Removal of the Polyglutamine Repeat of Ataxin-3 by Redirecting pre-mRNA Processing


Abstract
1. Introduction
2. Results
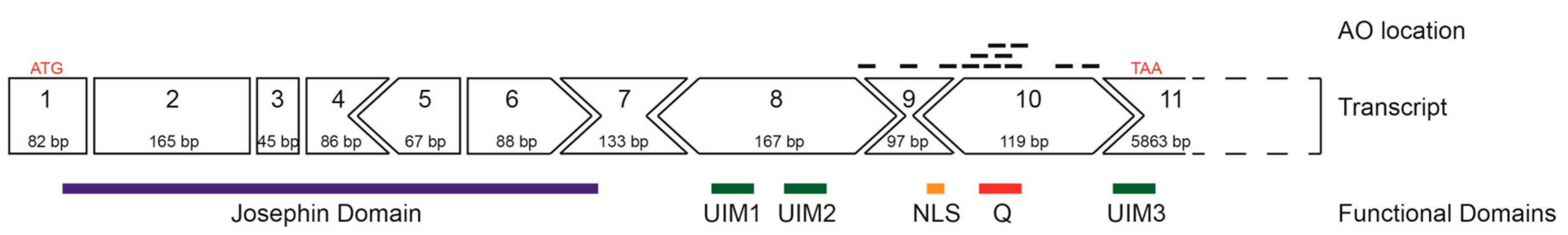
2.1. ATXN3 Transcript and Strategic Removal of Exons
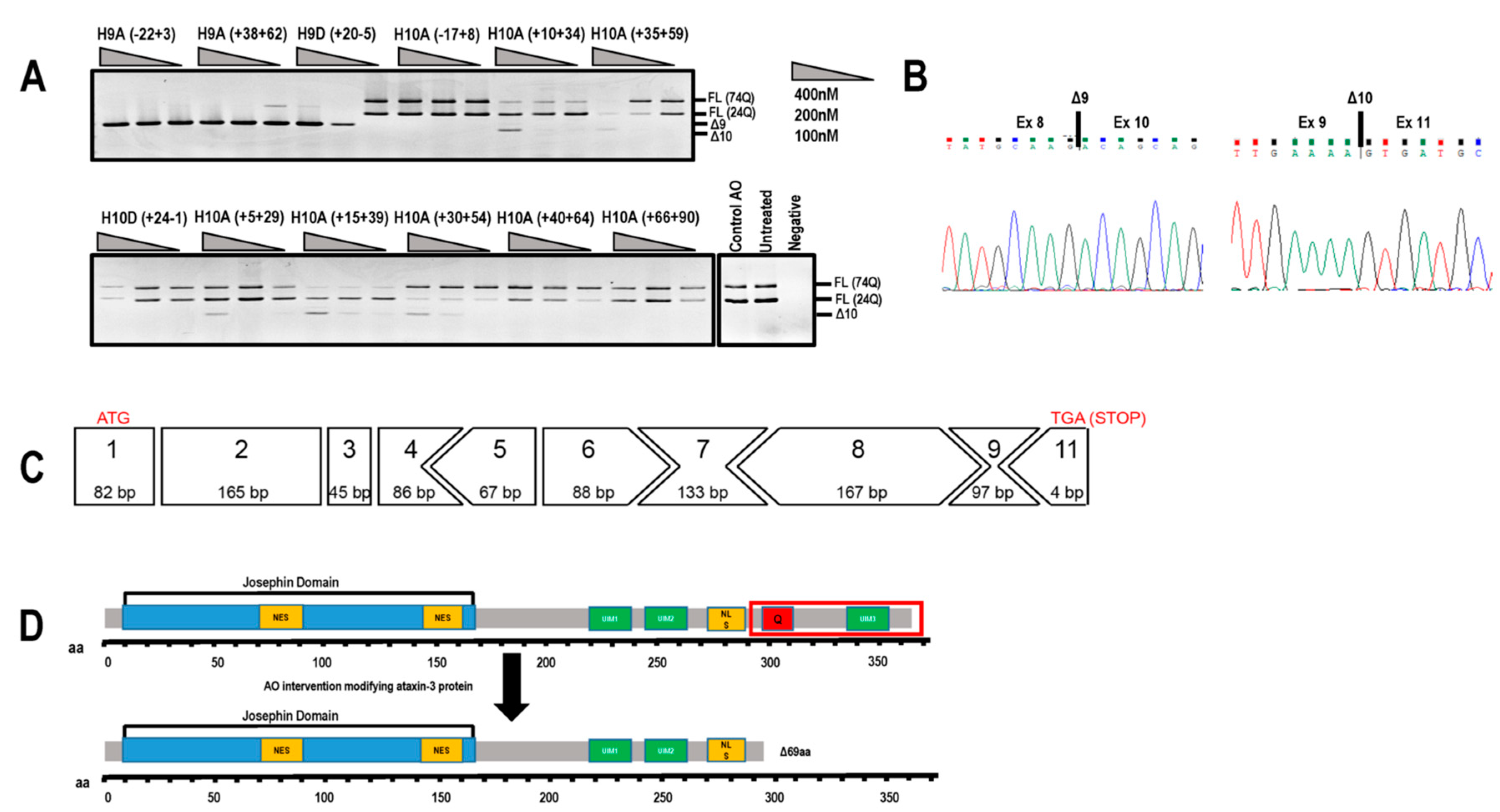
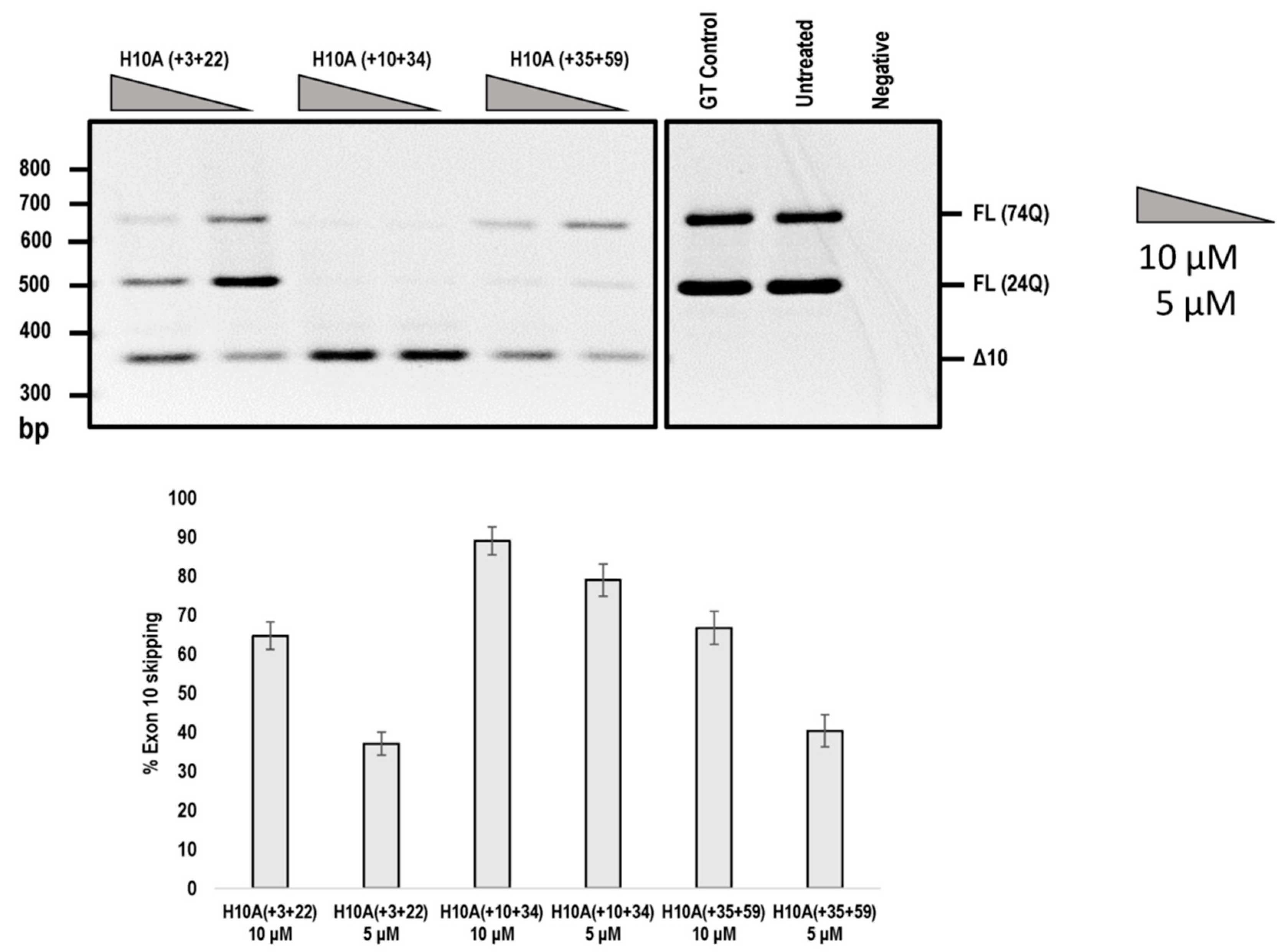
2.2. Evaluation of AOs to Induce Exon 9 and 10 Skipping from the ATNX3 Transcript
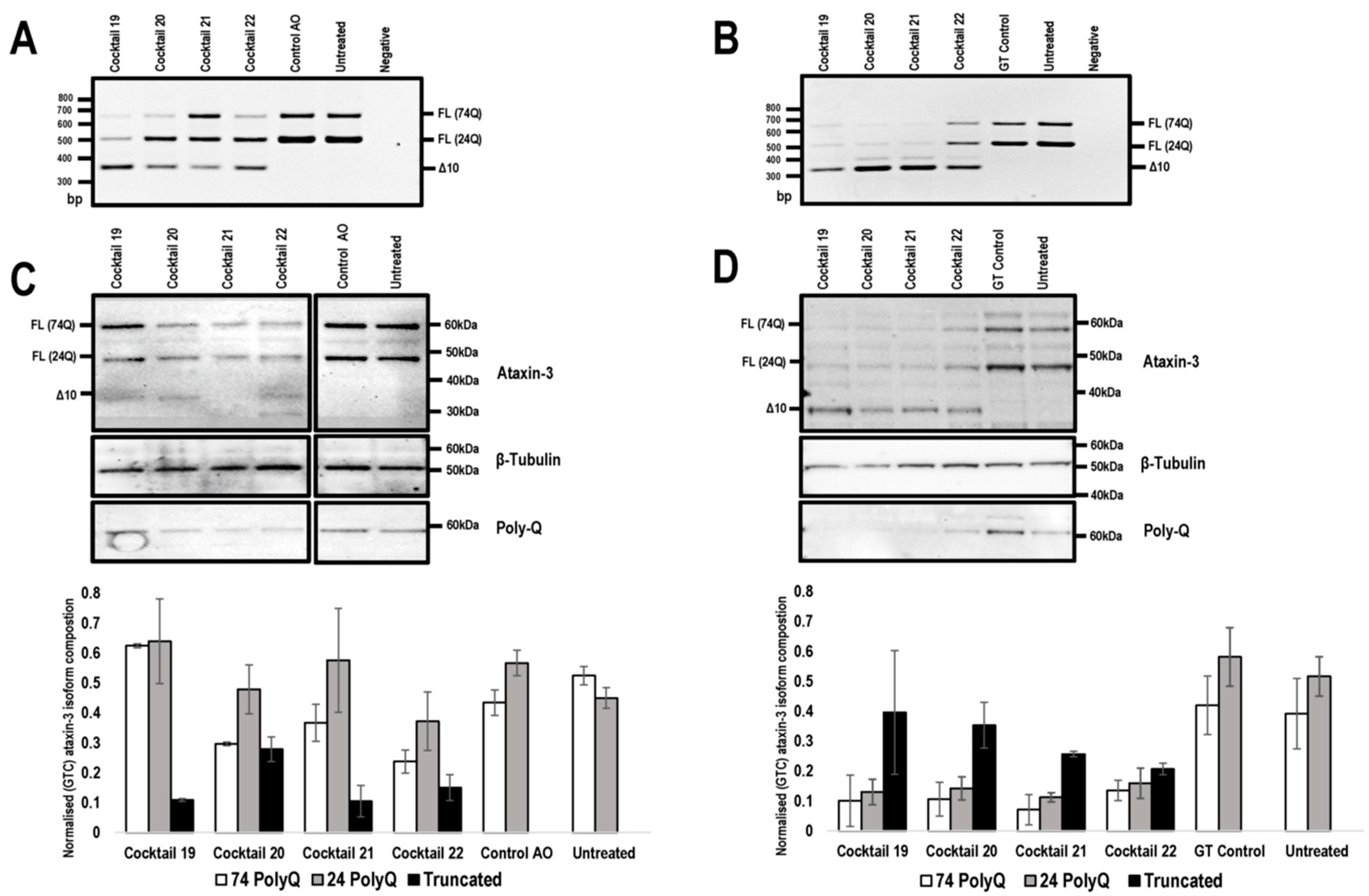
2.3. PMO Mediated Exon Skipping Reduces Full-Length Ataxin-3 Proteins and Induces a Truncated Ataxin-3 Isoform
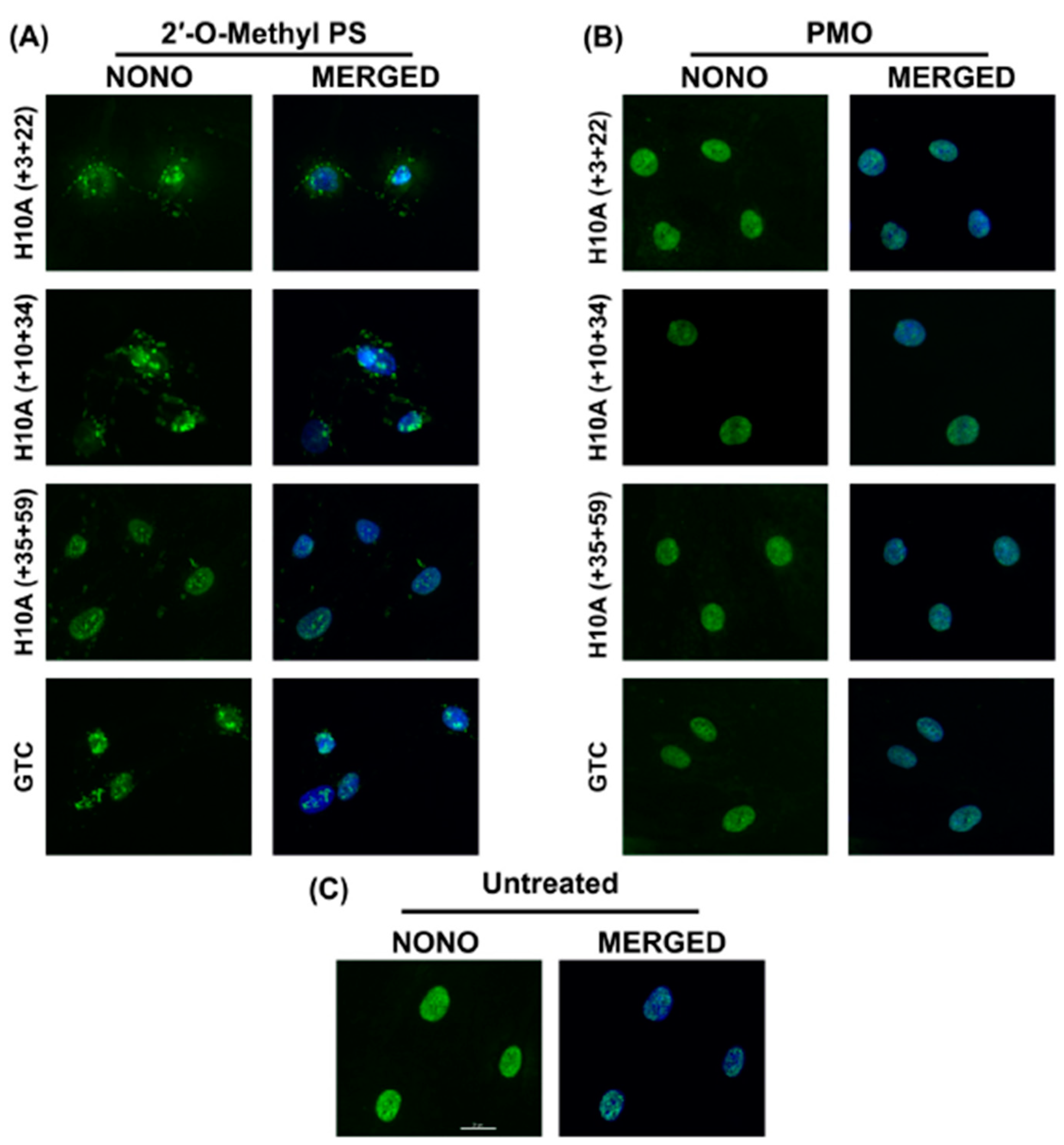
2.4. 2′-O-Methyl PS AOs Induce Sequence Independent Sequestration of Paraspeckle Protein NONO
3. Discussion
4. Materials and Methods
4.1. AO Design and Synthesis
4.2. Cell Culture
4.3. Transfection
4.4. RNA Extraction and RT-PCR Assays
4.5. Western Blotting
4.6. Immunofluorescence
4.7. Densitometric and Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 2′-Me | 2′-O-methyl |
| 2′-MOE | 2′-O-methoxy-ethyl |
| AO | Antisense oligonucleotide |
| DMD | Duchenne muscular dystrophy |
| PMO | Phosphorodiamidate morpholino oligomer |
| PolyQ | Polyglutamine |
| PS | Phosphorothioate |
| SCA3 | Spinocerebellar ataxia type 3 |
| UIM | Ubiquitin interacting motif |
References
- Bettencourt, C.; Lima, M. Machado-joseph disease: From first descriptions to new perspectives. Orphanet J. Rare Dis. 2011, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Matos, C.A.; de Macedo-Ribeiro, S.; Carvalho, A.L. Polyglutamine diseases: The special case of ataxin-3 and Machado–Joseph disease. Prog. Neurobiol. 2011, 95, 26–48. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, C.; Aung-Htut, M.; Fletcher, S.; Wilton, S. Polyglutamine ataxias: From clinical and molecular features to current therapeutic strategies. J. Genet. Syndr. Gene Ther. 2017, 8, 2. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Okamoto, T.; Taniwakiz, M.; Aizawa, M. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat. Genet. 1994, 8, 221. [Google Scholar] [CrossRef]
- Ashley, C.T.; Warren, S.T. Trinucleotide repeat expansion and human disease. Annu. Rev. Genet. 1995, 29, 703–728. [Google Scholar] [CrossRef]
- Tsou, W.-L.; Ouyang, M.; Hosking, R.R.; Sutton, J.R.; Blount, J.R.; Burr, A.A.; Todi, S.V. The deubiquitinase ataxin-3 requires rad23 and dnaj-1 for its neuroprotective role in drosophila melanogaster. Neurobiol. Dis. 2015, 82, 12–21. [Google Scholar] [CrossRef]
- Wen, J.; Scoles, D.R.; Facelli, J.C. Effects of the enlargement of polyglutamine segments on the structure and folding of ataxin-2 and ataxin-3 proteins. J. Biomol. Struct. Dyn. 2016, 35, 504–519. [Google Scholar] [CrossRef]
- Toonen, L.J.; Rigo, F.; van Attikum, H.; van Roon-Mom, W.M. Antisense oligonucleotide-mediated removal of the polyglutamine repeat in spinocerebellar ataxia type 3 mice. Mol. Ther.-Nucleic Acids 2017, 8, 232–242. [Google Scholar] [CrossRef]
- Evers, M.M.; Tran, H.-D.; Zalachoras, I.; Pepers, B.A.; Meijer, O.C.; den Dunnen, J.T.; van Ommen, G.-J.B.; Aartsma-Rus, A.; van Roon-Mom, W.M. Ataxin-3 protein modification as a treatment strategy for spinocerebellar ataxia type 3: Removal of the CAG containing exon. Neurobiol. Dis. 2013, 58, 49–56. [Google Scholar] [CrossRef][Green Version]
- Evers, M.M.; Pepers, B.A.; van Deutekom, J.C.; Mulders, S.A.; den Dunnen, J.T.; Aartsma-Rus, A.; van Ommen, G.-J.B.; van Roon-Mom, W.M. Targeting several CAG expansion diseases by a single antisense oligonucleotide. PLoS ONE 2011, 6, e24308. [Google Scholar] [CrossRef]
- Shen, W.; De Hoyos, C.L.; Sun, H.; Vickers, T.A.; Liang, X.-h.; Crooke, S.T. Acute hepatotoxicity of 2′ fluoro-modified 5–10–5 gapmer phosphorothioate oligonucleotides in mice correlates with intracellular protein binding and the loss of dbhs proteins. Nucleic Acids Res. 2018, 46, 2204–2217. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Liang, X.-h.; Crooke, S.T. Phosphorothioate oligonucleotides can displace Neat1 RNA and form nuclear paraspeckle-like structures. Nucleic Acids Res. 2014, 42, 8648–8662. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Liang, X.-h.; Sun, H.; Crooke, S.T. 2′-fluoro-modified phosphorothioate oligonucleotide can cause rapid degradation of p54nrb and psf. Nucleic Acids Res. 2015, 43, 4569–4578. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Baker, B.F.; Kwoh, T.J.; Cheng, W.; Schulz, D.J.; Xia, S.; Salgado, N.; Bui, H.-H.; Hart, C.E.; Burel, S.A. Integrated safety assessment of 2′-O-methoxyethyl chimeric antisense oligonucleotides in nonhuman primates and healthy human volunteers. Mol. Ther. 2016, 24, 1771–1782. [Google Scholar] [CrossRef]
- Toonen, L.J.; Casaca-Carreira, J.; Pellisé-Tintoré, M.; Mei, H.; Temel, Y.; Jahanshahi, A.; van Roon-Mom, W.M. Intracerebroventricular administration of a 2′-O-methyl phosphorothioate antisense oligonucleotide results in activation of the innate immune system in mouse brain. Nucleic Acid Ther. 2018, 28, 63–73. [Google Scholar] [CrossRef]
- Chi, X.; Gatti, P.; Papoian, T. Safety of antisense oligonucleotide and siRNA-based therapeutics. Drug Discov. Today 2017, 22, 823–833. [Google Scholar] [CrossRef]
- Crooke, S.T.; Baker, B.F.; Witztum, J.L.; Kwoh, T.J.; Pham, N.C.; Salgado, N.; McEvoy, B.W.; Cheng, W.; Hughes, S.G.; Bhanot, S. The effects of 2′-O-methoxyethyl containing antisense oligonucleotides on platelets in human clinical trials. Nucleic Acid Ther. 2017, 27, 121–129. [Google Scholar] [CrossRef]
- Flynn, L.L.; Li, R.; Aung-Htut, M.T.; Pitout, I.L.; Cooper, J.; Hubbard, A.; Griffiths, L.; Bond, C.; Wilton, S.D.; Fox, A.H. Interaction of modified oligonucleotides with nuclear proteins, formation of novel nuclear structures and sequence-independent effects on RNA processing. BioRxiv 2018, 446773. [Google Scholar] [CrossRef]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef]
- Cirak, S.; Arechavala-Gomeza, V.; Guglieri, M.; Feng, L.; Torelli, S.; Anthony, K.; Abbs, S.; Garralda, M.E.; Bourke, J.; Wells, D.J. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation study. Lancet 2011, 378, 595–605. [Google Scholar] [CrossRef]
- Kinali, M.; Arechavala-Gomeza, V.; Feng, L.; Cirak, S.; Hunt, D.; Adkin, C.; Guglieri, M.; Ashton, E.; Abbs, S.; Nihoyannopoulos, P. Local restoration of dystrophin expression with the morpholino oligomer avi-4658 in Duchenne muscular dystrophy: A single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009, 8, 918–928. [Google Scholar] [CrossRef]
- Adams, A.M.; Harding, P.L.; Iversen, P.L.; Coleman, C.; Fletcher, S.; Wilton, S.D. Antisense oligonucleotide induced exon skipping and the dystrophin gene transcript: Cocktails and chemistries. BMC Mol. Biol. 2007, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Mitrpant, C.; Adams, A.M.; Meloni, P.L.; Muntoni, F.; Fletcher, S.; Wilton, S.D. Rational design of antisense oligomers to induce dystrophin exon skipping. Mol. Ther. 2009, 17, 1418–1426. [Google Scholar] [CrossRef]
- Carver, M.P.; Charleston, J.S.; Shanks, C.; Zhang, J.; Mense, M.; Sharma, A.K.; Kaur, H.; Sazani, P. Toxicological characterization of exon skipping phosphorodiamidate morpholino oligomers (PMOs) in non-human primates. J. Neuromuscul. Dis. 2016, 3, 381–393. [Google Scholar] [CrossRef]
- McClorey, G.; Moulton, H.; Iversen, P.; Fletcher, S.; Wilton, S. Antisense oligonucleotide-induced exon skipping restores dystrophin expression in vitro in a canine model of DMD. Gene Ther. 2006, 13, 1373. [Google Scholar] [CrossRef]
- Saute, J.A.M.; Jardim, L.B. Machado-Joseph disease: Clinical and genetic aspects, and current treatment. Expert Opin. Orphan Drugs 2015, 3, 517–535. [Google Scholar] [CrossRef]
- Matos, C.; Pereira de Almeida, L.; Nóbrega, C. Machado-Joseph disease/spinocerebellar ataxia type 3: Lessons from disease pathogenesis and clues into therapy. J. Neurochem. 2018, 148, 8–28. [Google Scholar] [CrossRef]
- Weishäupl, D.; Schneider, J.; Pinheiro, B.P.; Ruess, C.; Dold, S.M.; von Zweydorf, F.; Gloeckner, C.J.; Schmidt, J.; Riess, O.; Schmidt, T. Physiological and pathophysiological characteristics of ataxin-3 isoforms. J. Biol. Chem. 2018, 294, 644–661. [Google Scholar] [CrossRef]
- Silverman, R.H. Viral encounters with 2′, 5′-oligoadenylate synthetase and RNAse l during the interferon antiviral response. J. Virol. 2007, 81, 12720–12729. [Google Scholar] [CrossRef]
- Tokarev, A.; Skasko, M.; Fitzpatrick, K.; Guatelli, J. Antiviral activity of the interferon-induced cellular protein bst-2/tetherin. Aids Res. Hum. Retrovir. 2009, 25, 1197–1210. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Zhao, L.; Zhang, T.; Qi, Y.; Wang, T.; Liu, K.; Wang, H.; Feng, H.; Jin, H.; Qin, C. Innate immune response gene expression profiles in central nervous system of mice infected with rabies virus. Comp. Immunol. Microbiol. Infect. Dis. 2011, 34, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Sahenk, Z.; Rodino-Klapac, L.R. Clinical trials of exon skipping in Duchenne muscular dystrophy. Expert Opin. Orphan Drugs 2017, 5, 683–690. [Google Scholar] [CrossRef]
- Janas, M.M.; Jiang, Y.; Schlegel, M.K.; Waldron, S.; Kuchimanchi, S.; Barros, S.A. Impact of oligonucleotide structure, chemistry, and delivery method on in vitro cytotoxicity. Nucleic Acid Ther. 2017, 27, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.; Stessl, M.; Amartey, J.; Noe, C.R. Off-target effects related to the phosphorothioate modification of nucleic acids. ChemMedChem 2010, 5, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Goemans, N.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Shao, J.; Kaye, E.M.; Mercuri, E.; Group, E.S.; Network, T.F.D.I.; et al. Longitudinal effect of Eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 2016, 79, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Pitout, I.; Flynn, L.L.; Wilton, S.D.; Fletcher, S. Antisense-mediated splice intervention to treat human disease: The odyssey continues. F1000Research 2019, 8. [Google Scholar] [CrossRef]
- Summerton, J.; Stein, D.; Huang, S.B.; Matthews, P.; Weller, D.; Partridge, M. Morpholino and phosphorothioate antisense oligomers compared in cell-free and in-cell systems. Antisense Nucleic Acid Drug Dev. 1997, 7, 63–70. [Google Scholar] [CrossRef]
- Summerton, J.; Weller, D. Morpholino antisense oligomers: Design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997, 7, 187–195. [Google Scholar] [CrossRef]
- Summerton, J.E. Morpholino, siRNA, and s-DNA compared: Impact of structure and mechanism of action on off-target effects and sequence specificity. Curr. Top. Med. Chem. 2007, 7, 651–660. [Google Scholar] [CrossRef]
- Flynn, L.L.; Mitrpant, C.; Pitout, I.L.; Fletcher, S.; Wilton, S.D. Antisense oligonucleotide-mediated terminal intron retention of the SMN2 transcript. Mol. Ther.-Nucleic Acids 2018, 11, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Amantana, A.; Iversen, P.L. Pharmacokinetics and biodistribution of phosphorodiamidate morpholino antisense oligomers. Curr. Opin. Pharmacol. 2005, 5, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Arora, V.; Devi, G.R.; Iversen, P.L. Neutrally charged phosphorodiamidate morpholino antisense oligomers: Uptake, efficacy and pharmacokinetics. Curr. Pharm. Biotechnol. 2004, 5, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Miyatake, S.; Mizobe, Y.; Tsoumpra, M.K.; Lim, K.R.Q.; Hara, Y.; Shabanpoor, F.; Yokota, T.; Takeda, S.i.; Aoki, Y. Scavenger receptor class A1 mediates uptake of morpholino antisense oligonucleotide into dystrophic skeletal muscle. Mol. Ther.-Nucleic Acids 2019, 14, 520–535. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Nagata, T.; Yokota, T.; Nakamura, A.; Wood, M.J.; Partridge, T.; Takeda, S.i. Highly efficient in vivo delivery of PMO into regenerating myotubes and rescue in laminin-α2 chain-null congenital muscular dystrophy mice. Hum. Mol. Genet. 2013, 22, 4914–4928. [Google Scholar] [CrossRef] [PubMed]
- Miyatake, S.; Mizobe, Y.; Takizawa, H.; Hara, Y.; Yokota, T.; Takeda, S.i.; Aoki, Y. Exon skipping therapy using phosphorodiamidate morpholino oligomers in the mdx52 mouse model of Duchenne muscular dystrophy. In Duchenne Muscular Dystrophy; Springer: Berlin/Heidelberg, Germany, 2018; pp. 1231–1241. [Google Scholar]
- Toonen, L.J.; Schmidt, I.; Luijsterburg, M.S.; van Attikum, H.; van Roon-Mom, W.M. Antisense oligonucleotide-mediated exon skipping as a strategy to reduce proteolytic cleavage of ataxin-3. Sci. Rep. 2016, 6, 35200. [Google Scholar] [CrossRef]
- Switonski, P.M.; Fiszer, A.; Kazmierska, K.; Kurpisz, M.; Krzyzosiak, W.J.; Figiel, M. Mouse ataxin-3 functional knock-out model. Neuromol. Med. 2011, 13, 54–65. [Google Scholar] [CrossRef]
- Evers, M.M.; Toonen, L.J.; van Roon-Mom, W.M. Ataxin-3 protein and RNA toxicity in spinocerebellar ataxia type 3: Current insights and emerging therapeutic strategies. Mol. Neurobiol. 2014, 49, 1513–1531. [Google Scholar] [CrossRef][Green Version]
- Li, X.; Liu, H.; Fischhaber, P.L.; Tang, T.-S. Toward therapeutic targets for sca3: Insight into the role of Machado–Joseph disease protein ataxin-3 in misfolded proteins clearance. Prog. Neurobiol. 2015, 132, 34–58. [Google Scholar] [CrossRef]
- Schmitt, I.; Linden, M.; Khazneh, H.; Evert, B.O.; Breuer, P.; Klockgether, T.; Wuellner, U. Inactivation of the mouse atxn3 (ataxin-3) gene increases protein ubiquitination. Biochem. Biophys. Res. Commun. 2007, 362, 734–739. [Google Scholar] [CrossRef]
- Moore, L.R.; Rajpal, G.; Dillingham, I.T.; Qutob, M.; Blumenstein, K.G.; Gattis, D.; Hung, G.; Kordasiewicz, H.B.; Paulson, H.L.; McLoughlin, H.S. Evaluation of antisense oligonucleotides targeting Atxn3 in SCA3 mouse models. Mol. Ther.-Nucleic Acids 2017, 7, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Boy, J.; Schmidt, T.; Wolburg, H.; Mack, A.; Nuber, S.; Böttcher, M.; Schmitt, I.; Holzmann, C.; Zimmermann, F.; Servadio, A. Reversibility of symptoms in a conditional mouse model of spinocerebellar ataxia type 3. Hum. Mol. Genet. 2009, 18, 4282–4295. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zhang, D.; McLoughlin, H.S.; Zalon, A.J.; Aravind, L.; Paulson, H.L. Loss of the spinocerebellar ataxia type 3 disease protein atxn3 alters transcription of multiple signal transduction pathways. PLoS ONE 2018, 13, e0204438. [Google Scholar] [CrossRef] [PubMed]
- McLoughlin, H.S.; Moore, L.R.; Chopra, R.; Komlo, R.; McKenzie, M.; Blumenstein, K.G.; Zhao, H.; Kordasiewicz, H.B.; Shakkottai, V.G.; Paulson, H.L. Oligonucleotide therapy mitigates disease in spinocerebellar ataxia type 3 mice. Ann. Neurol. 2018, 84, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.-O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human splicing finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed]
- Aung-Htut, M.T.; McIntosh, C.S.; Ham, K.A.; Pitout, I.L.; Flynn, L.L.; Greer, K.; Fletcher, S.; Wilton, S.D. Systematic approach to developing splice modulating antisense oligonucleotides. Int. J. Mol. Sci. 2019, 20, 5030. [Google Scholar] [CrossRef]
- Aung-Htut, M.T.; McIntosh, C.S.; West, K.A.; Fletcher, S.; Wilton, S.D. In vitro validation of phosphorodiamidate morpholino oligomers. Molecules 2019, 24, 2922. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. Nih image to imagej: 25 years of image analysis. Nat. Methods 2012, 9, 671. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5′–3′) |
|---|---|
| ATXN3 H9A (− 22 + 03) | UACCUGAAAACAAAACACAACACAA |
| ATXN3 H9A (+ 38 + 62) | UUCUGAAGUAAGAUUUGUACCUGAU |
| ATXN3 H9D (+ 20 − 05) | UUUACUUUUCAAAGUAGGCUUCUCG |
| ATXN3 H10A (− 17 + 08) | GCUGCUGUCUGAAACAUUCAAAAGU |
| ATXN3 H10A (+ 10 + 34) * | CUGCUGCUGCUGCUGUUGCUGCUUU |
| ATXN3 H10A (+ 35 + 59) * | GUCCUGAUAGGUCCCCCUGCUGCUG |
| ATXN3 H10D (+ 24 − 01) * | CCUAGAUCACUCCCAAGUGCUCCUG |
| ATXN3 H10A (+ 05 + 29) | GCUGCUGCUGUUGCUGCUUUUGCUG |
| ATXN3 H10A (+ 15 + 39) * | UGCUGCUGCUGCUGCUGCUGUUGCU |
| ATXN3 H10A (+ 30 + 54) | GAUAGGUCCCCCUGCUGCUGCUGCU |
| ATXN3 H10A (+ 40 + 64) | ACUCUGUCCUGAUAGGUCCCCCUGC |
| ATXN3 H10A (+ 66 + 90) | GUGGCUGGCCUUUCACAUGGAUGUG |
| ATXN3 H10A (+ 03 + 22) #/* | CUGUUGCUGCUUUUGCUGCU |
| Control AO @ | GGAUGUCCUGAGUCUAGACCCUCCG |
| Gene Tools Control | CCTCTTACCTCAGTTACAATTTATA |
| Cocktail Number | AO Combination |
|---|---|
| Cocktail 19 | ATXN3 H10A (+ 10 + 34) |
| ATXN3 H10A (+ 35 + 59) | |
| Cocktail 20 | ATXN3 H10A (+ 10 + 34) |
| ATXN3 H10D (+ 24 − 01) | |
| Cocktail 21 | ATXN3 H10A (+ 35 + 59) |
| ATXN3 H10D (+ 24 − 01) | |
| Cocktail 22 | ATXN3 H10A (+ 15 + 39) |
| ATXN3 H10D (+ 24 − 01) | |
| Cocktail 23 | ATXN3 H10A (+ 05 + 29) |
| ATXN3 H10D (+ 24 − 01) | |
| Cocktail 24 | ATXN3 H10A (+ 30 + 54) |
| ATXN3 H10D (+ 24 − 01) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McIntosh, C.S.; Aung-Htut, M.T.; Fletcher, S.; Wilton, S.D. Removal of the Polyglutamine Repeat of Ataxin-3 by Redirecting pre-mRNA Processing. Int. J. Mol. Sci. 2019, 20, 5434. https://doi.org/10.3390/ijms20215434
McIntosh CS, Aung-Htut MT, Fletcher S, Wilton SD. Removal of the Polyglutamine Repeat of Ataxin-3 by Redirecting pre-mRNA Processing. International Journal of Molecular Sciences. 2019; 20(21):5434. https://doi.org/10.3390/ijms20215434
Chicago/Turabian StyleMcIntosh, Craig S., May Thandar Aung-Htut, Sue Fletcher, and Steve D. Wilton. 2019. "Removal of the Polyglutamine Repeat of Ataxin-3 by Redirecting pre-mRNA Processing" International Journal of Molecular Sciences 20, no. 21: 5434. https://doi.org/10.3390/ijms20215434
APA StyleMcIntosh, C. S., Aung-Htut, M. T., Fletcher, S., & Wilton, S. D. (2019). Removal of the Polyglutamine Repeat of Ataxin-3 by Redirecting pre-mRNA Processing. International Journal of Molecular Sciences, 20(21), 5434. https://doi.org/10.3390/ijms20215434