1. Introduction
In recent years, multidrug resistance (MDR) of bacteria has become a global health problem, and there is an increasing demand for new antimicrobial agents and antibiotics. One of the most promising approaches in antibiotic development involves antimicrobial peptides (AMPs). AMPs are cationic amphipathic peptides representing an ancient host defense mechanism and are ubiquitous in living organisms. Many of them have a high affinity to components of the cell membrane of Gram-negative and/or Gram-positive bacteria. Therefore, compared to many conventional antibiotics which impair the bacterial metabolism, AMPs are less prone to the development of bacterial resistance since they interact directly with the bacterial membrane, which is an evolutionary highly conserved structure, and is not able to perform major structural changes without losing its various vital functions. The interaction of AMPs with the bacterial membrane initiates a change in the membrane curvature and leads to a destabilization and permeabilization of the membrane, finally resulting in the loss of the barrier function [
1,
2]. In order to improve the antimicrobial activity of some AMPs, their hydrophobic properties were increased by targeted N-acylation. This led to increased antimicrobial activity against
Escherichia coli, which correlates with the degree of permeability of bacterial membranes of these peptides [
3,
4]. Some AMPs have a specifically high affinity to lipopolysaccharides (LPS, endotoxins) from Gram-negative bacteria and are able to suppress an LPS-induced release of tumor necrosis factor alpha (TNF-α) and could protect mice from lethal endotoxemia [
3,
4]. Among these is Polymyxin B (PMB), a naturally occurring AMP produced by
Bacillus polymyxa. Due to its high affinity to LPS, it is used for a hemoperfusion adsorbent called Toraymyxin, which consists of a polystyrene-based woven fiber with covalently immobilized PMB. The manufacturer claims that Toraymyxin is able to remove LPS from patients suffering from endotoxemia using extracorporeal perfusion of the adsorbent cartridge. However, it has been shown recently that Toraymyxin acts as a delivery system of PMB by releasing the non-covalently bound fraction into the patients’ blood stream, and LPS is neutralized by PMB, which results in a prevention of the activation of pattern recognition receptors. Also, other adsorbents intended for the removal of LPS do not adsorb LPS sufficiently in in vitro experiments [
5]. Consequently, there is currently no membrane/adsorption based blood purification system available that is able to remove LPS from the patients’ blood.
AMPs based on lactoferrin (LF), a transferrin in mammals and some invertebrates that has endotoxin-neutralizing properties [
6], have proven to be potential candidates for use in extracorporeal therapies. They showed promising results by neutralizing endotoxins in the mouse model as well as by reducing late-onset sepsis in low-birth-weight neonates [
7]. However, there is still a lack of knowledge of the substantial properties of these AMPs in order to facilitate their use in extracorporeal blood purification. In this study, LF11-based peptides derived from the pepsin cleavage product of human LF were characterized. Human LF is a multifunctional, iron-binding glycoprotein, which occurs in mammalian exocrine secretions and neutrophil granules. It has antimicrobial and LPS-binding properties [
8,
9]. Bovine lactoferricin (LFcin) has an α-helical structure, which is lost in an isolated form [
10,
11]. Human lactoferricin (hLFcin) comprises amino acid residues 1-45 of the N-terminus of human lactoferrin (hLF) and has increased antibacterial activity compared to intact hLF [
12]. LFcin has animicrobial properties against Gram-positive bacteria, Gram-negative bacteria, and filamentous yeasts, including some antibiotic-resistant pathogens [
13]. Wakabayashi et al. [
13,
14] and Strom et al. [
15] were able to show that N-acylation of lactoferrin-based peptides could increase their antimicrobial activity. However, this usually leads to higher toxicity on eukaryotic cells, leading to loss of target cell selectivity [
16,
17].
The aim of this in vitro study was to characterize lactoferrin-based AMPs for their potential use in extracorporeal therapies with special attention to their biocompatibility, their binding affinity to LPS, and the capability to suppress cytokine release. Furthermore, it was examined whether the strongly polyanionic heparin has an influence on the antimicrobial effect of cationic AMPs. Selected AMPs were covalently immobilized on sepharose beads, and the resulting AMP-immobilized adsorbents were characterized for their antimicrobial properties. For this study, two synthetic LF11-based AMPs were selected, which showed good antimicrobial and LPS-binding properties in previous studies [
18,
19].
3. Discussion
David et al. suggested that a clear separation of cationic and apolar domains (amphiphilic character) is crucial for the design of a molecule for LPS sequestration [
27]. To neutralize endotoxins, the binding of peptides to the LPS molecule alone is not sufficient [
28]. Studies from other groups have shown that for certain cationic AMPs, a fatty acid substitution of ≥ C
8 is necessary for efficient LPS neutralization [
29,
30]. For this reason, only lipid acylations ≥ C
8 were selected for this study. According to the results of the NPN permeability study, the optimal lipid chain length of LF11 peptides for antimicrobial properties is C
12. This confirms the results of scientific studies carried out by other groups [
31,
32]. The results show that the necessary acyl chain length in order to maximize the binding affinity to LPS depends not only on the AMP, but also on the medium in which the experiment is carried out and the origin of LPS (
E. coli vs
P. aeruginosa). Some of the lipid substituted peptides were able to reduce significantly the endotoxin activity of
P. aeruginosa in serum, but showed no effect against LPS from
E. coli. This can be attributed to the fact that bacterial strains differ in the acylation (number, length) of the lipid A moiety of their endotoxins. In contrast, PMB is able to neutralize both LPS types. The cytokine release study confirms that the tested LF11 peptides are not able to neutralize the endotoxins in serum. Only PMB can form a stable LPS-PMB complex in whole blood and thus reduce the release of pro-inflammatory mediators of leukocytes [
20].
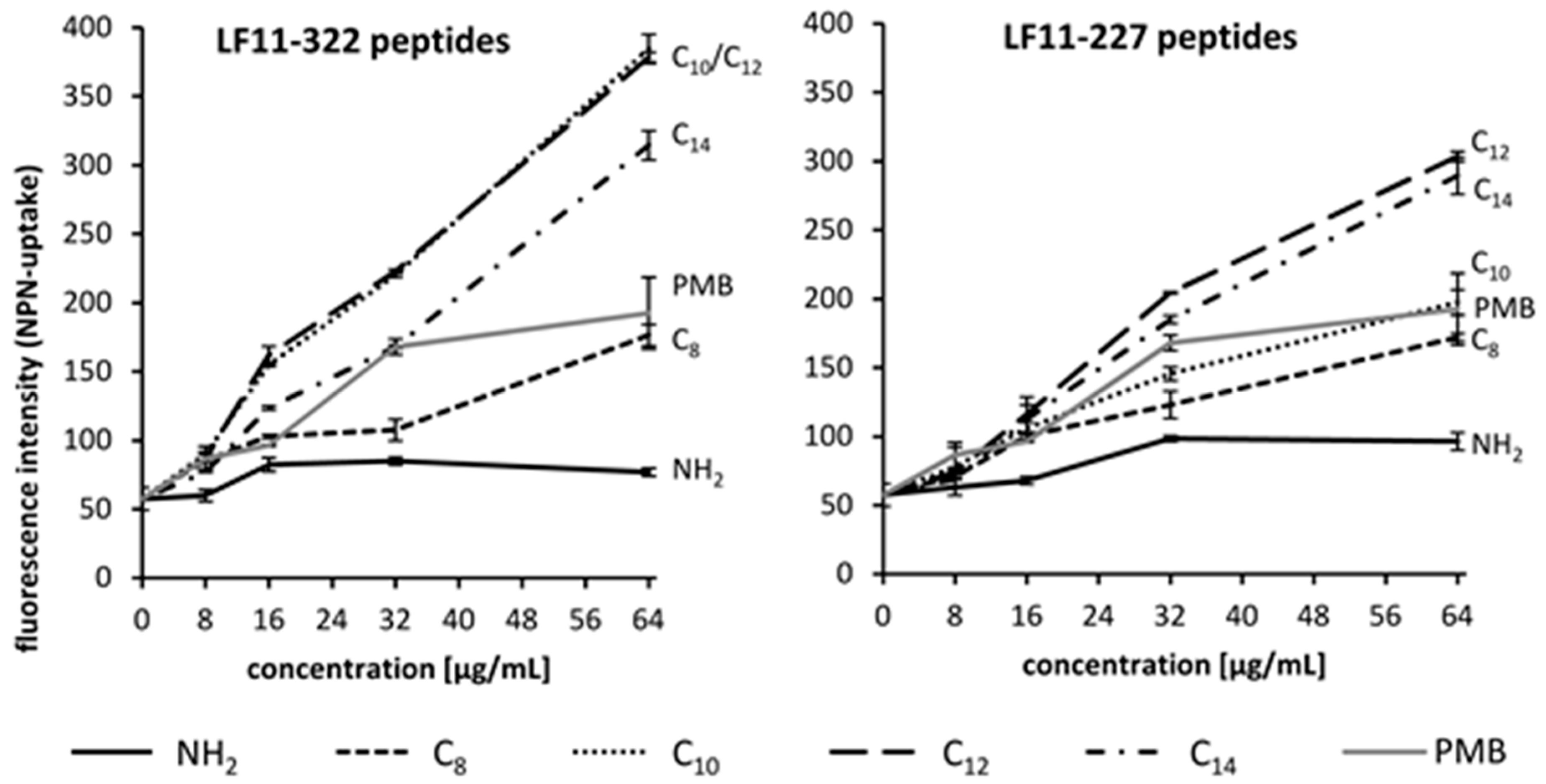
When determining the MIC, it can be clearly seen that the antimicrobial effect of AMPs strongly depends on the medium to be tested. The antimicrobial properties of the LF11-322 peptides against
E. coli and
S. aureus are reduced in albumin solution, whereas the acylated LF11-227 peptides show only an antimicrobial effect in protein solution. In general, the MIC of
S. aureus is slightly lower than that of
E. coli. This suggests that unlike PMB, the antimicrobial effect is not based on LPS binding and that the peptides do not selectively act against a bacterial group. The polyanionic heparin is able to cancel the antimicrobial effect of the AMPs in our MIC study. A possible explanation for this could be that the anionic heparin binds the cationic AMPs and thus inactivates them. Previous works have shown interactions between heparin and natural cationic AMPs such as LL-37 and protamine [
33,
34]. These studies also assume an ionic interaction between the anionic heparin and the cationic peptides. Further and especially targeted studies would be necessary to clarify this observed effect. This fact must be taken into account for clinical applications of AMPs, especially for patients who are subject to extracorporeal blood purification therapy. Their blood must be sufficiently anticoagulated before and during treatment and the most frequently used anticoagulant in intensive care medicine is heparin. LL-37 is a human antimicrobial peptide from the group of cathelicidins. It is mainly produced in immune cells and is part of the innate immune response as well as the apoptosis of endogenous cells. In the MIC study, LL-37 shows an antimicrobial effect only against
E. coli, which is also lost in the presence of heparin. PMB loses its antimicrobial effect induced by heparin only against Gram-positive bacteria. This shows that the affinity of PMB to LPS is not only selective, but almost specific and thus is significantly higher than to heparin. Although some synthetic AMPs have shown promising results in in vitro tests, especially in MIC determinations, none have yet been approved for clinical use [
30,
32,
35]. The difficulty lies in synthesizing a peptide that possesses antimicrobial properties against a broad spectrum of multiresistant germs on the one hand and on the other hand has good blood compatibility. Toxicological tests with cell-lines cultured in culture media or hemolysis tests with erythrocytes are usually performed as biocompatibility tests [
30,
31,
36,
37]. However, several and more complex tests are required to test the blood compatibility of antibiotics. In addition to the hemolysis test, we also investigated the influence of the LF11 peptides on the coagulation and complement system. Furthermore, it was investigated whether the AMPs only attack the cell membrane of bacteria or whether they also attack the membrane of erythrocytes, leukocytes and thrombocytes. The determination of hemolysis is a marker for the selectivity of a membrane-destabilizing AMP. The profound difference between bacterial and eukaryotic cell membranes is the fact that bacterial membranes are negatively charged due to embedded negatively charged lipids, while membranes from eukaryotic cells are neutral. This fact should allow appropriate AMPs to destabilize selectively the bacterial membrane, while leaving the eukaryotic cell widely unaffected. Since for the destabilization process of bacterial membranes amphipathic AMPs are advantageous [
38], only moderately acylated AMPs should be used in order to reduce the risk of destabilizing eukaryotic cells. In the presence of a membrane mimetic environment, the short (nine amino-acids) LF11-322 folds into a loop comprising a short helical segment [
39], which separates the cationic and hydrophobic residues along the molecular axis of the peptide. The secondary structure prediction using the program PEP-FOLD indicated a similar structure of LF11-227 (
http://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD/). The structure of both peptides is in accordance with the structure of the parent peptide LF11. N-acylation forced the peptide chain to wrap around the acyl chain, resulting in an even better defined fold [
40]. It can be expected that N-acylation of LF11-322 and LF11-227 follows the same principle. In our blood compatibility test we could show that with increasing acyl chain length of the LF11-322 peptide also leukocytes, RBC and platelets are lysed. The same tendency could be observed with the activation of the complement system and the plasmatic coagulation. In whole blood, where the blood cells are suspended in plasma, no hemolysis could be detected by acylated LF11 peptides. This indicates that most of these peptides are bound to plasma proteins. The SEM images confirm our results that PMB selectively destabilizes Gram-negative cell membranes, while Gram-positive bacteria and blood cells are not lysed or destabilized by PMB. In contrast, AMPs destabilize Gram-positive as well as Gram-negative cells. Together with the results of the blood compatibility tests, this suggests that depending on the concentration, acylated AMPs interact less selectively with all biological membranes, resulting in poor blood compatibility. In this study, the immobilization of the AMPs was conducted via their amino-groups. Since the AMPs used in this study are arginine-rich, it can be assumed that each AMP was immobilized via different amino-groups, leading to an undefined, varying orientation of the AMP on the adsorbents surface. However, the orientation of the AMP plays an important role in its efficiency [
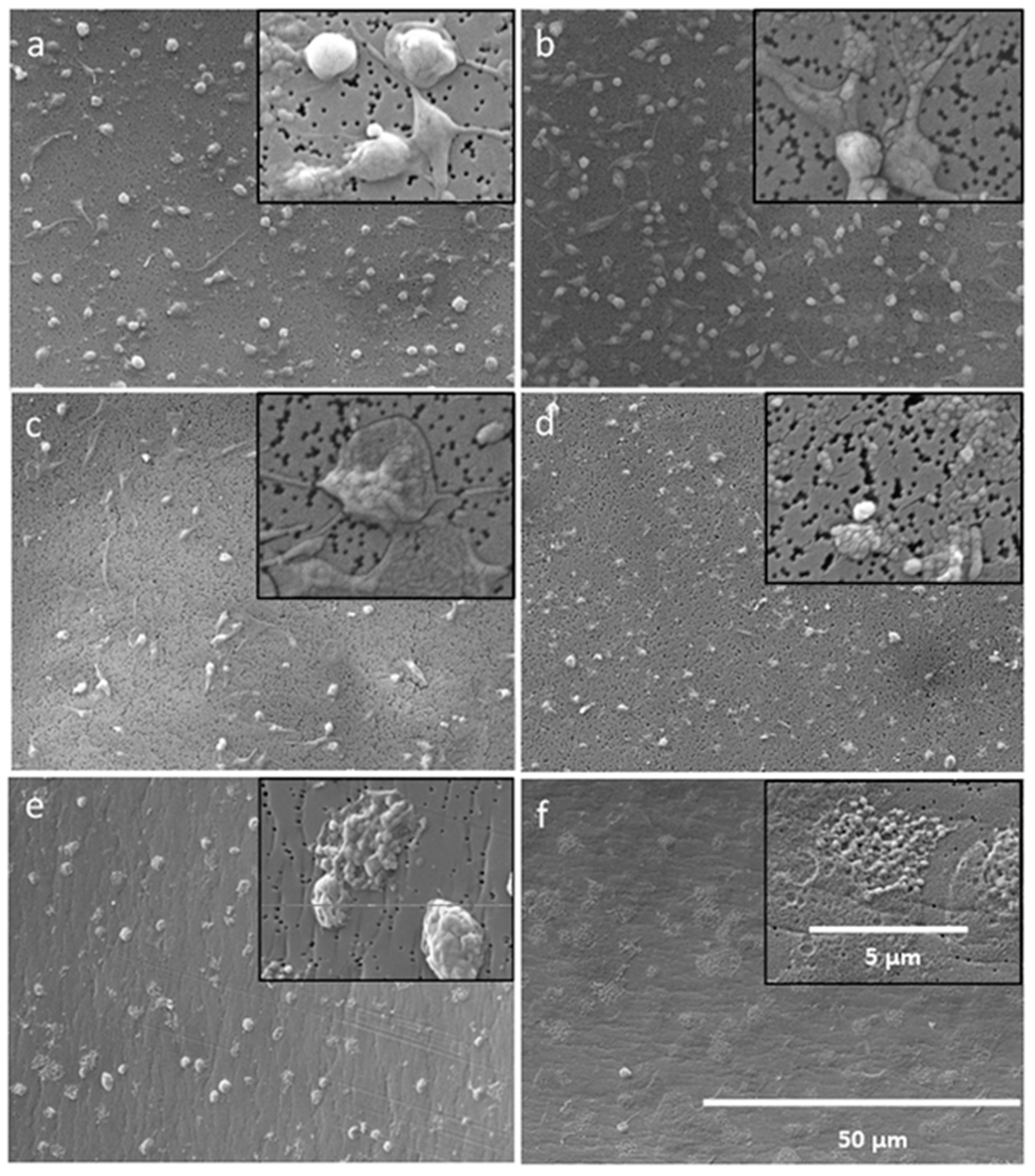
41], and the random orientation might lead to suboptimal adsorption characteristics of the adsorbent. Overall, it can be said that the antimicrobial property of AMPs and also of PMB is lost through immobilization on a surface. This can be explained by the fact that the peptides lose the ability to diffuse into the bacterial membrane to make it permeable due to immobilization on a surface.
5. Materials and Methods
5.1. Materials and Bacterial Strain
LPS from E. coli 055: B5 and P. aeruginosa 10 as well as PMB and the human host defense peptide LL-37 (LL-37 trifluoroacetate salt) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Ciprofloxacin was purchased from Sigma-Aldrich (St. Louis, MO, USA). For the MIC study, bacterial strains from E. coli (ATCC 11303) and S. aureus (ATCC 12600) were purchased from American Type Culture Collection, Manassas, VA, USA. Cytokine quantification was carried out using a Magnetic Luminex Assay from R&D Systems (Minneapolis, MN, USA) on a Bio-plex-200 Analyzer (Bio-Rad Laboratories, Hercules, CA, USA).
5.2. Animicrobial Peptides
The AMPs used in this study are shown in
Table 6. Different acylations were chosen in order to optimize their binding affinity to LPS as well as their capability to neutralize LPS. Peptides were purchased from PolyPeptide Laboratories (San Diego, CA, USA) and were synthesized using FMOC-chemistry. Purity of the peptides were >96% as determined by RP-HPLC and MS. The AMPs were obtained in lyophilized form and dissolved in 0.1 M 2-(
N-Morpholino)ethansulfonacid (MES) buffer at a concentration of 1 mg/mL. The peptide solutions were prepared weekly and stored at 4 °C.
5.3. Blood Donation
The blood donations were approved by the ethics committee of Danube University Krems (21/01/2012) and were taken from healthy volunteers who had to sign an informed consent form. Tubes (Vacuettes) for blood donation were purchases from Greiner Holding AG (Kremsmünster, Austria). Plasma or serum was obtained by centrifuging blood or coagulated blood at 3500 g for 10 min.
5.4. Endotoxin Binding Affinity
The characterization of the affinity of AMPs to LPS from
E. coli and
P. aeruginosa was carried out by a fluorescent displacement assay based on BODIPY TR cadaverine (BC) [
42] in microtiter plates. BC is a fluorescent reagent that tightly binds via ionic bonds to the lipid A moiety of LPS and forms a complex. Complex formation quenches the fluorescence of BC. When molecules that interact with LPS are added, BC will be displaced from the complex, with concomitant dequenching of its fluorescence emission. BC was purchased from ThermoFischer Scientific (Waltham, MA, USA). In this study, the assay solution consisted of 7.5 μg/mL LPS and 2.1 μM Bodipy TR cadaverine in TRIS buffer (50 mM, pH 7).
AMPs were added at concentrations ranging from 565 to 3387 ng/mL to the assay solution. After 30 min of incubation, fluorescence measurements were carried out using a Synergy 2 microplate reader (BioTek, VT, USA) at excitation and emission wavelengths of 580 and 620 nm, respectively. The LPS binding affinity was calculated from the area under the curve, which was drawn as fluorescence intensity vs. AMP concentration using Microsoft Excel for Windows 2016.
5.5. Endotoxin Inactivation
The LPS neutralization study in serum was conducted using a chromogenic Limulus Amebocyte Lysate (LAL) test (Charles River Laboratories, MA, USA). The LAL test is the most established method for quantifying LPS. This method uses amebocyte lysate from Limulus polyphemus, wherein the presence of LPS triggers a defense mechanism that induces coagulation. The LAL test is used successfully for detection of LPS in a wide variety of industrial, pharmaceutical, and research applications, such as water supplies, parenteral fluids, drugs for intravenous administration, and certain biologic fluids (e.g., cerebrospinal fluid). Therefore, serum, which was obtained from freshly drawn blood, was spiked with 500 ng/mL AMPs, PMB or LL-37 before adding 5 ng/mL LPS from E. coli or P. aeruginosa. As positive and negative controls, native serum with and without LPS was used, respectively. Endotoxin activity was measured using the LAL assay according to the manufacturer’s specifications.
5.6. Inhibition of Cytokine Release
By adding LPS to the blood, leukocytes are stimulated to produce inflammatory mediators (cytokines). In this experiment it was tested whether, similar to PMB, the activation of leukocytes by LPS can be reduced by AMP. The capability of AMPs to reduce or inhibit cytokine release by neutralizing endotoxins was evaluated in fresh heparinized (5 IU/mL) blood spiked with 150 ng/mL AMPs, PMB, or LL-37 followed by adding 0.5 ng/mL endotoxin from E. coli. Blood without any additive and blood spiked with endotoxins was used as the negative and positive control, respectively. After 4 h of incubation on a roller mixer at 37 °C, the plasma was separated by centrifugation and cytokine quantification (TNF-α, IL-1β, IL-6, and IL-8) was carried out.
5.7. Minimum Inhibitory Concentration (MIC)
The determination of the MIC was carried out in aqueous solution (0.01 M phosphate buffer, 0.0027 M potassium chloride and 0.137 M sodium chloride, pH 7.4) as well as in 4% human serum albumin (HSA; Kedrion Biopharmaceuticals, Gallicano, Italy) based on the standardized method described in DIN 58940-8 [
43]. Additionally, the MIC testing was also performed in aqueous solution containing 5 IU/mL heparin. For bacterial growth, a Müller-Hinton broth (Carl Roth, Karlsruhe, Germany) was used. The inoculum with 2 × 10
5–8 × 10
5 colony forming units (CFU) per mL was spiked with AMPs, PMB, LL-37, and commercially available antibiotics at concentrations from 0.0625 to 128 μg/mL. After overnight incubation at 37 °C, the MIC was determined by turbidity measurement at 620 nm using an Anthos ht3 plate reader (Biochrom, Cambridge, UK).
5.8. 1-N-Phenylnaphtylamine (NPN) Uptake Assay
The uptake of the fluorescent dye 1-
N-phenylnaphthylamine (NPN; Merck, Darmstadt, Germany) allows the measurement of permeability changes of the outer membrane of Gram-negative bacteria [
23,
44]. Hydrophobic substances such as NPN are excluded by an intact cell membrane. However, once damaged, the entry of NPN to the phospholipid layer is possible, resulting in prominent fluorescence. According to Koh et al. [
30], a bacterial suspension of
E. coli with an optical density of 0.4 at 600 nm (OD
600) was incubated with 40 μM NPN dye in equal volumes. AMPs were added to a final concentration ranging from 8 to 64 μg/mL to the bacteria suspension followed by incubation for 1 h at 37 °C. After incubation, fluorescence was measured at excitation and emission wavelengths of 335 nm and 405 nm, respectively, using a Synergy2 microplate reader (BioTek, Winooski, VT, USA).
5.9. Scanning Electron Microscopy (SEM)
In order to verify and to visualize the impact of AMPs on the bacterial membrane, electron microscopy was conducted. Therefore, bacterial suspensions of E. coli and S. aureus with a concentration of 2 × 105–8 × 105 CFU/mL were incubated with 128 μg/mL of one AMP (LF11-322-C8) and PMB in a microplate over night at 37 °C. After overnight culture, the bacteria were prepared for SEM. To achieve this, the bacteria suspensions were filtered through a sterile filter (Swinnex filter holders, Merck Millipore, Burlington, MA, USA). The filters with the collected bacteria were than washed with PBS (10 mM, pH 7.2) and fixed with a 2.5% glutaraldehyde solution (Carl Roth, Karlsruhe, Deutschland). The fixed specimens were gradually dehydrated by increasing ethanol solutions (10% to 100%) and sputtered with gold (Q150R ES Sputter Coater, Quorum Technologies Ltd., East Sussex, UK) prior to examination with a scanning electron microscope (FlexSEM 1000, Hitachi, Tokyo, Japan).
5.10. Hemolysis
Hemolysis was tested with fresh citrate anticoagulated blood from healthy volunteers as well as with blood cells which were suspended in phosphate buffered saline (10 mM pH 7.4). Afterwards, 100 μL AMP solution was added to 500 μL blood to finally achieve AMP concentrations of 8, 32, and 128 μg/mL. As the negative and positive control, 100 μL PBS buffer and 100 μL 12% Triton-X buffer was added to 500 μL blood, respectively. After incubation on an orbital shaker for 60 min at 37 °C, the cells were removed by centrifugation and 100 μL of the supernatant were transferred to a 96 well microplate for hemolysis measurement. The hemolysis was calculated from the extinction at the wavelengths 562, 578, and 598 nm with a Synergy 2 microplate reader (BioTek, Winooski, VT, USA) according to the method published by Kahn [
24].
5.11. Activation of the Clotting Cascade
To check the influence of AMPs on heparin and citrate anticoagulation, freshly heparinized (0.5 IU/mL) and citrated (17 mM) plasma was spiked with 128 μg/mL AMP, PMB, and LL-37. In addition, conventional antibiotics with a final plasma level of 128 μg/mL were included in the test. Plasma spiked with MES buffer without AMP was used as the negative control. Plasma with 128 μg/mL protamine, which is used as antidote for heparin and 2 μL calcium (500 mM)- magnesium (250 mM) chloride solution for citrate binding, was used as the positive control. Samples were incubated at 37 °C for 120 min. Coagulation was determined by turbidity measurement at 600 nm using a well plate reader.
5.12. Clotting Batch Test in Citrate Plasma
Since the acylated LF11-322 peptides cause plasmatic coagulation of citrate anticoagulated plasma, separate clotting experiments were performed with this peptide group. The aim was to check whether the coagulation activation is dependent on the AMP concentration and on the acyl chain length. For this purpose, plasma from freshly drawn citrated blood was spiked with different amounts (0, 8, 32, 128, and 256 μg/mL) of LF11-322 peptides, LL-37, and PMB. Plasma without additives served as the negative control, whereas plasma spiked with CaCl2/MgCl2 (500/250 mM) solution to restore the physiological calcium and magnesium level in plasma was used as the positive control. Incubation was performed at 37 °C for 60 min. Clot formation was determined by turbidity measurement at 600 nm using a plate reader. In order to ensure that the measured turbidity of the plasma is caused by fibrin formation, the fibrinogen and D-Dimer concentrations were additionally determined. During plasmatic clotting, the fibrinogen level decreases and the D-Dimer level in serum increases. Both were analyzed on a Sysmex CA560 with the corresponding reagents (Siemens Healthineers, Erlangen, Germany).
5.13. Platelets Activation
Since it was observed that some AMPs are able to activate the plasmatic coagulation system of heparinized and citrate anticoagulated blood, the influence of this peptides on platelets was investigated. For this purpose, fresh heparinized and citrated blood were collected from three donors. The blood was centrifuged at 500 g for 5 min and platelet rich plasma (PRP) was obtained. The PRP was incubated at 37 °C for 2 h with 128 μg/mL AMP, PMB, and LL-37. PRP without any additives served as the control. After incubation, the platelet number was determined by a Sysmex Automated Hematology Analyzer KX-21-N (Kobe, Japan). In addition, the PRP samples were prepared for SEM to visualize the morphology of the thrombocytes. After incubation, 10 μL PRP were diluted with 990 μL PBS (0.01 M) and filtered through a 0.2 μm membrane (Membrane Filter Isopore 0.2 μm, Merck, Darmstadt, Germany). The platelets were retained on the membrane, washed with 1 mL PBS, and fixed with 1 mL glutaraldehyde solution (2.5% glutaraldehyde in physiological saline solution). After an incubation time of 60 min, the samples were dehydrated using a series of ethanol (30%, 50%, 60%, 70%, 80%, 90%, and 100%). Afterwards the samples were dried overnight at RT, sputtered with gold and visualized with a scanning electron microscope (FLEXSem 1000, Hitachi, Japan).
5.14. Flow Cytometry
The measurement of the membrane destabilization of leukocytes by AMPs was carried out by flow cytometry. In order to determine the cytotoxic properties of the tested AMPs on human leukocytes, fresh citrate anticoagulated blood was mixed with 128 μg/mL AMP and incubated for 60 min at 37 °C. In addition, blood samples were incubated with 128 μg/mL PMB and ciprofloxacin. Whole blood without AMPs or antibiotics was used as a control. After incubation, the proportion of apoptotic and dead cells was determined by flow cytometry. To achieve this, blood samples were diluted 1:100 with 0.01 M phosphate buffered saline and centrifuged at 300 g for 10 min. The supernatant was removed, and cells were stained with 10 μL CD45 pacific blue (leukocyte marker; Beckman coulter, Brea, CA) and 10 μL 7-AAD (a marker for dead cells; Sigma-Aldrich, Steinheim, Germany) for 15 min at room temperature in the dark. After incubation, 1.8 mL lysis buffer (1:10 dilution in distilled water) was added, mixed, and incubated for 10 min at room temperature in the dark. This was followed by two washing steps (2300 g, 5 min, room temperature) and the cells were resuspended in a final volume of 200 μL 0.01 M PBS for measurement. Flow cytometry was performed using a Gallios flow cytometer (Beckman Coulter, Brea, CA, USA) equipped with 405 nm, 488 nm, and 638 nm lasers and the Kaluza Analysis Software (Beckman Coulter, Brea, CA, USA), which was used for measurement and data analysis. Blood cells which were incubated in 70% methanol before staining served as the positive control and blood cells without any additives were used as the negative control.
5.15. Complement Activation
The complement system is part of the innate immunity and consists of plasma proteins that can be activated in the course of the immune response on numerous foreign surfaces. Separate tests were performed to check whether the AMPs used in this study are able to activate the human complement system. Plasma from fresh donated blood, anticoagulated with citrate (5 mM), was spiked with 128 μg/mL AMP, PMB and LL-37 and incubated for 60 min at 37 °C on a roller mixer. Plasma spiked with 1 μg/mL LPS from E. coli served as the positive control and plasma without any additive was used as the negative control. After incubation, the complement system was stopped by adding 100 μL ice cold EDTA solution (10%, w/v) per mL plasma. Samples were stored at −80 °C until quantification of the complement factor C3a by ELISA (C3a des Arg ELISA kit, Abcam, Cambridge, UK) was performed.
5.16. Antimicrobial Properties of Immobilized AMPs
In order to determine whether the AMPs retain their antimicrobial property after immobilizing onto a surface, selected AMPs and PMB were covalently bound to agarose beads under sterile conditions. Therefore, the most hydrophilic and the most hydrophobic AMP from each peptide family were chosen, namely the non-acylated LF11-322 and LF11-227 and the C14-acylated M-LF11-322 and M-LF11-227 peptides. The immobilization was achieved by adding 400 μL peptide solution (1 mg/mL) on N-hydroxysuccinimid (NHS) activated agarose beads (Pierce spin columns, ThermoFisher Scientific, MA, USA) followed by two hours of incubation at room temperature in an overhead shaker. The activated agarose contains NHS ester functional groups that react with primary amines on AMPs to form stable amide linkages. The coupling reaction is performed in an amine-free buffer at pH 7.4 (PBS). After incubation, the peptide content of the supernatant was quantified (Pierce 660 nm Protein Assay, ThermoFisher Scientific, Waltham, MA, USA) in order to determine the coupling efficiency.
To check the antimicrobial properties, 200 μL bacteria suspensions of
E. coli (2 × 10
5–8 × 10
5 CFU/mL) were incubated with 5%, 10%, and 20% (
v/v) of the AMP-immobilized adsorbent in 96 microplate wells over night at 37 °C. After incubation, bacterial growth of the supernatant was determined by OD
620 nm measurement using an Anthos HT3 microplate reader. To determine whether the antimicrobial effect is caused by a peptide release from the adsorbent surface, additional release samples were performed according to the protocol of Rapsch et al. [
37]. For this purpose, 20% (
v/v) AMP adsorbent was incubated in Müller-Hinton broth for 2 h. The adsorbent was then centrifuged and the supernatant was incubated with the same bacteria suspension under the same conditions as described above. In addition, adsorbent with Tris deactivated surface was used as control adsorbent and the bacteria suspension without adsorbent was used as the positive control.
5.17. Statistics
All tests were carried out at least in triplicate. Calculations of standard deviations and area under the curve were carried out using Microsoft Excel 2010 (Microsoft, WA, USA). All other statistical calculations were carried out with SigmaStat for Windows 2.03. The Kolmogorov-Smirnov Test was applied in order to check the data for normal distribution. Normal distributed data were compared using the t-Test. For data showing no normal distribution, the Mann-Whitney Rank Sum Test was used. p-values of ≤ 0.05 were considered as statistically significant.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}