]
- Van Kaer, L.; Wu, L. Therapeutic Potential of Invariant Natural Killer T Cells in Autoimmunity. Front. Immunol. 2018, 9, 519. [Google Scholar] [CrossRef]
- Miyake, S.; Yamamura, T. Therapeutic potential of glycolipid ligands for natural killer (NK) T cells in the suppression of autoimmune diseases. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2005, 5, 315–322. [Google Scholar] [CrossRef]
- Mariette, X.; Seror, R.; Quartuccio, L.; Baron, G.; Salvin, S.; Fabris, M.; Desmoulins, F.; Nocturne, G.; Ravaud, P.; De Vita, S. Efficacy and safety of belimumab in primary Sjögren’s syndrome: Results of the BELISS open-label phase II study. Ann. Rheum. Dis. 2015, 74, 526–531. [Google Scholar] [CrossRef]
- Grigoriadou, S.; Chowdhury, F.; Pontarini, E.; Tappuni, A.; Bowman, S.J.; Bombardieri, M. B cell depletion with rituximab in the treatment of primary Sjogren’s syndrome: What have we learnt? Clin. Exp. Rheumatol. 2019, 37 (Suppl. 118), 217–224. [Google Scholar]
- Ciccia, F.; Giardina, A.; Rizzo, A.; Guggino, G.; Cipriani, P.; Carubbi, F.; Giacomelli, R.; Triolo, G. Rituximab modulates the expression of IL-22 in the salivary glands of patients with primary Sjogren’s syndrome. Ann. Rheum. Dis. 2013, 72, 782–783. [Google Scholar] [CrossRef]
- Fasano, S.; Isenberg, D.A. Present and novel biologic drugs in primary Sjogren’s syndrome. Clin. Exp. Rheumatol. 2019, 37 (Suppl. 118), 167–174. [Google Scholar] [PubMed]
- Kroese, F.G.M.; Haacke, E.A.; Bombardieri, M. The role of salivary gland histopathology in primary Sjogren’s syndrome: Promises and pitfalls. Clin. Exp. Rheumatol. 2018, 36, 222–233. [Google Scholar] [PubMed]
- Porcelli, S.A.; Modlin, R.L. The CD1 system: Antigen-presenting molecules for T cell recognition of lipids and glycolipids. Annu. Rev. Immunol. 1999, 17, 297–329. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rizzo, C.; La Barbera, L.; Lo Pizzo, M.; Ciccia, F.; Sireci, G.; Guggino, G. Invariant NKT Cells and Rheumatic Disease: Focus on Primary Sjogren Syndrome. Int. J. Mol. Sci. 2019, 20, 5435. https://doi.org/10.3390/ijms20215435
Rizzo C, La Barbera L, Lo Pizzo M, Ciccia F, Sireci G, Guggino G. Invariant NKT Cells and Rheumatic Disease: Focus on Primary Sjogren Syndrome. International Journal of Molecular Sciences. 2019; 20(21):5435. https://doi.org/10.3390/ijms20215435
Chicago/Turabian StyleRizzo, Chiara, Lidia La Barbera, Marianna Lo Pizzo, Francesco Ciccia, Guido Sireci, and Giuliana Guggino. 2019. "Invariant NKT Cells and Rheumatic Disease: Focus on Primary Sjogren Syndrome" International Journal of Molecular Sciences 20, no. 21: 5435. https://doi.org/10.3390/ijms20215435
APA StyleRizzo, C., La Barbera, L., Lo Pizzo, M., Ciccia, F., Sireci, G., & Guggino, G. (2019). Invariant NKT Cells and Rheumatic Disease: Focus on Primary Sjogren Syndrome. International Journal of Molecular Sciences, 20(21), 5435. https://doi.org/10.3390/ijms20215435