Exploring the Binding Mechanism and Dynamics of EndoMS/NucS to Mismatched dsDNA


Abstract
:1. Introduction
2. Results and Discussion
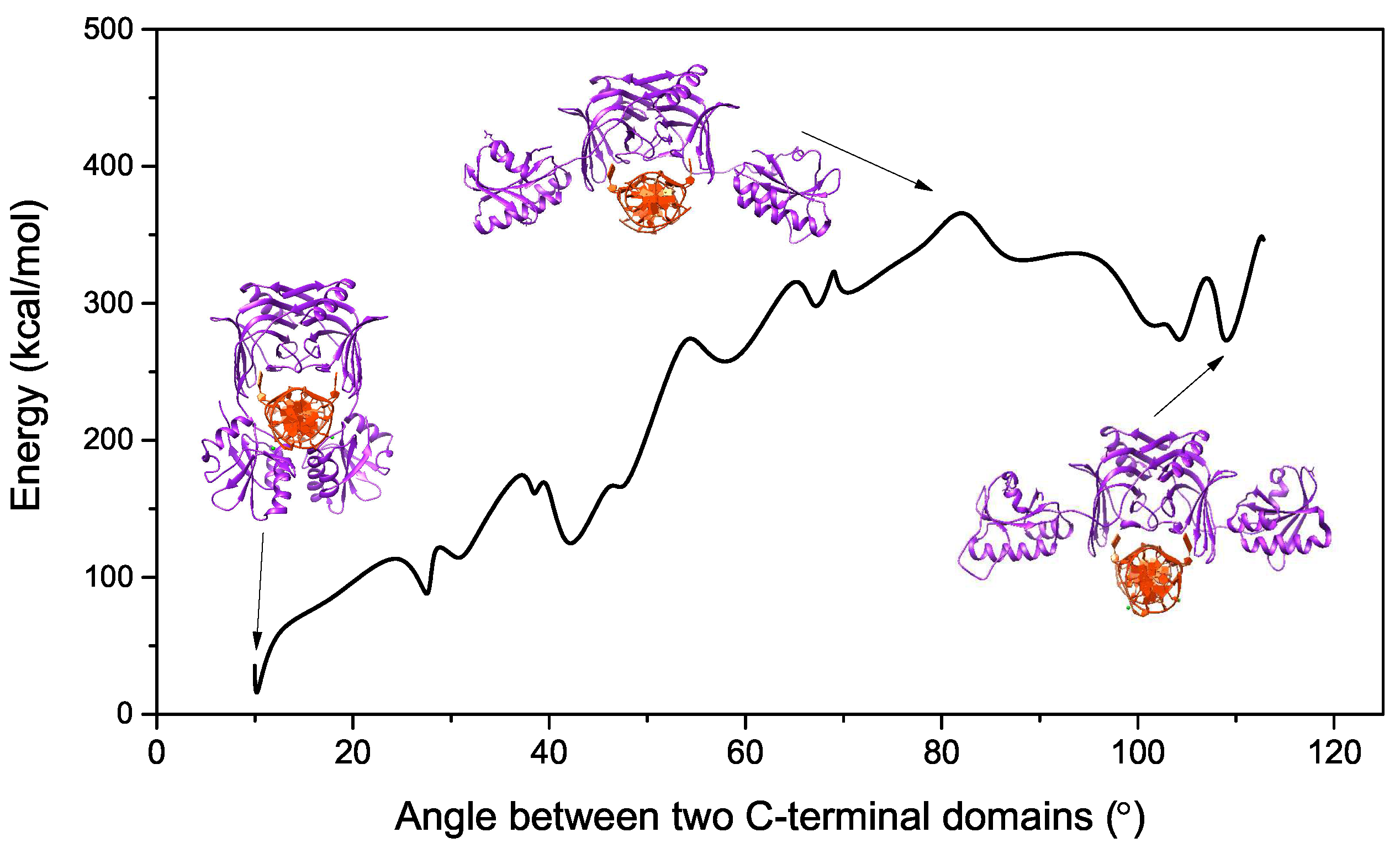
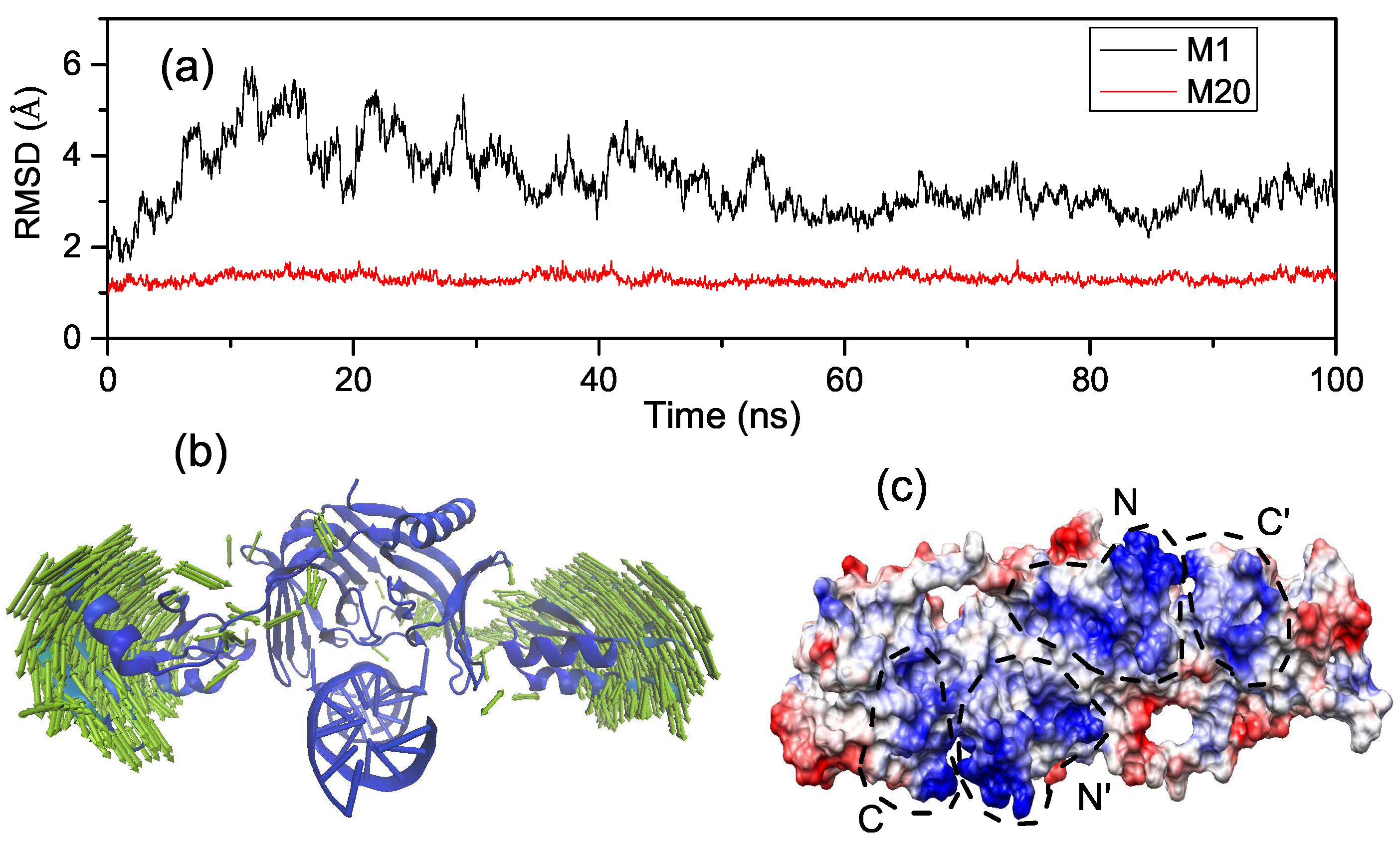
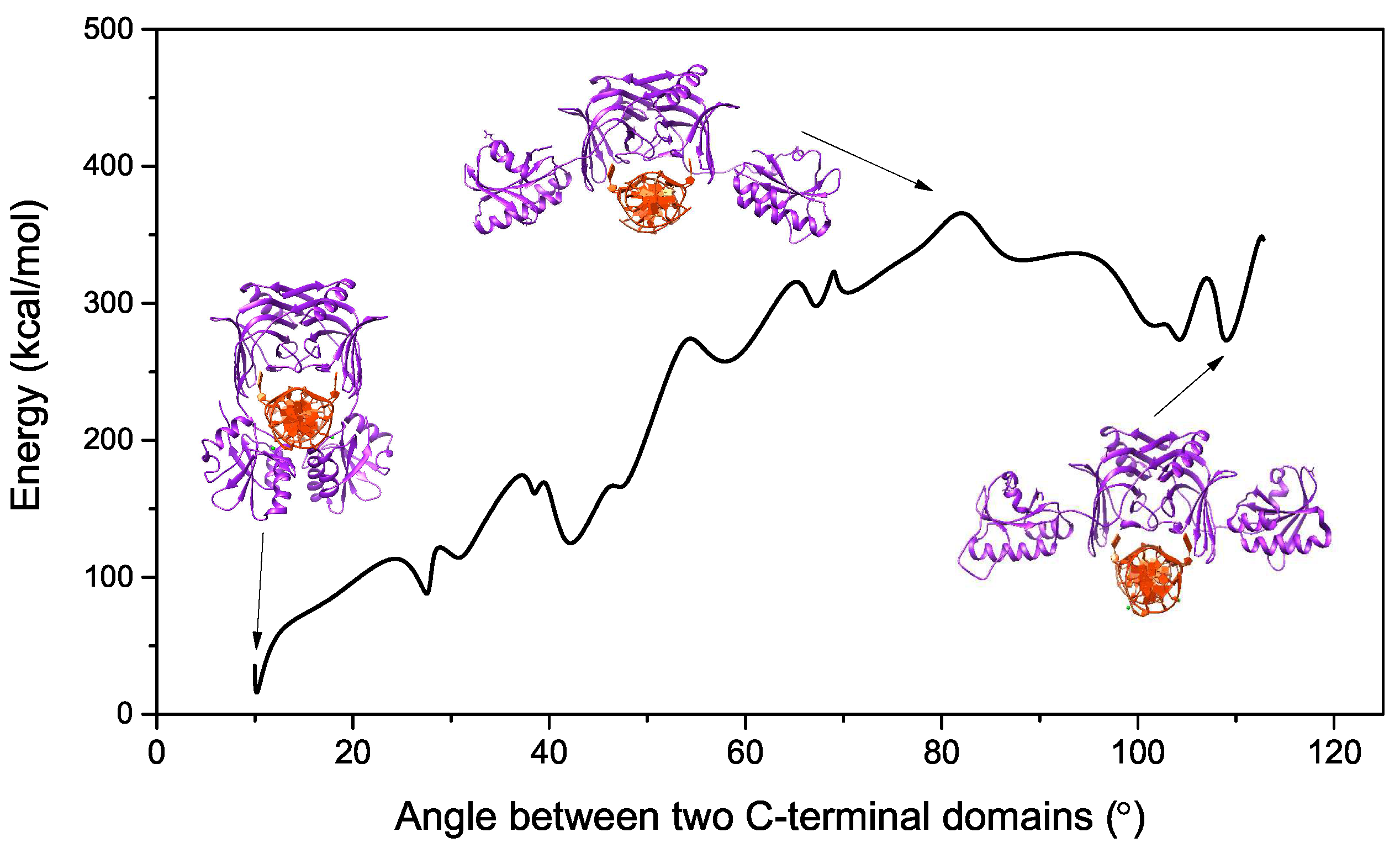
2.1. Energy Curve of EndoMS/NucS from the Open State to the Closed State
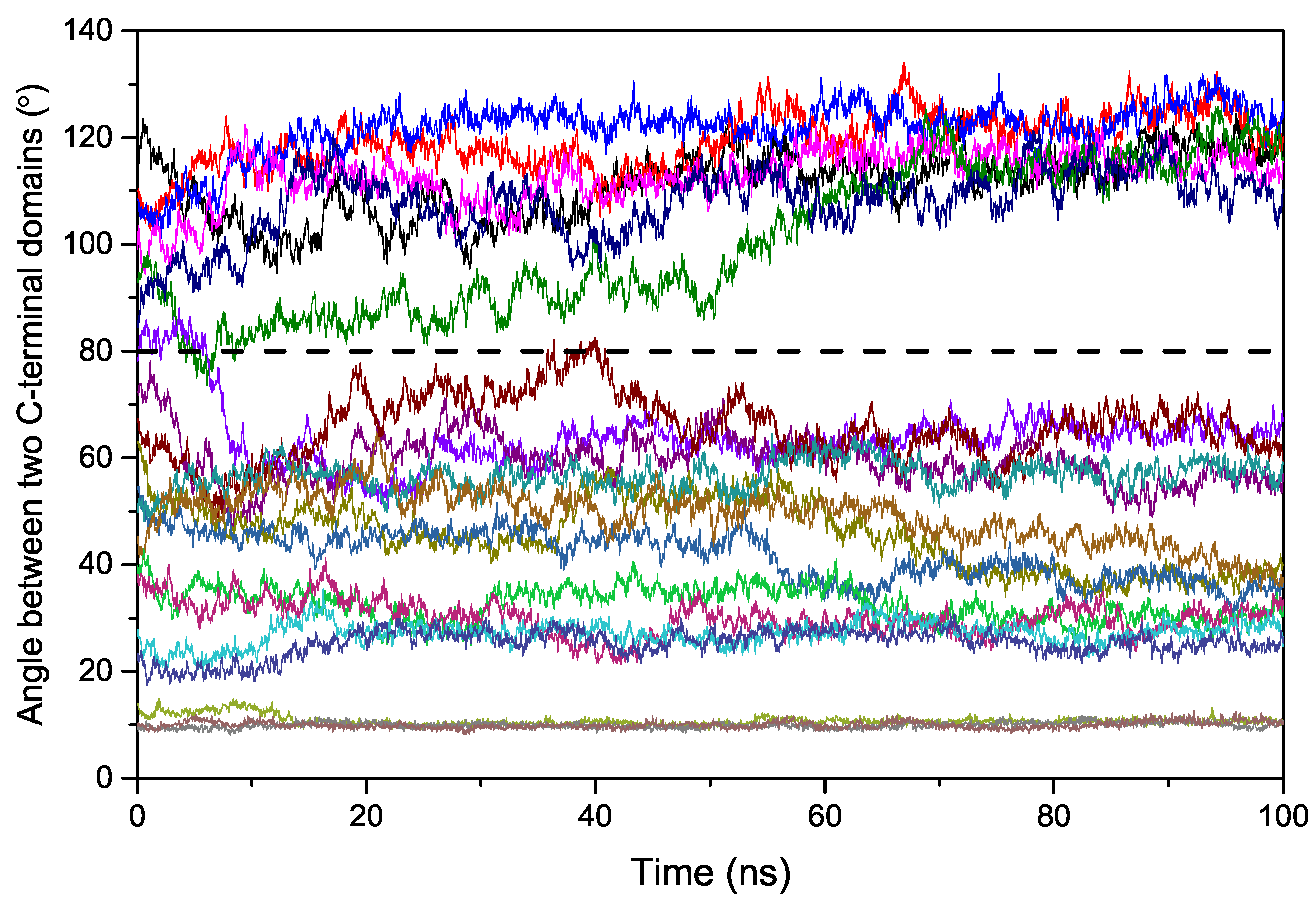
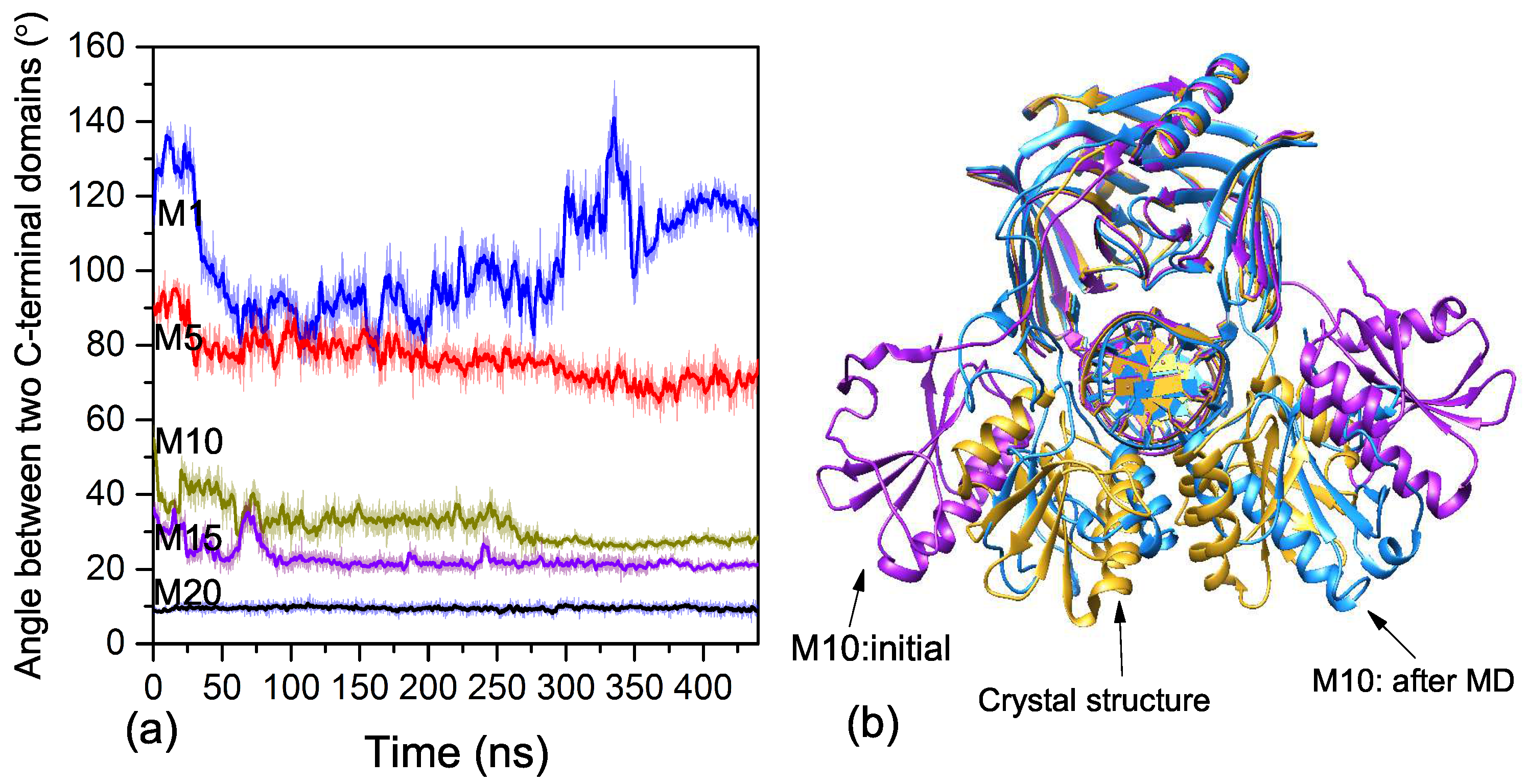
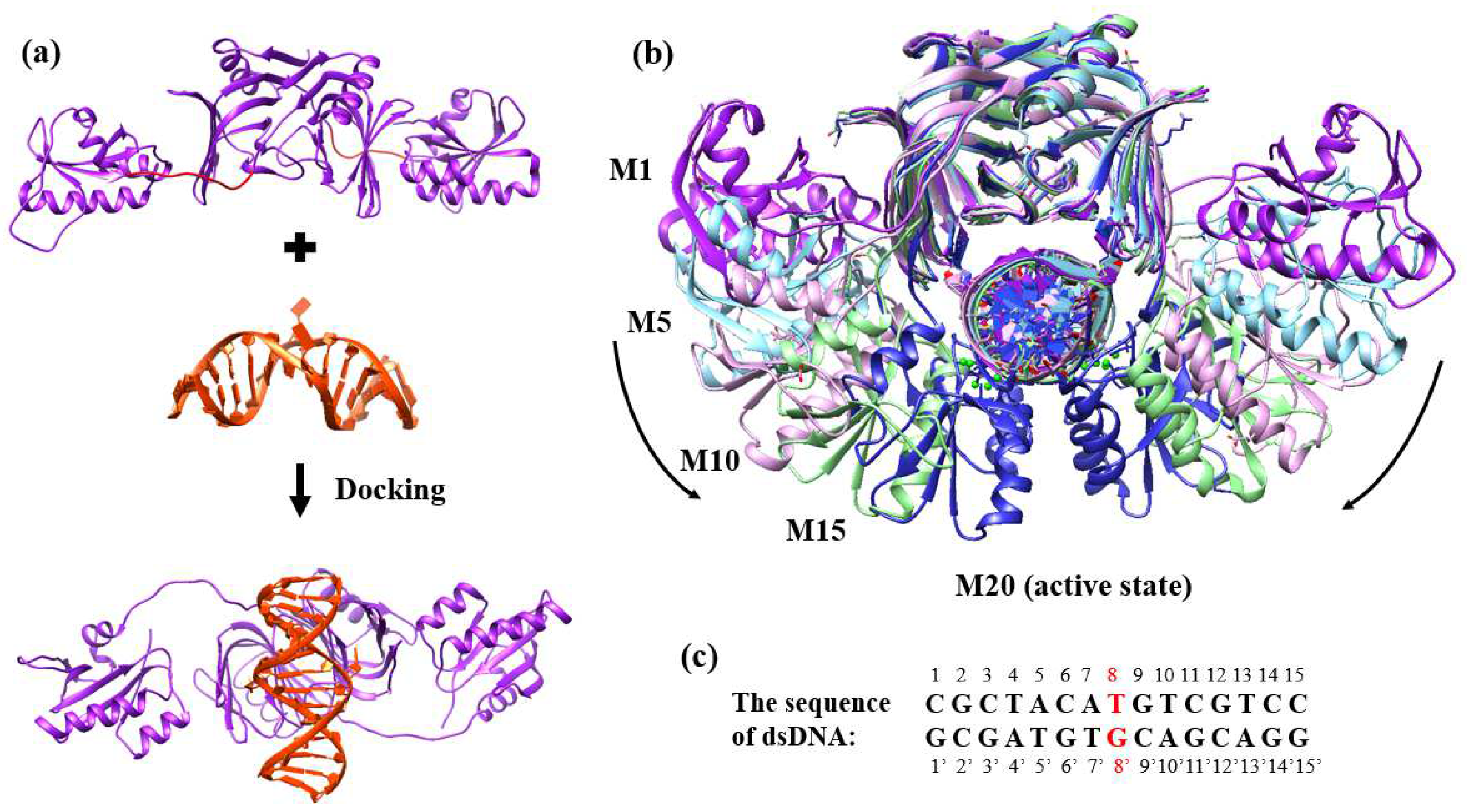
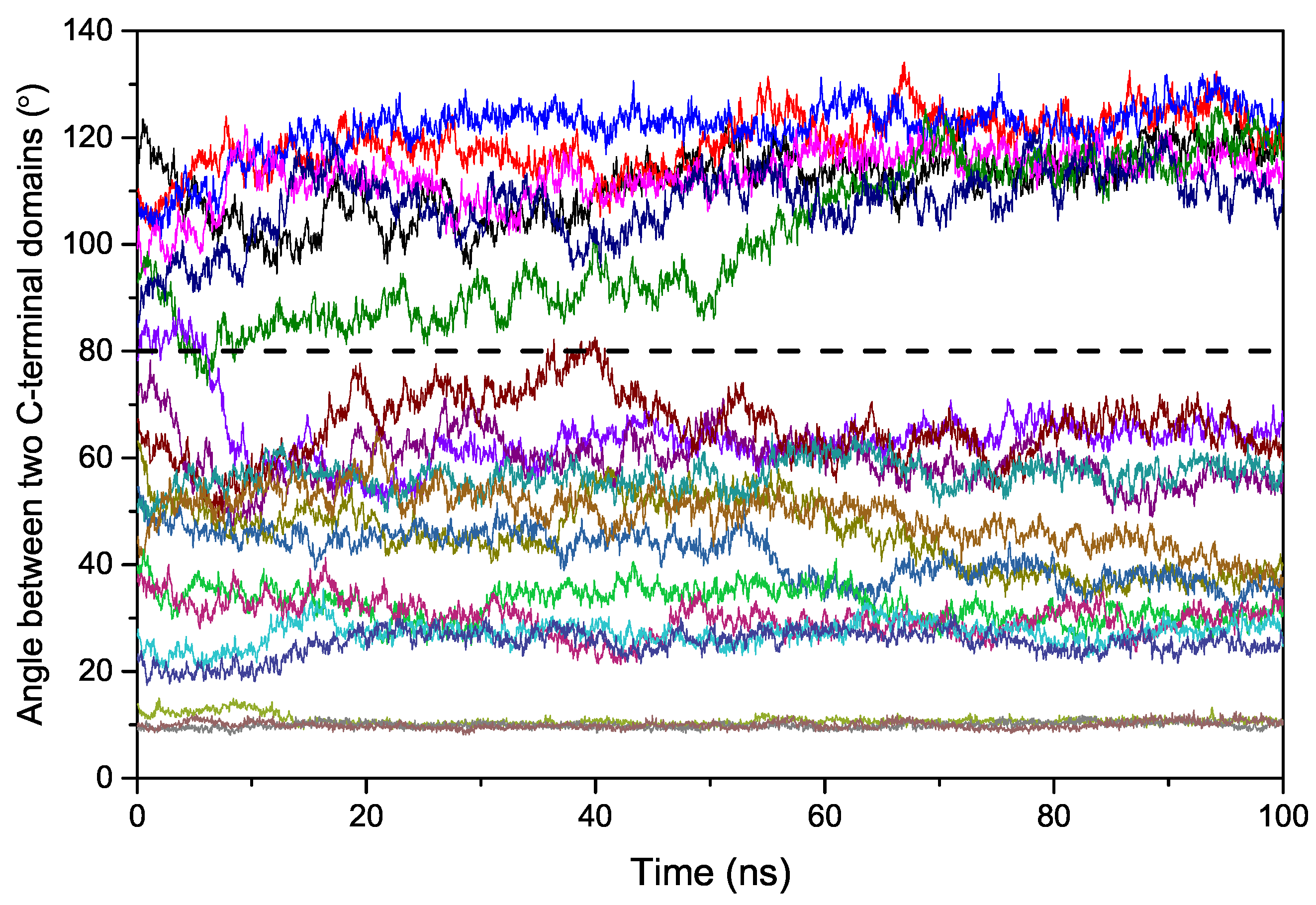
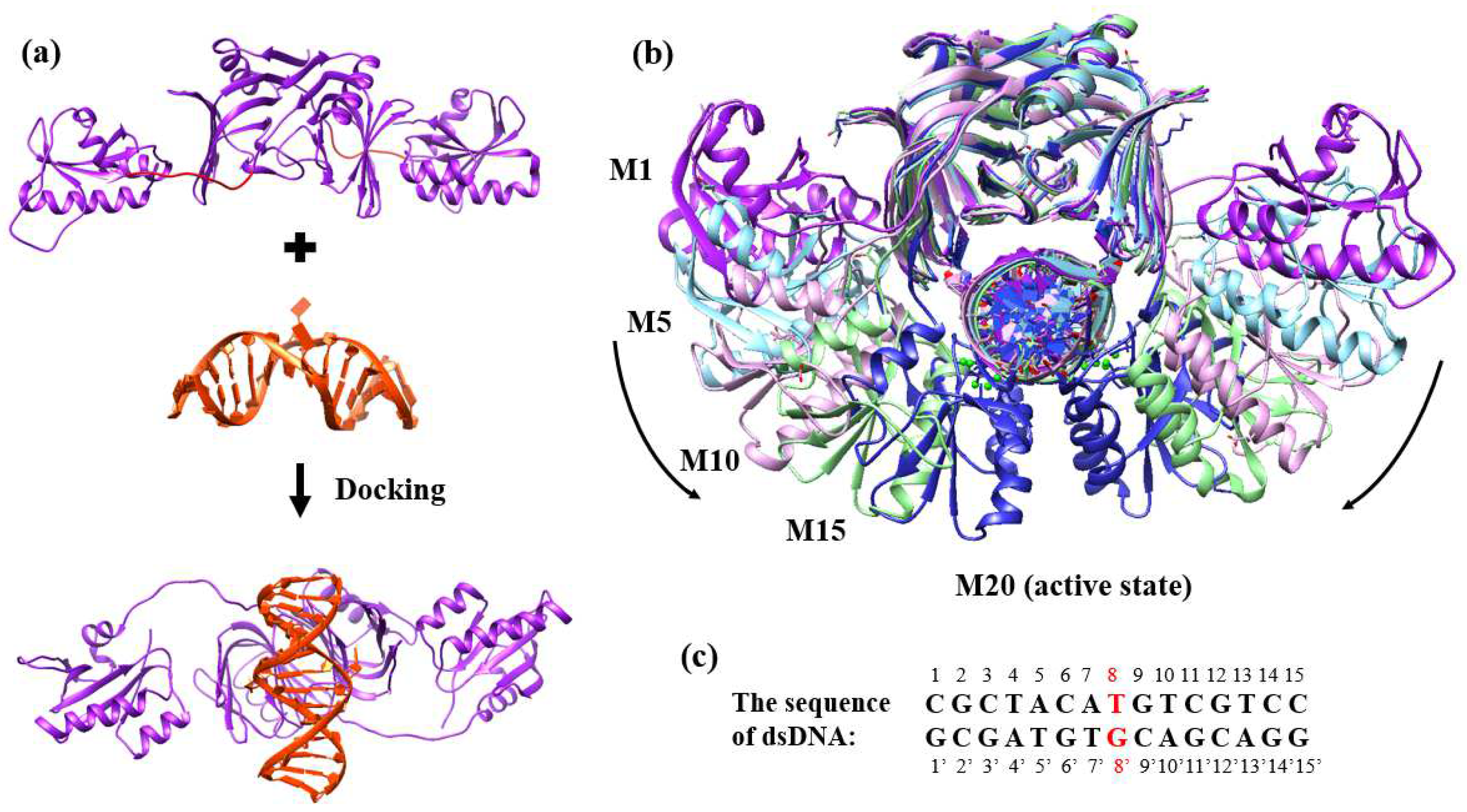
2.2. Binding Process of EndoMS/NucS to the Mismatched dsDNA
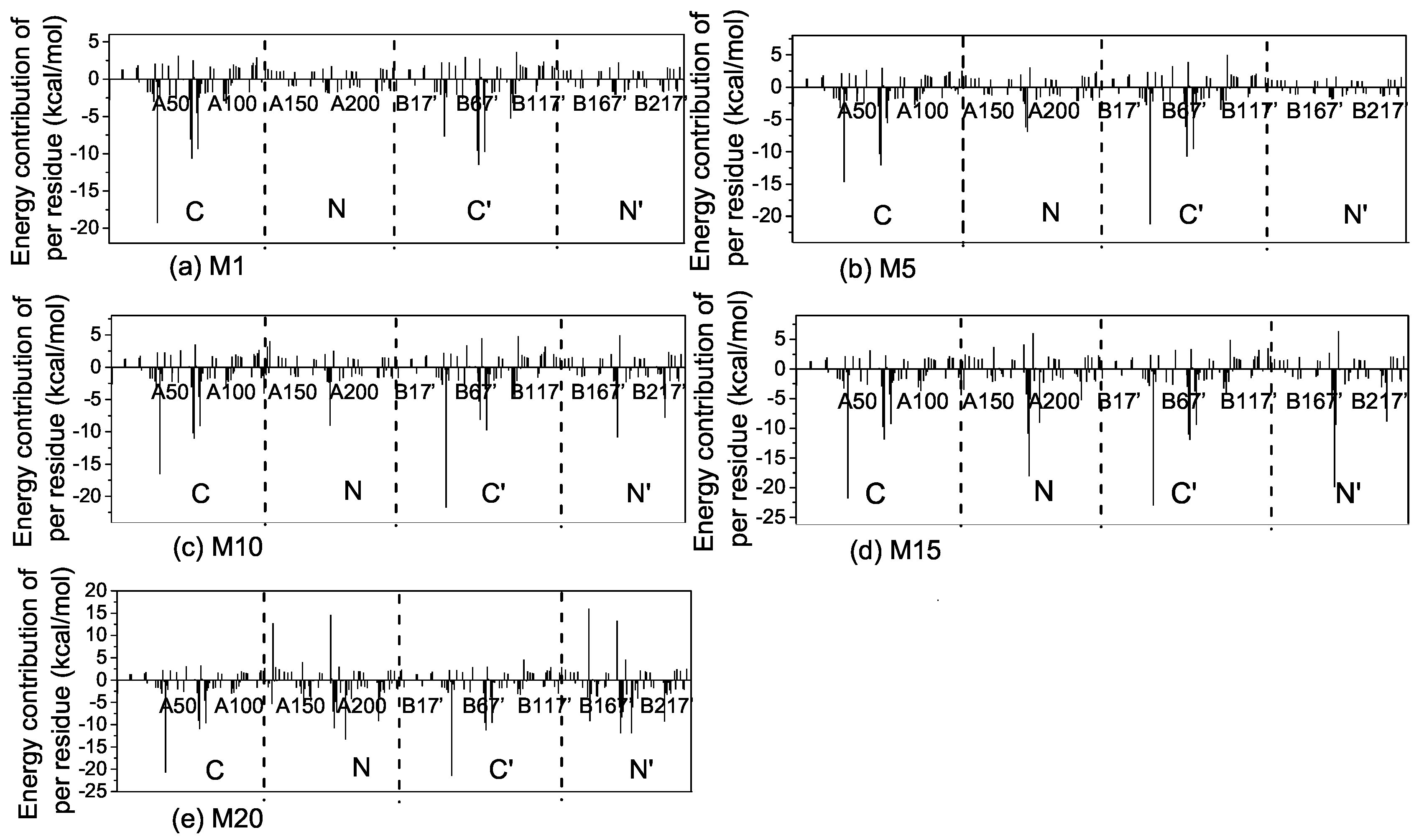
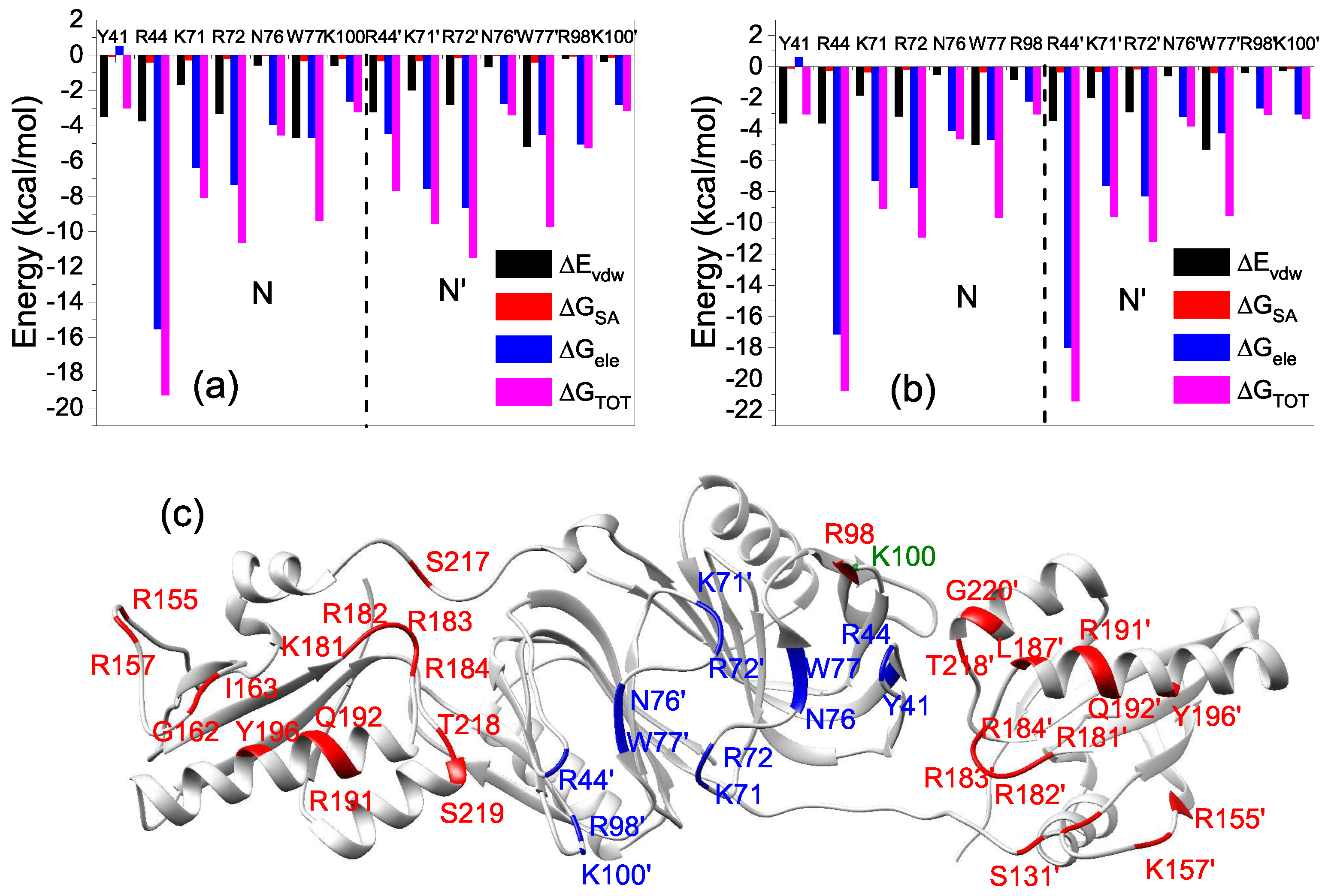
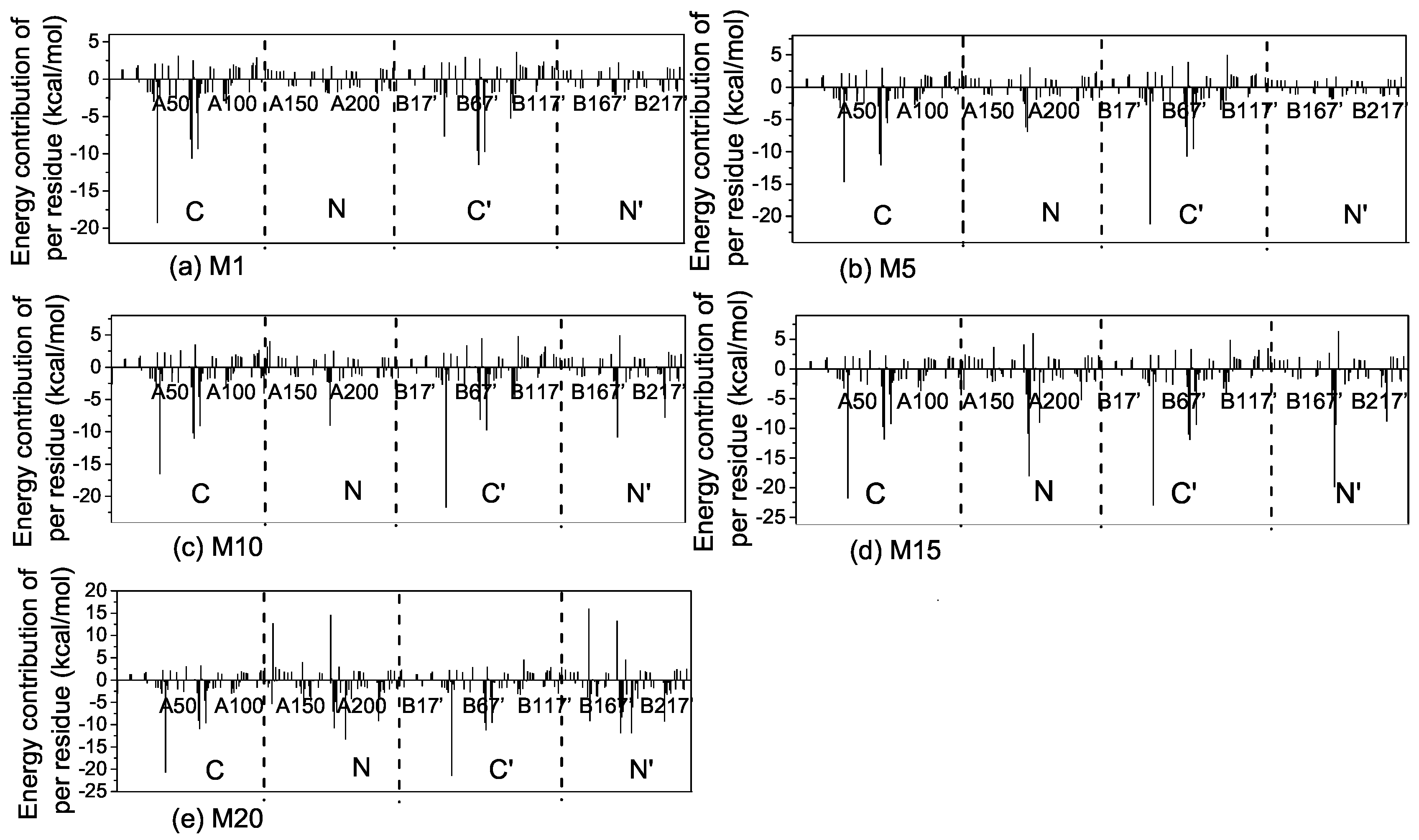
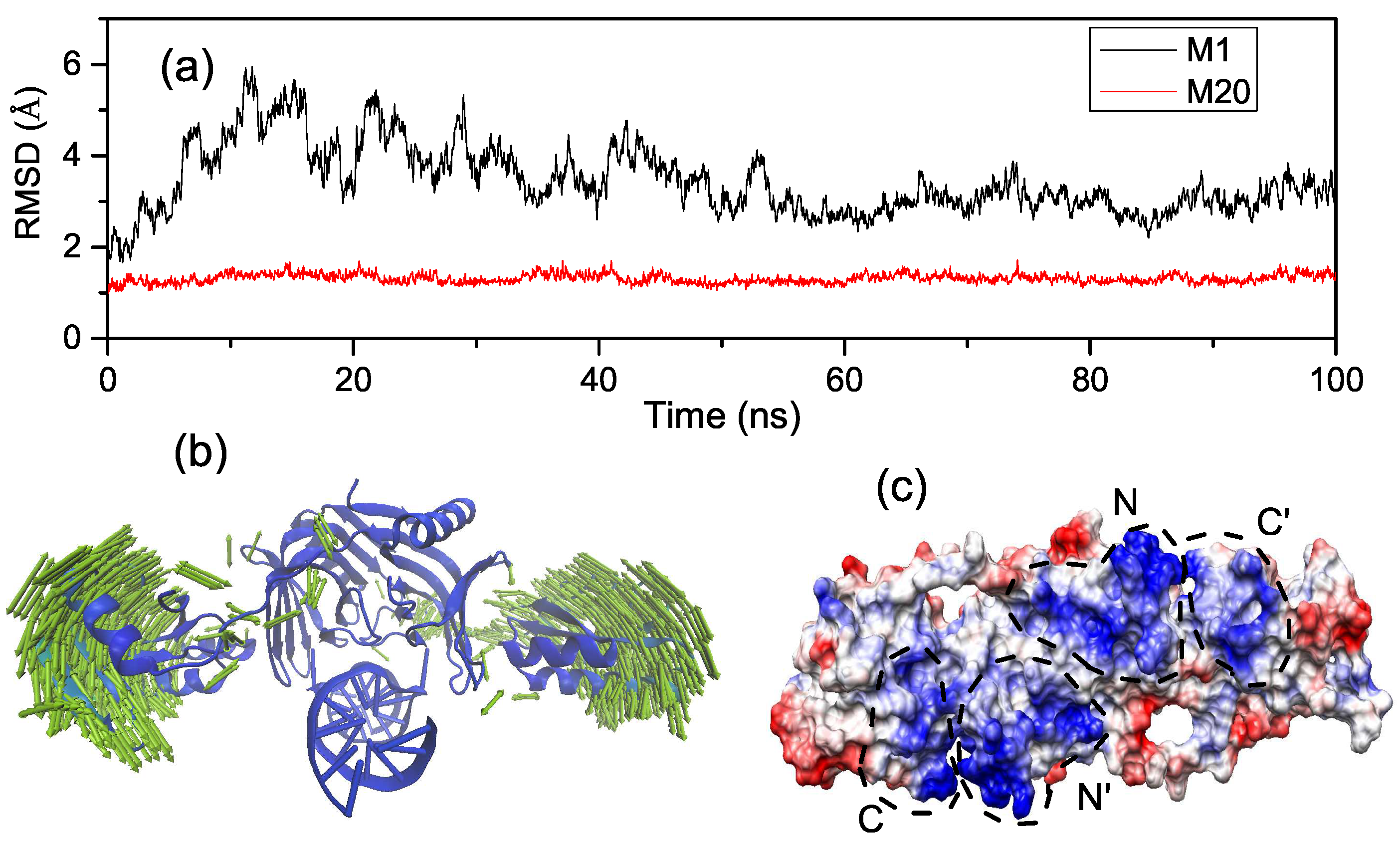
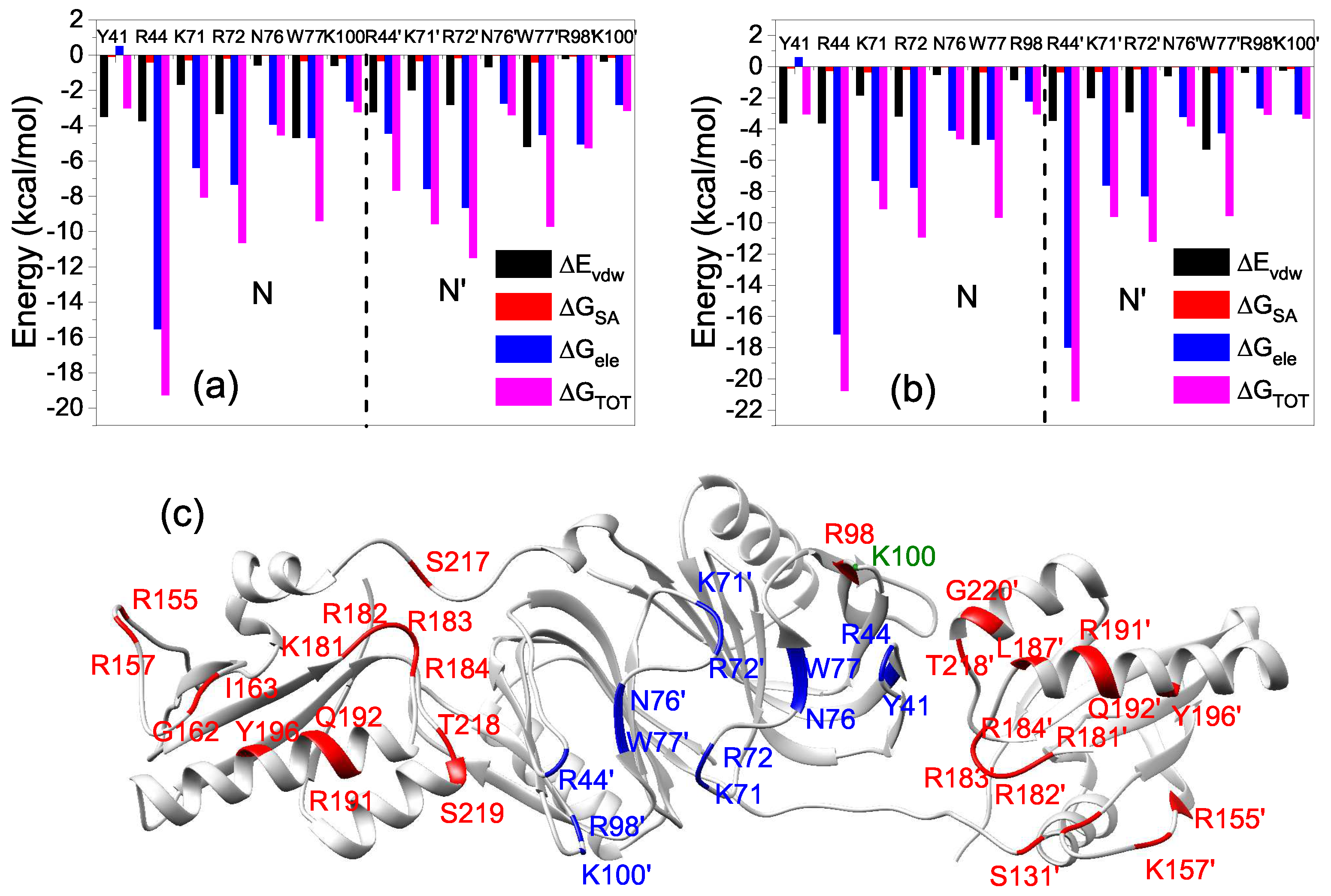
2.3. Interactions of the Open State and the Closed State with Mismatched dsDNA
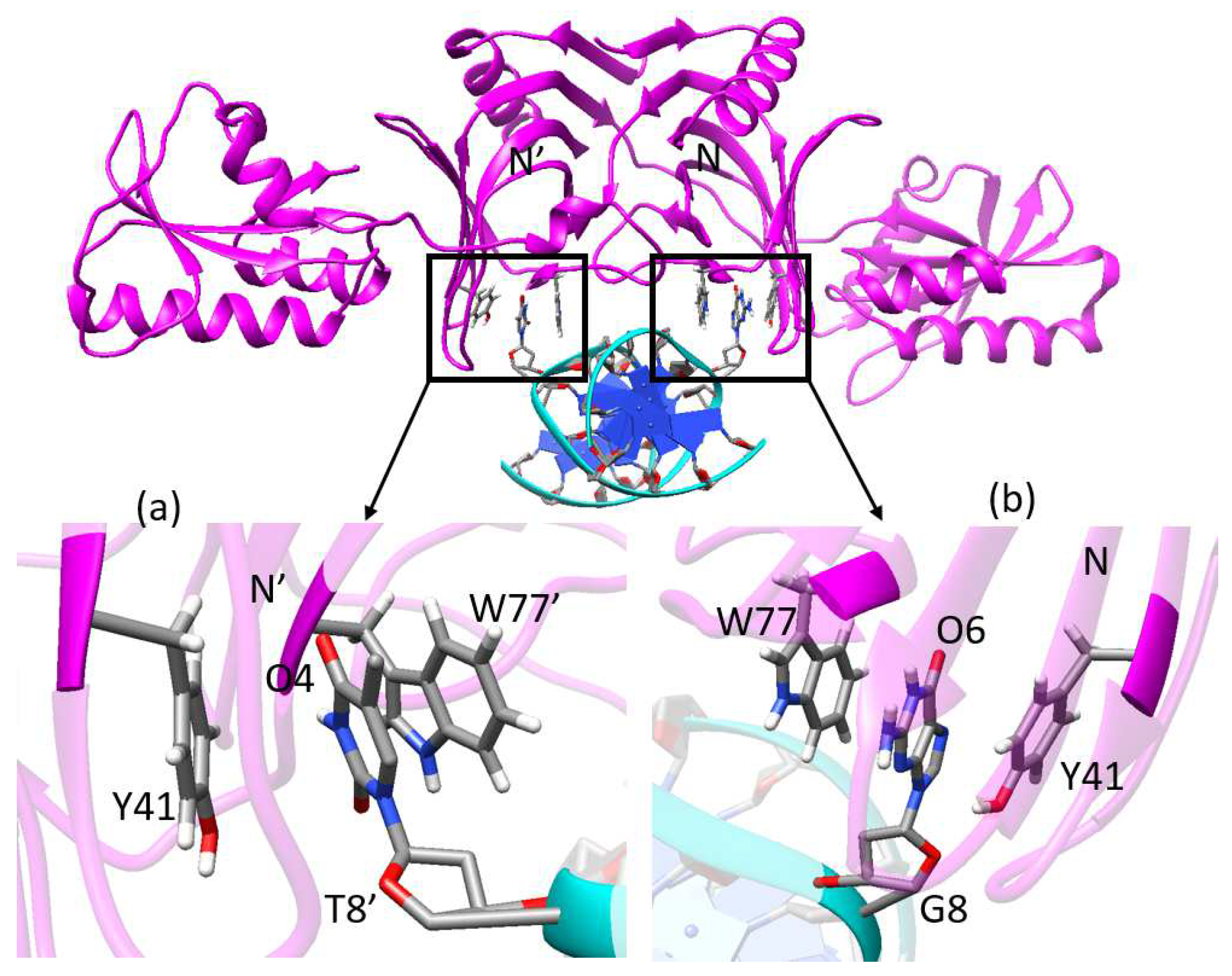
2.4. Hydrogen Bonds Analysis
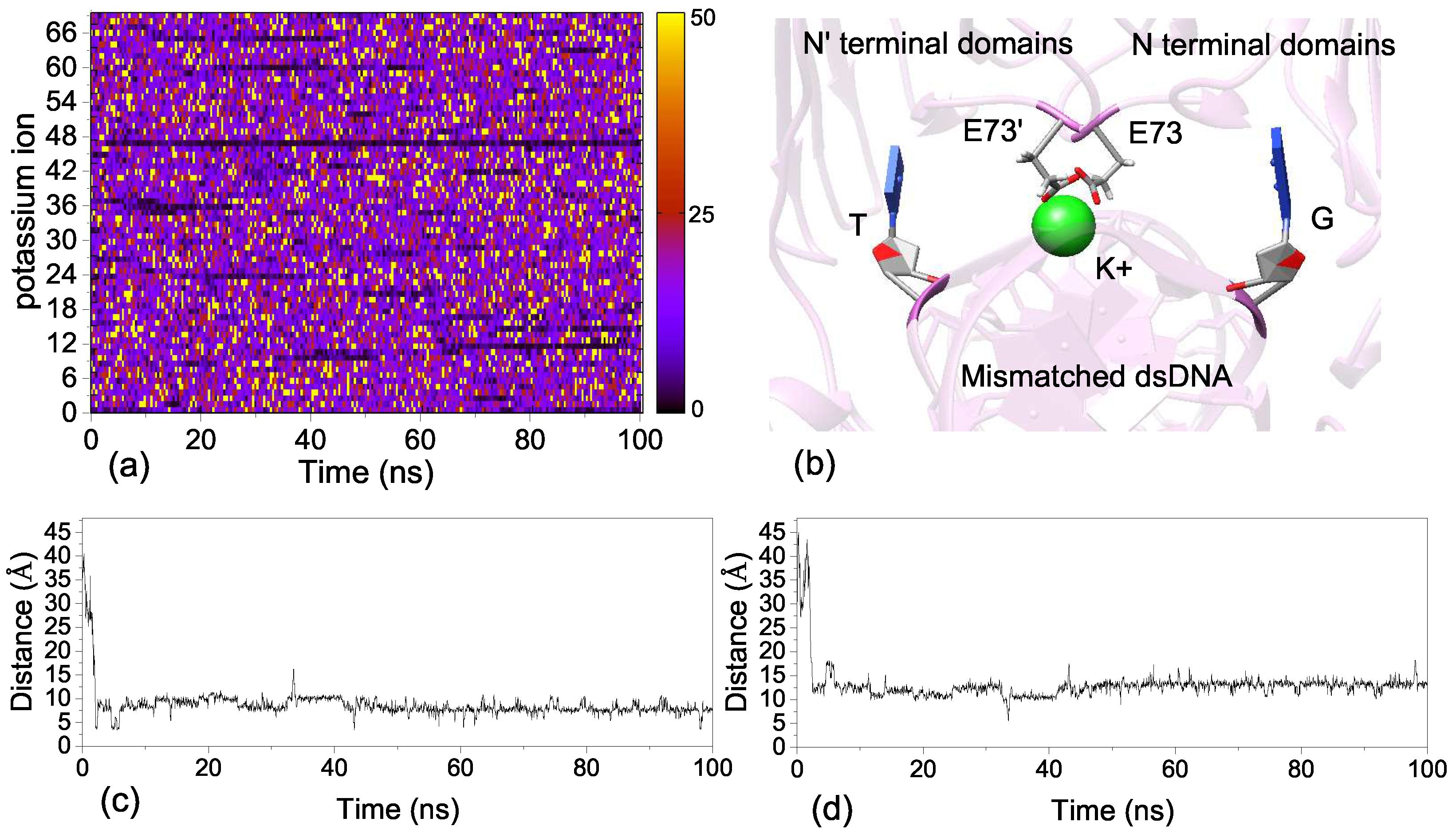
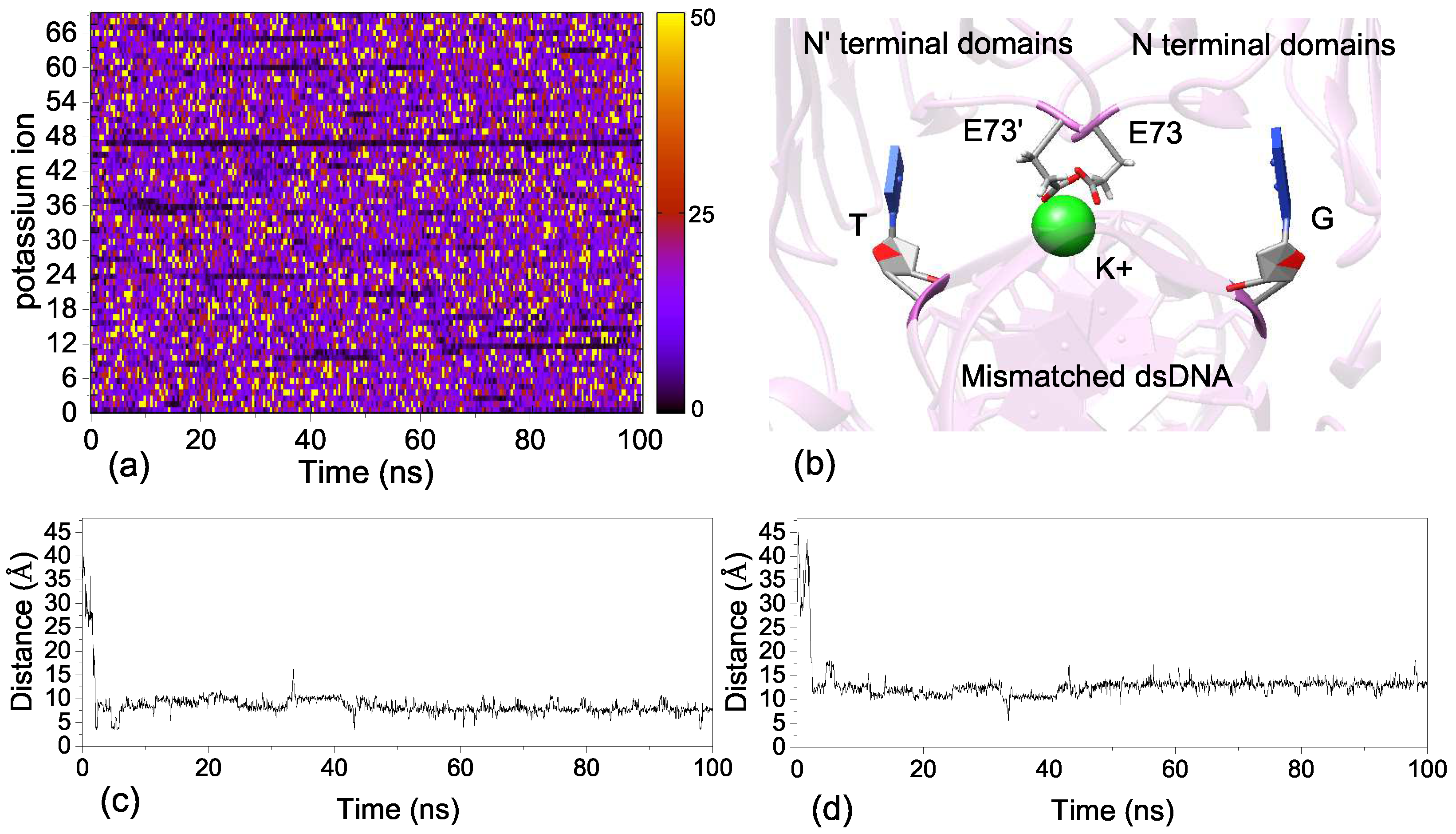
2.5. Ion Binding Sites
3. Materials and Methods
3.1. Protein Models Preparation
3.2. Molecular Dynamics Simulation Protocol
3.3. Conformational and Environmental Analysis
3.4. Free Energy Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helena, J.M.; Joubert, A.M.; Grobbelaar, S.; Nolte, E.M.; Nel, M.; Pepper, M.S.; Coetzee, M.; Mercier, A.E. Deoxyribonucleic Acid Damage and Repair: Capitalizing on Our Understanding of the Mechanisms of Maintaining Genomic Integrity for Therapeutic Purposes. Int. J. Mol. Sci. 2018, 19, 1148. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.E.; Lindahl, T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu. Rev. Genet. 2004, 38, 445–476. [Google Scholar] [CrossRef] [PubMed]
- Iyer, R.R.; Pluciennik, A.; Burdett, V.; Modrich, P.L. DNA mismatch repair: Functions and mechanisms. Chem. Rev. 2006, 106, 302–323. [Google Scholar] [CrossRef]
- Skucha, A.; Ebner, J.; Grebien, F. Roles of SETD2 in LeukemiaTranscription, DNA-Damage, and Beyond. Int. J. Mol. Sci. 2019, 20, 1029. [Google Scholar] [CrossRef]
- Gomes, L.R.; Menck, C.F.M.; Leandro, G.S. Autophagy Roles in the Modulation of DNA Repair Pathways. Int. J. Mol. Sci. 2017, 18, 21. [Google Scholar] [CrossRef]
- Kim, J.H. Chromatin Remodeling and Epigenetic Regulation in Plant DNA Damage Repair. Int. J. Mol. Sci. 2019, 20, 4093. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Erie, D.A. DNA mismatch repair. Annu. Rev. Biochem. 2005, 74, 681–710. [Google Scholar] [CrossRef]
- Eisen, J.A.; Hanawalt, P.C. A phylogenomic study of DNA repair genes, proteins, and processes. Mutat. Res.-DNA Repair 1999, 435, 171–213. [Google Scholar] [CrossRef] [Green Version]
- Lenhart, J.S.; Schroeder, J.W.; Walsh, B.W.; Simmons, L.A. DNA Repair and Genome Maintenance in Bacillus subtilis. Microbiol. Mol. Biol. Rev. 2012, 76, 530–564. [Google Scholar] [CrossRef] [Green Version]
- Su, S.S.; Modrich, P. Escherichia coli mutS-encoded protein binds to mismatched DNA base pairs. Proc. Natl. Acad. Sci. USA 1986, 83, 5057–5061. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Popodi, E.; Tang, H.X.; Foster, P.L. Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, E2774–E2783. [Google Scholar] [CrossRef] [PubMed]
- Putnam, C.D. Evolution of the methyl directed mismatch repair system in Escherichia coli. DNA Repair 2016, 38, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Castaneda-Garcia, A.; Prieto, A.I.; Rodriguez-Beltran, J.; Alonso, N.; Cantillon, D.; Costas, C.; Perez-Lago, L.; Zegeye, E.D.; Herranz, M.; Plocinski, P.; et al. A non-canonical mismatch repair pathway in prokaryotes. Nat. Commun. 2017, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Ishino, S.; Nishi, Y.; Oda, S.; Uemori, T.; Sagara, T.; Takatsu, N.; Yamagami, T.; Shirai, T.; Ishino, Y. Identification of a mismatch-specific endonuclease in hyperthermophilic Archaea. Nucleic Acids Res. 2016, 44, 2977–2986. [Google Scholar] [CrossRef] [Green Version]
- Banasik, M.; Sachadyn, P. Conserved motifs of MutL proteins. Mutat. Res.-Fundam. Mol. Mech. Mutagen. 2014, 769, 69–79. [Google Scholar]
- Ford, C.B.; Lin, P.L.; Chase, M.R.; Shah, R.R.; Iartchouk, O.; Galagan, J.; Mohaideen, N.; Ioerger, T.R.; Sacchettini, J.C.; Lipsitch, M.; et al. Use of whole genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection. Nat. Genet. 2011, 43, 482. [Google Scholar] [CrossRef]
- Lin, Z.; Nei, M.; Ma, H. The origins and early evolution of DNA mismatch repair genesmultiple horizontal gene transfers and co-evolution. Nucleic Acids Res. 2007, 35, 7591–7603. [Google Scholar] [CrossRef]
- Grogan, D.W.; Carver, G.T.; Drake, J.W. Genetic fidelity under harsh conditions: Analysis of spontaneous mutation in the thermoacidophilic archaeon Sulfolobus acidocaldarius. Proc. Natl. Acad. Sci. USA 2001, 98, 7928–7933. [Google Scholar] [CrossRef]
- Grogan, D.W. Stability and repair of DNA in hyperthermophilic archaea. Curr. Issues Mol. Biol. 2004, 6, 137–144. [Google Scholar]
- Busch, C.R.; DiRuggiero, J. MutS and MutL Are Dispensable for Maintenance of the Genomic Mutation Rate in the Halophilic Archaeon Halobacterium salinarum NRC-1. PLoS ONE 2010, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Ishino, S.; Skouloubris, S.; Kudo, H.; I’Hermitte-Stead, C.; Es-Sadik, A.; Lambry, J.C.; Ishino, Y.; Myllykallio, H. Activation of the mismatch-specific endonuclease EndoMS/NucS by the replication clamp is required for high fidelity DNA replication. Nucleic Acids Res. 2018, 46, 6206–6217. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, N.; Numata, I.; Su’etsugu, M.; Miyoshi-Akiyama, T. Bacterial EndoMS/NucS acts as a clamp-mediated mismatch endonuclease to prevent asymmetric accumulation of replication errors. Nucleic Acids Res. 2018, 46, 6152–6165. [Google Scholar] [CrossRef] [PubMed]
- Nakae, S.; Hijikata, A.; Tsuji, T.; Yonezawa, K.; Kouyama, K.; Mayanagi, K.; Ishino, S.; Ishino, Y.; Shirai, T. Structure of the EndoMS-DNA Complex as Mismatch Restriction Endonuclease. Structure 2016, 24, 1960–1971. [Google Scholar] [CrossRef] [Green Version]
- Ariyoshi, M.; Morikawa, K. A Dual Base Flipping Mechanism for Archaeal Mismatch Repair. Structure 2016, 24, 1859–1861. [Google Scholar] [CrossRef] [Green Version]
- Creze, C.; Lestini, R.; Kuhn, J.; Ligabue, A.; Becker, H.F.; Czjzek, M.; Flament, D.; Myllykallio, H. Structure and function of a novel endonuclease acting on branched DNA substrates. Biochem. Soc. Trans. 2011, 39, 145–149. [Google Scholar] [CrossRef]
- Fishel, R.; Ewel, A.; Lescoe, M.K. Purified human MSH2 protein binds to DNA containing. Cancer Res. 1994, 54, 5539–5542. [Google Scholar]
- Su, S.S.; Lahue, R.S.; Au, M.K.; Modrich, P. Mispair specificity of methyl-directed DNA mismatch correction in vitro. Biol. Chem. 1988, 263, 6829–6835. [Google Scholar]
- Natrajan, G.; Lamers, M.H.; Enzlin, J.H.; Winterwerp, H.K.; Perrakis, A.; Sixma, T.K. Structure of Escherichia coli DNA mismatch repair enzyme MutS in complex with different mismatches: a common recognition mode for diverse substrates. Nucleic Acids Res. 2003, 31, 4814–4821. [Google Scholar] [CrossRef]
- Kok, D.B.; Groothuizen, F.S.; Fish, A.; Dharadhar, S.; Winterwerp, H.K.; Sixma, T.K. Sharp kinking of a coiled-coil in MutS allows DNA binding and release. Nucleic Acids Res. 2019, 47, 8888–8898. [Google Scholar]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-Y. Exploring the potential of global protein-protein docking: An overview and critical assessment of current programs for automatic ab initio docking. Drug Discov. Today 2015, 20, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-Y.; Zou, X. A knowledge-based scoring function for protein-RNA interactions derived from a statistical mechanics-based iterative method. Nucleic Acids Res. 2014, 42, e55. [Google Scholar] [CrossRef]
- Yan, Y.; Wen, Z.; Wang, X.; Huang, S.-Y. Addressing recent docking challenges: A hybrid strategy to integrate template-based and free protein-protein docking. Proteins 2017, 85, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.-Y. HDOCK: A web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Tao, H.; Huang, S.-Y. Protein-ensemble-RNA docking by efficient consideration of protein flexibility through homology models. Bioinformatics 2019. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pK(a)s and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Myers, J.; Grothaus, G.; Narayanan, S.; Onufriev, A. A simple clustering algorithm can be accurate enough for use in calculations of pKs in macromolecules. Proteins 2006, 63, 928–938. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Nadine, H.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Tom Darden, D.Y.; Lee, P. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Ding, J.N.; Zhong, H.; Han, J.G. Exploration micromechanism of VP35 IID interaction and recognition dsRNA: A molecular dynamics simulation. Proteins 2017, 85, 1008–1023. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Ding, J.N.; Feng, T.T.; Han, J.G. Exploring interaction mechanisms of the inhibitor binding to the VP35 IID region of Ebola virus by all atom molecular dynamics simulation method. Proteins 2015, 83, 2263–2278. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Tao, H.Y.; Huang, S.Y. Dynamics and Mechanisms in the Recruitment and Transference of Histone Chaperone CIA/ASF1. Int. J. Mol. Sci. 2019, 20, 3325. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.H.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Accounts Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. Free Energy Calculations by the Molecular Mechanics Poisson-Boltzmann Surface Area Method. Mol. Inf. 2012, 31, 114–122. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compoment | E | E | G | G | G | −TS | G |
|---|---|---|---|---|---|---|---|
| M1 | −112.32 | −2512.7 | −8.76 | 2513.51 | −120.16 | 51.91 | −68.25 |
| M20 | −265.49 | −3777 | −23.47 | 3858.14 | −207.77 | 84.83 | −122.94 |
| Acceptor | DonorH | Donor | Occupancy (%) | Distance (Å) |
|---|---|---|---|---|
| M1 (the open state) N-terminal | ||||
| DG_10’@OP2 | ARG_44@HH12 | ARG_44@NH1 | 100 | 2.78 |
| DG_8’@O6 | TRP_77@H | TRP_77@N | 100 | 2.93 |
| DT_8@O4 | TRP_77’@H | TRP_77’@N | 99.8 | 2.97 |
| DT_10@OP1 | TRP_77’@HE1 | TRP_77’@NE1 | 99 | 2.81 |
| DG_8’@O6 | ASN_76@HD22 | ASN_76@ND2 | 99 | 2.89 |
| DG_10’@OP1 | ARG_44@HH22 | ARG_44@NH2 | 98.8 | 2.88 |
| DT_8@O4 | ASN_76’@HD22 | ASN_76’@ND2 | 97.2 | 2.99 |
| DG_10’@OP1 | TRP_77’@HE1 | TRP_77’@NE1 | 87.8 | 2.87 |
| DT_11’@OP1 | ARG_72’@H | ARG_72’@N | 86.4 | 2.85 |
| DT_8@O2 | ARG_98’@HH22 | ARG_98’@NH2 | 77.8 | 2.80 |
| DC_11@OP1 | GLU_73@H | GLU_73@N | 74.4 | 3.00 |
| M20 (the closed state) N-terminal | ||||
| DG_10’@OP2 | ARG_44@HH12 | ARG_44@NH1 | 100 | 2.77 |
| DG_10’@OP1 | ARG_44@HH22 | ARG_44@NH2 | 100 | 2.88 |
| DG_8’@O6 | TRP_77@H | TRP_77@N | 100 | 2.96 |
| DT_10@OP2 | ARG_44’@HH12 | ARG_44’@NH1 | 99.8 | 2.80 |
| DT_10@OP1 | TRP_77’@HE1 | TRP_77’@NE1 | 99.6 | 2.83 |
| DT_8@O4 | TRP_77’@H | TRP_77’@N | 99.6 | 2.99 |
| DG_8’@O6 | ASN_76@HD22 | ASN_76@ND2 | 99 | 2.91 |
| DT_10@OP1 | ARG_44’@HH22 | ARG_44’@NH2 | 98.2 | 2.93 |
| DT_8@O4 | ASN_76’@HD22 | ASN_76’@ND2 | 97.6 | 2.97 |
| DT_11’@OP1 | ARG_72’@H | ARG_72’@N | 94.2 | 2.83 |
| DG_10’@OP1 | TRP_77@HE1 | TRP_77@NE1 | 91 | 2.84 |
| DG_9@OP1 | ARG_44’@HH11 | ARG_44’@NH1 | 78.6 | 2.99 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Huang, S. Exploring the Binding Mechanism and Dynamics of EndoMS/NucS to Mismatched dsDNA. Int. J. Mol. Sci. 2019, 20, 5142. https://doi.org/10.3390/ijms20205142
Zhang Y, Huang S. Exploring the Binding Mechanism and Dynamics of EndoMS/NucS to Mismatched dsDNA. International Journal of Molecular Sciences. 2019; 20(20):5142. https://doi.org/10.3390/ijms20205142
Chicago/Turabian StyleZhang, Yanjun, and Shengyou Huang. 2019. "Exploring the Binding Mechanism and Dynamics of EndoMS/NucS to Mismatched dsDNA" International Journal of Molecular Sciences 20, no. 20: 5142. https://doi.org/10.3390/ijms20205142
APA StyleZhang, Y., & Huang, S. (2019). Exploring the Binding Mechanism and Dynamics of EndoMS/NucS to Mismatched dsDNA. International Journal of Molecular Sciences, 20(20), 5142. https://doi.org/10.3390/ijms20205142