Atherosclerosis and Coenzyme Q10

 ,
, 

Abstract

1. Methods
2. Atherosclerosis: Old and New Approaches
3. Atherosclerosis Treatment
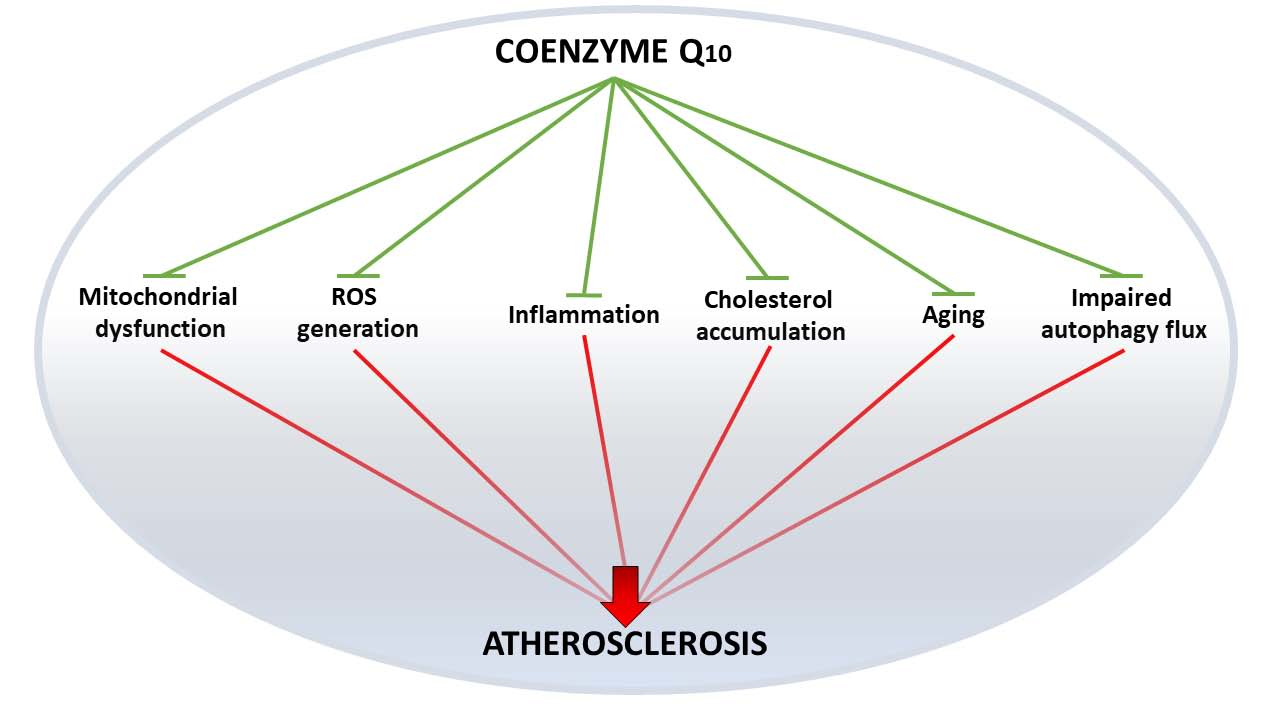
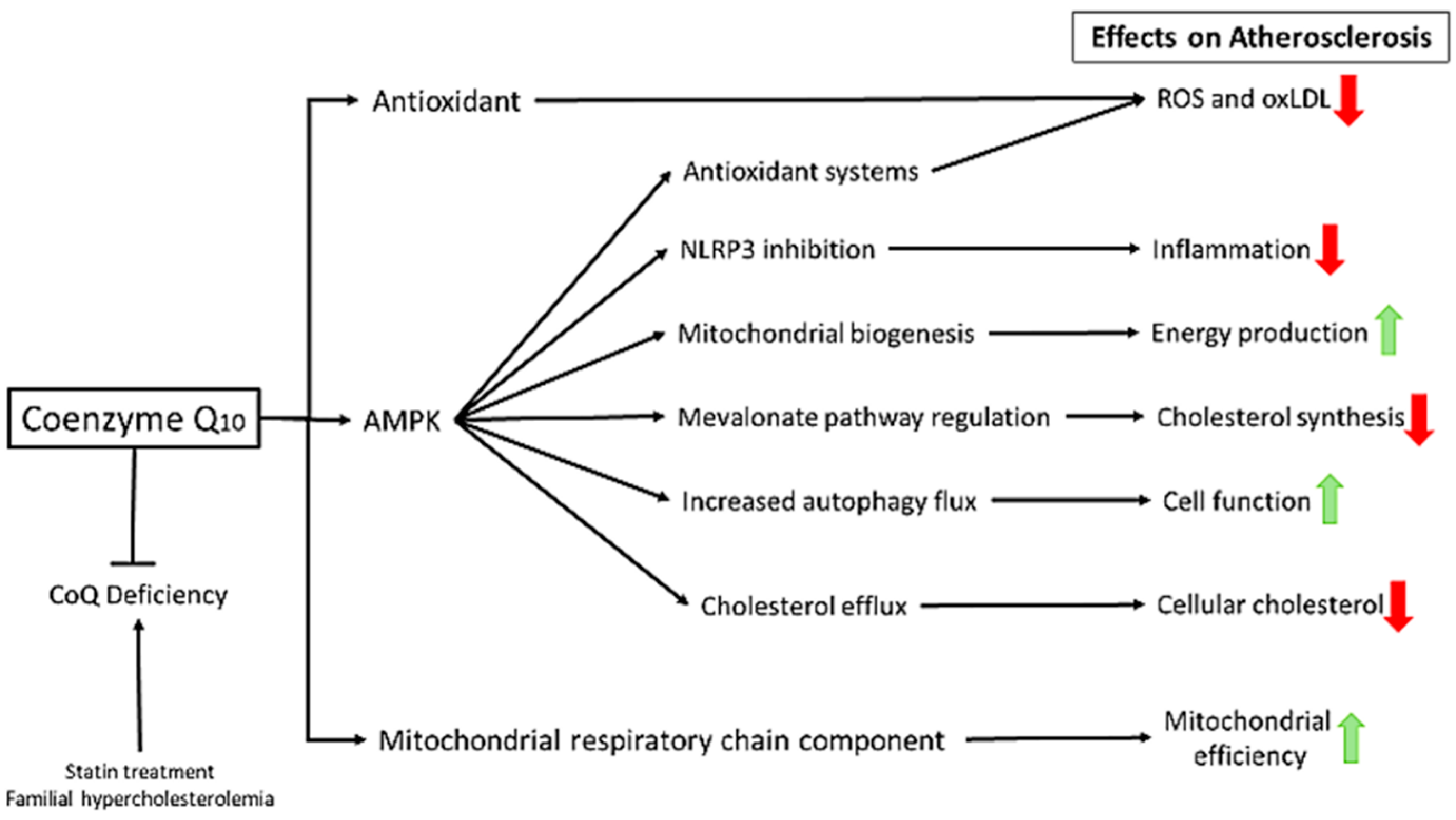
4. Coenzyme Q10: A Panacea?
5. Familial Hypercholesterolemia and Atherosclerosis
6. CoQ and Inflammation
7. CoQ and Statin Myopathy
8. CoQ Supplementation
9. Looking behind Atherosclerosis: Aging
10. Future Research Directions
Funding
Conflicts of Interest
Abbreviations
| ABCA1 | ATP-binding cassette transport A1 |
| ABCG1 | ATP-binding cassette transport G1 |
| AMPK | AMP-activated protein kinase |
| ASO | Antisense Oligonucleotide |
| CoQ | Coenzyme Q10 |
| CVD | Cardiovascular diseases |
| EC | Endothelial Cell |
| FPP | Farnesyl Pyrophosphate |
| HDL | High Density Lipoprotein |
| HMGCR | 3-Hydroxy-3-Methyl-Glutaryl-Coenzyme A Reductase |
| IL | Interleukin |
| LDL | Low Density Lipoprotein |
| LDL-R | Low Density Lipoprotein Receptor |
| NLRP3 | Nucleotide-binding domain leucine-rich-containing family pyrin domain-containing-3 |
| mtDNA | Mitochondrial DNA |
| MRC | Mitochondrial Respiratory Chain |
| MRC | Mitochondrial Respiratory Chain |
| mtROS | Mitochondrial ROS |
| MTP | Microsomal Transfer Proteins |
| NLRP3 | Nucleotide-binding domain leucine-rich-containing family pyrin domain-containing-3 |
| NO | Nitric Oxid |
| NPC1L1 | N-terminal Niemann-Pick C1-like protein 1 |
| oxLDL | Oxidized LDL |
| OXPHOS | Mitochondrial Oxidative Phosphorylation System |
| PCSK9 | Proprotein Convertase Subtilisin/Kexin type 9 |
| PPAR-α | Peroxisome proliferator-activated receptor-alpha |
| ROS | Reactive Oxygen Species |
| RCT | Reverse Cholesterol Transport |
| SAMS | Statin-Associated Muscle Symptoms |
| SREBP-2 | Sterol regulatory element-binding proteins 2 |
| TNF-α | Tumor Necrosis Factor-α |
| VSMCs | Vascular Smooth Muscle Cells |
References
- Thomas, H.; Diamond, J.; Vieco, A.; Chaudhuri, S.; Shinnar, E.; Cromer, S.; Perel, P.; Mensah, G.A.; Narula, J.; Johnson, C.O.; et al. Global Atlas of Cardiovascular Disease 2000–2016. Glob. Heart 2018, 13, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Fisher, E.A.; Feig, J.E.; Hewing, B.; Hazen, S.L.; Smith, J.D. High-density lipoprotein function, dysfunction, and reverse cholesterol transport. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2813–2820. [Google Scholar] [CrossRef] [PubMed]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [PubMed]
- Komarova, Y.A.; Kruse, K.; Mehta, D.; Malik, A.B. Protein Interactions at Endothelial Junctions and Signaling Mechanisms Regulating Endothelial Permeability. Circ. Res. 2017, 120, 179–206. [Google Scholar] [CrossRef]
- Mestas, J.; Ley, K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc. Med. 2008, 18, 228–232. [Google Scholar] [CrossRef]
- Jones, D.P.; True, H.D.; Patel, J. Leukocyte Trafficking in Cardiovascular Disease: Insights from Experimental Models. Med. Inflamm. 2017, 2017, 9746169. [Google Scholar] [CrossRef]
- Insull, W. The pathology of atherosclerosis: Plaque development and plaque responses to medical treatment. Am. J. Med. 2009, 122, S3–S14. [Google Scholar] [CrossRef]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef]
- Woollard, K.J.; Geissmann, F. Monocytes in atherosclerosis: Subsets and functions. Nat. Rev. Cardiol. 2010, 7, 77–86. [Google Scholar] [CrossRef]
- Li, A.C.; Glass, C.K. The macrophage foam cell as a target for therapeutic intervention. Nat. Med. 2002, 8, 1235–1242. [Google Scholar] [CrossRef]
- Zhang, M.; Zhu, H.; Ding, Y.; Liu, Z.; Cai, Z.; Zou, M.H. AMP-activated protein kinase alpha1 promotes atherogenesis by increasing monocyte-to-macrophage differentiation. J. Biol. Chem. 2017, 292, 7888–7903. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Melnichenko, A.A.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Mechanisms of foam cell formation in atherosclerosis. J. Mol. Med. 2017, 95, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Hamada, M.; Nakamura, M.; Tran, M.T.; Moriguchi, T.; Hong, C.; Ohsumi, T.; Dinh, T.T.; Kusakabe, M.; Hattori, M.; Katsumata, T.; et al. MafB promotes atherosclerosis by inhibiting foam-cell apoptosis. Nat. Commun. 2014, 5, 3147. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.M.; Strong, A.; Tohyama, J.; Jin, X.; Morales, C.R.; Billheimer, J.; Millar, J.; Kruth, H.; Rader, D.J. Macrophage sortilin promotes LDL uptake, foam cell formation, and atherosclerosis. Circ. Res. 2015, 116, 789–796. [Google Scholar] [CrossRef]
- Shen, Z.X.; Chen, X.Q.; Sun, X.N.; Sun, J.Y.; Zhang, W.C.; Zheng, X.J.; Zhang, Y.Y.; Shi, H.J.; Zhang, J.W.; Li, C.; et al. Mineralocorticoid Receptor Deficiency in Macrophages Inhibits Atherosclerosis by Affecting Foam Cell Formation and Efferocytosis. J. Biol. Chem. 2017, 292, 925–935. [Google Scholar] [CrossRef]
- Tanaka, S.; Matsumoto, T.; Matsubara, Y.; Harada, Y.; Kyuragi, R.; Koga, J.I.; Egashira, K.; Nakashima, Y.; Yonemitsu, Y.; Maehara, Y. BubR1 Insufficiency Results in Decreased Macrophage Proliferation and Attenuated Atherogenesis in Apolipoprotein E-Deficient Mice. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef]
- Alexander, R.W.; Dzau, V.J. Vascular biology: The past 50 years. Circulation. 2000, 102. [Google Scholar] [CrossRef]
- Newby, A.C.; Zaltsman, A.B. Fibrous cap formation or destruction—The critical importance of vascular smooth muscle cell proliferation, migration and matrix formation. Cardiovasc. Res. 1999, 41, 345–360. [Google Scholar] [CrossRef]
- Badimon, L.; Vilahur, G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J. Intern. Med. 2014, 276, 618–632. [Google Scholar] [CrossRef]
- Badimon, L.; Padro, T.; Vilahur, G. Atherosclerosis, platelets and thrombosis in acute ischaemic heart disease. Eur. Heart J. Acute Cardiovasc. Care 2012, 1, 60–74. [Google Scholar] [CrossRef]
- Weber, C.; Badimon, L.; Mach, F.; van der Vorst, E.P.C. Therapeutic strategies for atherosclerosis and atherothrombosis: Past, present and future. Thromb. Haemost. 2017, 117, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Buhaescu, I.; Izzedine, H. Mevalonate pathway: A review of clinical and therapeutical implications. Clin. Biochem. 2007, 40, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Nußbaumer, B.; Glechner, A.; Kaminski-Hartenthaler, A.; Mahlknecht, P.; Gartlehner, G. Ezetimibe-Statin Combination Therapy: Efficacy and Safety as Compared with Statin Monotherapy. Deutsch. Ärztebl. Int. 2016, 113, 445–453. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stroes, E.; Colquhoun, D.; Sullivan, D.; Civeira, F.; Rosenson, R.S.; Watts, G.F.; Bruckert, E.; Cho, L.; Dent, R.; Knusel, B.; et al. Anti-PCSK9 Antibody Effectively Lowers Cholesterol in Patients With Statin Intolerance. J. Am. Coll. Cardiol. 2014, 63, 2541–2548. [Google Scholar] [CrossRef] [PubMed]
- Sirtori, C.R.; Pavanello, C.; Bertolini, S. Microsomal transfer protein (MTP) inhibition—A novel approach to the treatment of homozygous hypercholesterolemia. Ann. Med. 2014, 46, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Geary, R.S.; Baker, B.F.; Crooke, S.T. Clinical and Preclinical Pharmacokinetics and Pharmacodynamics of Mipomersen (Kynamro®): A Second-Generation Antisense Oligonucleotide Inhibitor of Apolipoprotein B. Clin. Pharmacokinet. 2015, 54, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Okopień, B.; Bułdak, Ł.; Bołdys, A. Current and future trends in the lipid lowering therapy. Pharmacol. Rep. 2016, 68, 737–747. [Google Scholar] [CrossRef]
- Thompson, P.D.; Panza, G.; Zaleski, A.; Taylor, B. Statin-Associated Side Effects. J. Am. Coll. Cardiol. 2016, 67, 2395–2410. [Google Scholar] [CrossRef]
- Nashimoto, S.; Sato, Y.; Takekuma, Y.; Sugawara, M. Inhibitory effect of ezetimibe can be prevented by an administration interval of 4 h between α-tocopherol and ezetimibe. Biopharmaceut. Drug Dispos. 2017, 38, 280–289. [Google Scholar] [CrossRef]
- Savarese, G.; de Ferrari, G.M.; Rosano, G.M.C.; Perrone-Filardi, P. Safety and efficacy of ezetimibe: A meta-analysis. Int. J. Cardiol. 2015, 201, 247–252. [Google Scholar] [CrossRef]
- Roth, E.M. A safety evaluation of evolocumab. Expert Opin. Drug Saf. 2018, 17, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Wallemacq, C. Evolocumab (Repatha®): A human monoclonal antibody against PCSK9 protein as potent cholesterol-lowering therapy. Revue Med. Liege 2017, 72, 505–512. [Google Scholar]
- Berberich, A.J.; Hegele, R.A. Lomitapide for the treatment of hypercholesterolemia. Expert Opin. Pharmacother. 2017, 18, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, R.; Matveyenko, A.; Thomas, T.; Pavlyha, M.; Ngai, C.; Holleran, S.; Ramakrishnan, R.; Ginsberg, H.N.; Karmally, W.; Marcovina, S.M.; et al. Effects of mipomersen, an apolipoprotein B100 antisense, on lipoprotein (a) metabolism in healthy subjects. J. Lipid Res. 2018, 59, 2397–2402. [Google Scholar] [CrossRef] [PubMed]
- Seneviratne, A.N.; Monaco, C. Role of inflammatory cells and toll-like receptors in atherosclerosis. Curr. Vasc. Pharmacol. 2015, 13, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Bäck, M.; Hansson, G.K. Anti-inflammatory therapies for atherosclerosis. Nat. Rev. Cardiol. 2015, 12, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Diamantis, E.; Kyriakos, G.; Quiles-Sanchez, L.V.; Farmaki, P.; Troupis, T. The Anti-Inflammatory Effects of Statins on Coronary Artery Disease: An Updated Review of the Literature. Curr. Cardiol. Rev. 2017, 13, 209–216. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef]
- Libby, P. Atherosclerosis: The New View. Sci. Am. 2002, 286, 46–55. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; di Raimondo, D.; Pecoraro, R.; Arnao, V.; Pinto, A.; Licata, G. Atherosclerosis as an inflammatory disease. Curr. Pharm. Des. 2012, 18, 4266–4288. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Assisted Living in the Atheroma: Elderly Macrophages Promote Plaques. Cell Metab. 2016, 24, 779–781. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-Y.; Li, C.-J.; Hou, M.-F.; Chu, P.-Y. New Insights into the Role of Inflammation in the Pathogenesis of Atherosclerosis. Int. J. Mol. Sci. 2017, 18, 2034. [Google Scholar] [CrossRef] [PubMed]
- Fredman, G.; Tabas, I. Boosting Inflammation Resolution in Atherosclerosis. Am. J. Pathol. 2017, 187, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Kavurma, M.M.; Rayner, K.J.; Karunakaran, D. The walking dead. Curr. Opin. Lipidol. 2017, 28, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Nowak, W.N.; Deng, J.; Ruan, X.Z.; Xu, Q. Reactive Oxygen Species Generation and Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e41–e52. [Google Scholar] [CrossRef]
- Peng, W.; Cai, G.; Xia, Y.; Chen, J.; Wu, P.; Wang, Z.; Li, G.; Wei, D. Mitochondrial Dysfunction in Atherosclerosis. DNA Cell Biol. 2019, 38, 597–606. [Google Scholar] [CrossRef]
- De Lavera, I.; Pavon, A.D.; Paz, M.V.; Oropesa-Avila, M.; de la Mata, M.; Alcocer-Gomez, E.; Garrido-Maraver, J.; Cotan, D.; Alvarez-Cordoba, M.; Sanchez-Alcazar, J.A. The Connections Among Autophagy, Inflammasome and Mitochondria. Curr. Drug Targets 2017, 18, 1030–1038. [Google Scholar] [CrossRef]
- Madamanchi, N.R.; Runge, M.S. Mitochondrial dysfunction in atherosclerosis. Circ. Res. 2007, 100, 460–473. [Google Scholar] [CrossRef]
- Yabal, M.; Calleja, D.J.; Simpson, D.S.; Lawlor, K.E. Stressing out the mitochondria: Mechanistic insights into NLRP3 inflammasome activation. J. Leukoc. Biol. 2019, 105, 377–399. [Google Scholar] [CrossRef] [PubMed]
- Baldrighi, M.; Mallat, Z.; Li, X. NLRP3 inflammasome pathways in atherosclerosis. Atherosclerosis 2017, 267, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Karasawa, T.; Takahashi, M. Role of NLRP3 Inflammasomes in Atherosclerosis. J. Atheroscler. Thromb. 2017, 24, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- An, N.; Gao, Y.; Si, Z.; Zhang, H.; Wang, L.; Tian, C.; Yuan, M.; Yang, X.; Li, X.; Shang, H.; et al. Regulatory Mechanisms of the NLRP3 Inflammasome, a Novel Immune-Inflammatory Marker in Cardiovascular Diseases. Front. Immunol. 2019, 10, 1592. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Turunen, M.; Olsson, J.; Dallner, G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta 2004, 1660, 171–199. [Google Scholar] [CrossRef]
- Gutierrez-Mariscal, F.M.; Yubero-Serrano, E.M.; Villalba, J.M.; Lopez-Miranda, J. Coenzyme Q10: From bench to clinic in aging diseases, a translational review. Crit. Rev. Food Sci. Nutr. 2018, 1–18. [Google Scholar] [CrossRef]
- De Barcelos, I.P.; Haas, R.H. CoQ10 and Aging. Biology 2019, 8, 28. [Google Scholar] [CrossRef]
- Rahmani, E.; Jamilian, M.; Samimi, M.; Zarezade Mehrizi, M.; Aghadavod, E.; Akbari, E.; Tamtaji, O.R.; Asemi, Z. The effects of coenzyme Q10 supplementation on gene expression related to insulin, lipid and inflammation in patients with polycystic ovary syndrome. Gynecol. Endocrinol. 2018, 34, 217–222. [Google Scholar] [CrossRef]
- Sharma, A.; Fonarow, G.C.; Butler, J.; Ezekowitz, J.A.; Felker, G.M. Coenzyme Q10 and Heart Failure: A State-of-the-Art Review. Circ. Heart Fail. 2016, 9, e002639. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.; Derosa, G.; Miconi, A.; Laghi, L.; Nascetti, S.; Gaddi, A. Treatment of Massive Hypertriglyceridemia Resistant to PUFA and Fibrates: A Possible Role for the Coenzyme Q10? Biofactors 2005, 23, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.G.; Fogacci, F.; Colletti, A. Food and plant bioactives for reducing cardiometabolic disease risk: An evidence based approach. Food Funct. 2017, 8, 2076–2088. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, R.; Akbari, M.; Sharifi, N.; Lankarani, K.B.; Moosazadeh, M.; Kolahdooz, F.; Taghizadeh, M.; Asemi, Z. The Effects of Coenzyme Q10 Supplementation on Blood Pressures Among Patients with Metabolic Diseases: A Systematic Review and Meta-analysis of Randomized Controlled Trials. High. Blood Press Cardiovasc. Prev. 2018, 25, 41–50. [Google Scholar] [CrossRef]
- Sarter, B. Coenzyme Q10 and cardiovascular disease: A review. J. Cardiovasc. Nurs. 2002, 16, 9–20. [Google Scholar] [CrossRef]
- Defesche, J.C.; Gidding, S.S.; Harada-Shiba, M.; Hegele, R.A.; Santos, R.D.; Wierzbicki, A.S. Familial hypercholesterolaemia. Nat. Rev. Dis. Primers 2017, 3, 17093. [Google Scholar] [CrossRef]
- Suárez-Rivero, J.M.; de la Mata, M.; Pavón, A.D.; Villanueva-Paz, M.; Povea-Cabello, S.; Cotán, D.; Álvarez-Córdoba, M.; Villalón-García, I.; Ybot-González, P.; Salas, J.J.; et al. Intracellular cholesterol accumulation and coenzyme Q10 deficiency in Familial Hypercholesterolemia. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 3697–3713. [Google Scholar] [CrossRef]
- Griffin, S.; Preta, G.; Sheldon, I.M. Inhibiting mevalonate pathway enzymes increases stromal cell resilience to a cholesterol-dependent cytolysin. Sci. Rep. 2017, 7, 17050. [Google Scholar] [CrossRef]
- Marzetti, E.; Csiszar, A.; Dutta, D.; Balagopal, G.; Calvani, R.; Leeuwenburgh, C. Role of mitochondrial dysfunction and altered autophagy in cardiovascular aging and disease: From mechanisms to therapeutics. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H459–H476. [Google Scholar] [CrossRef]
- Fiordelisi, A.; Iaccarino, G.; Morisco, C.; Coscioni, E.; Sorriento, D. NFkappaB is a Key Player in the Crosstalk between Inflammation and Cardiovascular Diseases. Int. J. Mol. Sci. 2019, 20, 1599. [Google Scholar] [CrossRef]
- Chokchaiwong, S.; Kuo, Y.T.; Lin, S.H.; Hsu, Y.C.; Hsu, S.P.; Liu, Y.T.; Chou, A.J.; Kao, S.H. Coenzyme Q10 serves to couple mitochondrial oxidative phosphorylation and fatty acid β-oxidation, and attenuates NLRP3 inflammasome activation. Free Radic. Res. 2018, 52, 1445–1455. [Google Scholar] [CrossRef] [PubMed]
- Cordero, M.D.; Alcocer-Gómez, E.; de Miguel, M.; Culic, O.; Carrión, A.M.; Alvarez-Suarez, J.M.; Bullón, P.; Battino, M.; Fernández-Rodríguez, A.; Sánchez-Alcazar, J.A. Can coenzyme q10 improve clinical and molecular parameters in fibromyalgia? Antioxid. Redox Signal. 2013, 19, 1356–1361. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yan, X.; Xia, M.; Yang, Y.; Li, D.; Li, X.; Song, F.; Ling, W. Coenzyme Q10 Promotes Macrophage Cholesterol Efflux by Regulation of the Activator Protein-1/miR-378/ATP-Binding Cassette Transporter G1–Signaling Pathway. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1860–1870. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, H.; Hao, Y.; Xu, L.; Zhang, T.; Liu, Y.; Guo, L.; Zhu, L.; Pei, Z. Coenzyme Q10 protects against hyperlipidemia-induced cardiac damage in apolipoprotein E-deficient mice. Lipids Health Dis. 2018, 17, 279. [Google Scholar] [CrossRef]
- Allen, R.M.; Vickers, K.C. Coenzyme Q10 Increases Cholesterol Efflux and Inhibits Atherosclerosis Through MicroRNAs. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1795–1797. [Google Scholar] [CrossRef]
- Chen, K.; Chen, X.; Xue, H.; Zhang, P.; Fang, W.; Chen, X.; Ling, W. Coenzyme Q10 attenuates high-fat diet-induced non-alcoholic fatty liver disease through activation of the AMPK pathway. Food Funct. 2019, 10, 814–823. [Google Scholar] [CrossRef]
- Ou, H.; Liu, C.; Feng, W.; Xiao, X.; Tang, S.; Mo, Z. Role of AMPK in atherosclerosis via autophagy regulation. Sci. China Life Sci. 2018, 61, 1212–1221. [Google Scholar] [CrossRef]
- Nussenzweig, S.C.; Verma, S.; Finkel, T. The role of autophagy in vascular biology. Circ. Res. 2015, 116, 480–488. [Google Scholar] [CrossRef]
- Shao, B.-Z.; Han, B.-Z.; Zeng, Y.-X.; Su, D.-F.; Liu, C. The roles of macrophage autophagy in atherosclerosis. Acta Pharmacol. Sin. 2016, 37, 150–156. [Google Scholar] [CrossRef]
- Wang, J.; Ma, A.; Zhao, M.; Zhu, H. AMPK activation reduces the number of atheromata macrophages in ApoE deficient mice. Atherosclerosis 2017, 258, 97–107. [Google Scholar] [CrossRef]
- Deshmukh, A.S.; Long, Y.C.; de Castro Barbosa, T.; Karlsson, H.K.; Glund, S.; Zavadoski, W.J.; Gibbs, E.M.; Koistinen, H.A.; Wallberg-Henriksson, H.; Zierath, J.R. Nitric oxide increases cyclic GMP levels, AMP-activated protein kinase (AMPK)alpha1-specific activity and glucose transport in human skeletal muscle. Diabetologia 2010, 53, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Ye, Z.X.; Wang, X.F.; Chang, J.; Yang, M.W.; Zhong, H.H.; Hong, F.F.; Yang, S.L. Nitric oxide bioavailability dysfunction involves in atherosclerosis. Biomed. Pharmacother. 2018, 97, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.-J.; Kim, J.W.; Chung, H.S.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Choi, K.M.; Baik, S.H.; Yoo, H.J. Knockdown of sestrin2 increases pro-inflammatory reactions and ER stress in the endothelium via an AMPK dependent mechanism. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 1436–1444. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.A.; Sung, J.Y.; Woo, C.-H.; Choi, H.C. Laminar shear stress suppresses vascular smooth muscle cell proliferation through nitric oxide-AMPK pathway. Biochem. Biophys. Res. Commun. 2017, 490, 1369–1374. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.; Moulis, M.; Roth, L.; Martinet, W.; Vindis, C.; Bennett, M.R.; De Meyer, G.R.Y. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neointima formation and atherogenesis. Autophagy 2015, 11, 2014–2032. [Google Scholar] [CrossRef]
- Taylor, B.A.; Thompson, P.D. Muscle-related side-effects of statins. Curr. Opin. Lipidol. 2015, 26, 221–227. [Google Scholar] [CrossRef]
- Serban, M.-C.; Colantonio, L.D.; Manthripragada, A.D.; Monda, K.L.; Bittner, V.A.; Banach, M.; Chen, L.; Huang, L.; Dent, R.; Kent, S.T.; et al. Statin Intolerance and Risk of Coronary Heart Events and All-Cause Mortality Following Myocardial Infarction. J. Am. Coll. Cardiol. 2017, 69, 1386–1395. [Google Scholar] [CrossRef]
- Phillips, P.S.; Haas, R.H. Statin myopathy as a metabolic muscle disease. Expert Rev. Cardiovasc. Ther. 2008, 6, 971–978. [Google Scholar] [CrossRef]
- Needham, M.; Mastaglia, F.L. Statin myotoxicity: A review of genetic susceptibility factors. Neuromuscul. Disord. 2014, 24, 4–15. [Google Scholar] [CrossRef]
- Baker, S.K.; Vladutiu, G.D.; Peltier, W.L.; Isackson, P.J.; Tarnopolsky, M.A. Metabolic myopathies discovered during investigations of statin myopathy. Can. J. Neurol. Sci. 2008, 35, 94–97. [Google Scholar] [CrossRef]
- Golomb, B.A.; Evans, M.A. Statin adverse effects: A review of the literature and evidence for a mitochondrial mechanism. Am. J. Cardiovasc. Drugs 2008, 8, 373–418. [Google Scholar] [CrossRef] [PubMed]
- Vladutiu, G.D.; Simmons, Z.; Isackson, P.J.; Tarnopolsky, M.; Peltier, W.L.; Barboi, A.C.; Sripathi, N.; Wortmann, R.L.; Phillips, P.S. Genetic risk factors associated with lipid-lowering drug-induced myopathies. Muscle Nerve 2006, 34, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Stroes, E.S.; Thompson, P.D.; Corsini, A.; Vladutiu, G.D.; Raal, F.J.; Ray, K.K.; Roden, M.; Stein, E.; Tokgözoğlu, L.; Nordestgaard, B.G.; et al. Statin-associated muscle symptoms: Impact on statin therapy-European Atherosclerosis Society Consensus Panel Statement on Assessment, Aetiology and Management. Eur. Heart J. 2015, 36, 1012–1022. [Google Scholar] [CrossRef] [PubMed]
- Vladutiu, G.D. Genetic predisposition to statin myopathy. Curr. Opin. Rheumatol. 2008, 20, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Brewer, H.B. Benefit-risk assessment of Rosuvastatin 10 to 40 milligrams. Am. J. Cardiol. 2003, 92, 23K–29K. [Google Scholar] [CrossRef]
- Taylor, B.A.; Lorson, L.; White, C.M.; Thompson, P.D. A randomized trial of coenzyme Q10 in patients with confirmed Statin Myopathy. Atherosclerosis 2015, 238, 329–335. [Google Scholar] [CrossRef]
- Tóth, Š.; Šajty, M.; Pekárová, T.; Mughees, A.; Štefanič, P.; Katz, M.; Spišáková, K.; Pella, J.; Pella, D. Addition of omega-3 fatty acid and coenzyme Q10 to statin therapy in patients with combined dyslipidemia. J. Basic Clin. Physiol. Pharmacol. 2017, 28, 327–336. [Google Scholar] [CrossRef]
- Mancini, G.B.J.; Baker, S.; Bergeron, J.; Fitchett, D.; Frohlich, J.; Genest, J.; Gupta, M.; Hegele, R.A.; Ng, D.; Pearson, G.J.; et al. Diagnosis, Prevention, and Management of Statin Adverse Effects and Intolerance: Canadian Consensus Working Group Update. Can. J. Cardiol. 2016, 32, S35–S65. [Google Scholar] [CrossRef]
- Spence, J.D.; Dresser, G.K. Overcoming Challenges with Statin Therapy. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef]
- Zhang, Y.; Aberg, F.; Appelkvist, E.L.; Dallner, G.; Ernster, L. Uptake of dietary coenzyme Q supplement is limited in rats. J. Nutr. 1995, 125, 446–453. [Google Scholar] [CrossRef]
- Bhagavan, H.N.; Chopra, R.K.; Craft, N.E.; Chitchumroonchokchai, C.; Failla, M.L. Assessment of coenzyme Q10 absorption using an in vitro digestion-Caco-2 cell model. Int. J. Pharm. 2007, 333, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Feng, Y.; Chen, G.C.; Qin, L.Q.; Fu, C.L.; Chen, L.H. Effects of coenzyme Q10 supplementation on inflammatory markers: A systematic review and meta-analysis of randomized controlled trials. Pharmacol. Res. 2017, 119, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, T.; Fujii, K.; Funahashi, I.; Fukutomi, N.; Hosoe, K. Safety Assessment of Coenzyme Q10 (CoQ10). Biofactors 2008, 32, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Liu, Y. Efficacy of coenzyme Q10 in patients with cardiac failure: A meta-analysis of clinical trials. BMC Cardiovasc. Disord. 2017, 17, 196. [Google Scholar] [CrossRef] [PubMed]
- Stojanovic, M.; Radenkovic, M. A meta-analysis of randomized and placebo-controlled clinical trials suggests that coenzyme Q10 at low dose improves glucose and HbA1c levels. Nutr. Res. 2017, 38, 1–12. [Google Scholar] [CrossRef]
- Zhu, Z.G.; Sun, M.X.; Zhang, W.L.; Wang, W.W.; Jin, Y.M.; Xie, C.L. The efficacy and safety of coenzyme Q10 in Parkinson’s disease: A meta-analysis of randomized controlled trials. Neurol. Sci. 2017, 38, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Masotta, N.E.; Martinefski, M.R.; Lucangioli, S.; Rojas, A.M.; Tripodi, V. High-dose coenzyme Q10-loaded oleogels for oral therapeutic supplementation. Int. J. Pharm. 2019, 556, 9–20. [Google Scholar] [CrossRef]
- Wang, Y.; Hekimi, S. Understanding Ubiquinone. Trends Cell Biol. 2016, 26, 367–378. [Google Scholar] [CrossRef]
- Huntington Study Group Pre2CARE Investigators. Safety and tolerability of high-dosage coenzyme Q10 in Huntington’s disease and healthy subjects. Mov. Disord. 2010, 25, 1924–1928. [Google Scholar] [CrossRef]
- Mehrabani, S.; Askari, G.; Miraghajani, M.; Tavakoly, R.; Arab, A. Effect of coenzyme Q10 supplementation on fatigue: A systematic review of interventional studies. Complement. Ther. Med. 2019, 43, 181–187. [Google Scholar] [CrossRef]
- Lyseng-Williamson, K.A. Idebenone: A Review in Leber’s Hereditary Optic Neuropathy. Drugs 2016, 76, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Kouga, T.; Takagi, M.; Miyauchi, A.; Shimbo, H.; Iai, M.; Yamashita, S.; Murayama, K.; Klein, M.B.; Miller, G.; Goto, T.; et al. Japanese Leigh syndrome case treated with EPI-743. Brain Dev. 2018, 40, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Zesiewicz, T.; Salemi, J.L.; Perlman, S.; Sullivan, K.L.; Shaw, J.D.; Huang, Y.; Isaacs, C.; Gooch, C.; Lynch, D.R.; Klein, M.B. Double-blind, randomized and controlled trial of EPI-743 in Friedreich’s ataxia. Neurodegener. Dis. Manag. 2018, 8, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Gueven, N. Idebenone for Leber’s hereditary optic neuropathy. Drugs Today 2016, 52, 173–181. [Google Scholar] [CrossRef]
- James, A.M.; Sharpley, M.S.; Manas, A.R.; Frerman, F.E.; Hirst, J.; Smith, R.A.; Murphy, M.P. Interaction of the Mitochondria-targeted Antioxidant MitoQ with Phospholipid Bilayers and Ubiquinone Oxidoreductases. J. Biol. Chem. 2007, 282, 14708–14718. [Google Scholar] [CrossRef]
- James, A.M.; Cocheme, H.M.; Murphy, M. Mitochondria-targeted redox probes as tools in the study of oxidative damage and ageing. Mech. Ageing Dev. 2005, 126, 982–986. [Google Scholar] [CrossRef]
- Onoue, S.; Uchida, A.; Kuriyama, K.; Nakamura, T.; Seto, Y.; Kato, M.; Hatanaka, J.; Tanaka, T.; Miyoshi, H.; Yamada, S. Novel solid self-emulsifying drug delivery system of coenzyme Q10 with improved photochemical and pharmacokinetic behaviors. Eur. J. Pharm. Sci. 2012, 46, 492–499. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, G.; Zhang, J.; Sun, N.; Duan, M.; Yan, Z.; Xia, Q. Novel lipid-free nanoformulation for improving oral bioavailability of coenzyme Q10. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef]
- Beg, S.; Javed, S.; Kohli, K. Bioavailability enhancement of coenzyme Q10: An extensive review of patents. Recent Patents Drug Deliv. Formul. 2010, 4, 245–255. [Google Scholar] [CrossRef]
- Li, H.; Chen, F. Preparation and quality evaluation of coenzyme Q10 long-circulating liposomes. Saudi J. Biol. Sci. 2017, 24, 797–802. [Google Scholar] [CrossRef]
- Head, T.; Daunert, S.; Goldschmidt-Clermont, P.J. The Aging Risk and Atherosclerosis: A Fresh Look at Arterial Homeostasis. Front. Genet. 2017, 8, 216. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Ogura, Y.; Koya, D. The protective role of Sirt1 in vascular tissue: Its relationship to vascular aging and atherosclerosis. Aging 2016, 8, 2290–2307. [Google Scholar] [CrossRef] [PubMed]
- Greene, M.A.; Loeser, R.F. Aging-related inflammation in osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1966–1971. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Maurya, P.K.; Noto, C.; Rizzo, L.B.; Rios, A.C.; Nunes, S.O.; Barbosa, D.S.; Sethi, S.; Zeni, M.; Mansur, R.B.; Maes, M.; et al. The role of oxidative and nitrosative stress in accelerated aging and major depressive disorder. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 65, 134–144. [Google Scholar] [CrossRef]
- Breitenbach, M.; Rinnerthaler, M.; Hartl, J.; Stincone, A.; Vowinckel, J.; Breitenbach-Koller, H.; Ralser, M. Mitochondria in ageing: There is metabolism beyond the ROS. FEMS Yeast Res. 2014, 14, 198–212. [Google Scholar] [CrossRef]
- Schniertshauer, D.; Gebhard, D.; Bergemann, J. Age-Dependent Loss of Mitochondrial Function in Epithelial Tissue Can Be Reversed by Coenzyme Q10. J. Aging Res. 2018, 2018, 6354680. [Google Scholar] [CrossRef]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 3565127. [Google Scholar] [CrossRef]
- Stefanatos, R.; Sanz, A. The role of mitochondrial ROS in the aging brain. FEBS Lett. 2018, 592, 743–758. [Google Scholar] [CrossRef]
- El-Kenawi, A.; Ruffell, B. Inflammation, ROS, and Mutagenesis. Cancer Cell 2017, 32, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Iurciuc, S.; Cimpean, A.M.; Mitu, F.; Heredea, R.; Iurciuc, M. Vascular aging and subclinical atherosclerosis: Why such a “never ending” and challenging story in cardiology? Clin. Interv. Aging 2017, 12, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.; Lukens, J.R.; Kanneganti, T.D. Mitochondria: Diversity in the regulation of the NLRP3 inflammasome. Trends Mol. Med. 2015, 21, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef]
- Cruz, C.S.D.; Kang, M.J. Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion 2018, 41, 37–44. [Google Scholar] [CrossRef]
- Hernández-Camacho, J.D.; Bernier, M.; López-Lluch, G.; Navas, P. Coenzyme Q10 Supplementation in Aging and Disease. Front. Physiol. 2018, 9, 44. [Google Scholar] [CrossRef]
- Marcheggiani, F.; Cirilli, I.; Orlando, P.; Silvestri, S.; Vogelsang, A.; Knott, A.; Blatt, T.; Weise, J.M.; Tiano, L. Modulation of Coenzyme Q10 content and oxidative status in human dermal fibroblasts using HMG-CoA reductase inhibitor over a broad range of concentrations. From mitohormesis to mitochondrial dysfunction and accelerated aging. Aging 2019, 11, 2565–2582. [Google Scholar] [CrossRef]
- Huo, J.; Xu, Z.; Hosoe, K.; Kubo, H.; Miyahara, H.; Dai, J.; Mori, M.; Sawashita, J.; Higuchi, K. Coenzyme Q10 Prevents Senescence and Dysfunction Caused by Oxidative Stress in Vascular Endothelial Cells. Oxid. Med. Cell. Longev. 2018, 2018, 3181759. [Google Scholar] [CrossRef]
- Katsuumi, G.; Shimizu, I.; Yoshida, Y.; Minamino, T. Vascular Senescence in Cardiovascular and Metabolic Diseases. Front. Cardiovasc. Med. 2018, 5, 18. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Strategy | Drug | Drawback | Source |
|---|---|---|---|
| To block endogenous cholesterol biosynthesis | Statins | Diabetes Myopathies | [27,28] |
| To block cholesterol intestinal absorption | Ezetimibe | Vitamin absorption inhibition | [29,30] |
| To reduce LDL receptor degradation | Evolocumab Alirocumab | Immunological response in rare cases High price | [31,32] |
| To disrupt LDL synthesis | Lomitapide Mipormesren | Hepatotoxicity Diarrhea Increased cardiovascular risk | [33,34] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; de la Mata, M.; Villanueva-Paz, M.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Suárez-Carrillo, A.; Talaverón-Rey, M.; Munuera, M.; et al. Atherosclerosis and Coenzyme Q10. Int. J. Mol. Sci. 2019, 20, 5195. https://doi.org/10.3390/ijms20205195
Suárez-Rivero JM, Pastor-Maldonado CJ, de la Mata M, Villanueva-Paz M, Povea-Cabello S, Álvarez-Córdoba M, Villalón-García I, Suárez-Carrillo A, Talaverón-Rey M, Munuera M, et al. Atherosclerosis and Coenzyme Q10. International Journal of Molecular Sciences. 2019; 20(20):5195. https://doi.org/10.3390/ijms20205195
Chicago/Turabian StyleSuárez-Rivero, Juan M., Carmen J. Pastor-Maldonado, Mario de la Mata, Marina Villanueva-Paz, Suleva Povea-Cabello, Mónica Álvarez-Córdoba, Irene Villalón-García, Alejandra Suárez-Carrillo, Marta Talaverón-Rey, Manuel Munuera, and et al. 2019. "Atherosclerosis and Coenzyme Q10" International Journal of Molecular Sciences 20, no. 20: 5195. https://doi.org/10.3390/ijms20205195
APA StyleSuárez-Rivero, J. M., Pastor-Maldonado, C. J., de la Mata, M., Villanueva-Paz, M., Povea-Cabello, S., Álvarez-Córdoba, M., Villalón-García, I., Suárez-Carrillo, A., Talaverón-Rey, M., Munuera, M., & Sánchez-Alcázar, J. A. (2019). Atherosclerosis and Coenzyme Q10. International Journal of Molecular Sciences, 20(20), 5195. https://doi.org/10.3390/ijms20205195