The Good and the Bad of Natural Killer Cells in Virus Control: Perspective for Anti-HBV Therapy

 ,
,  , ,
, , 
Abstract
1. Introduction
2. NK Cell Subsets
3. Liver NK Cells
4. NK Cells in HBV Infection
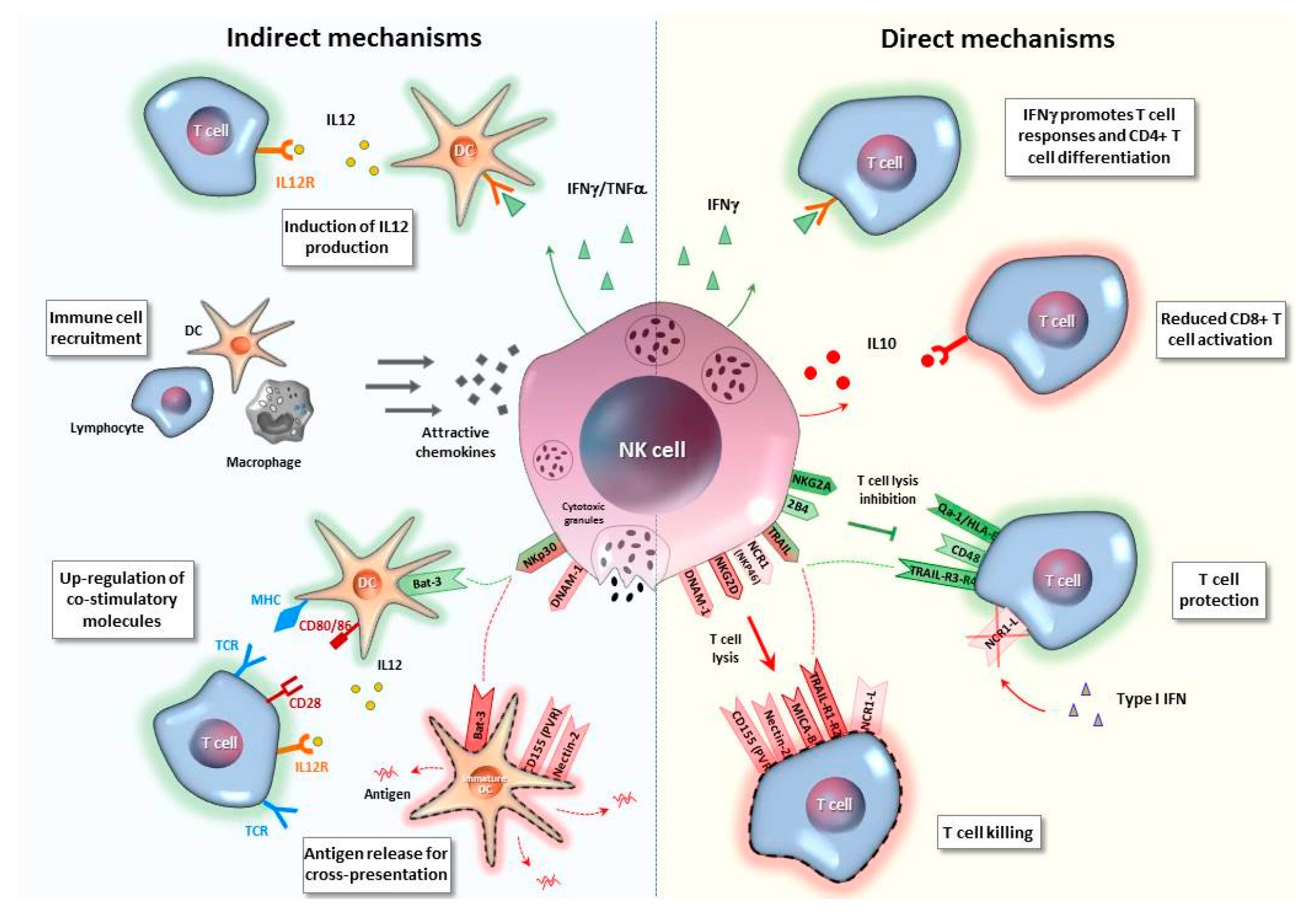
5. NK/T Cell Interplay
5.1. Indirect Mechanisms
5.2. Direct Mechanisms
5.2.1. Cytokine-Mediated Interaction
5.2.2. Receptor/Ligand NK-T Cell Cross-Talk
5.2.3. Checkpoint Inhibitory Pathways
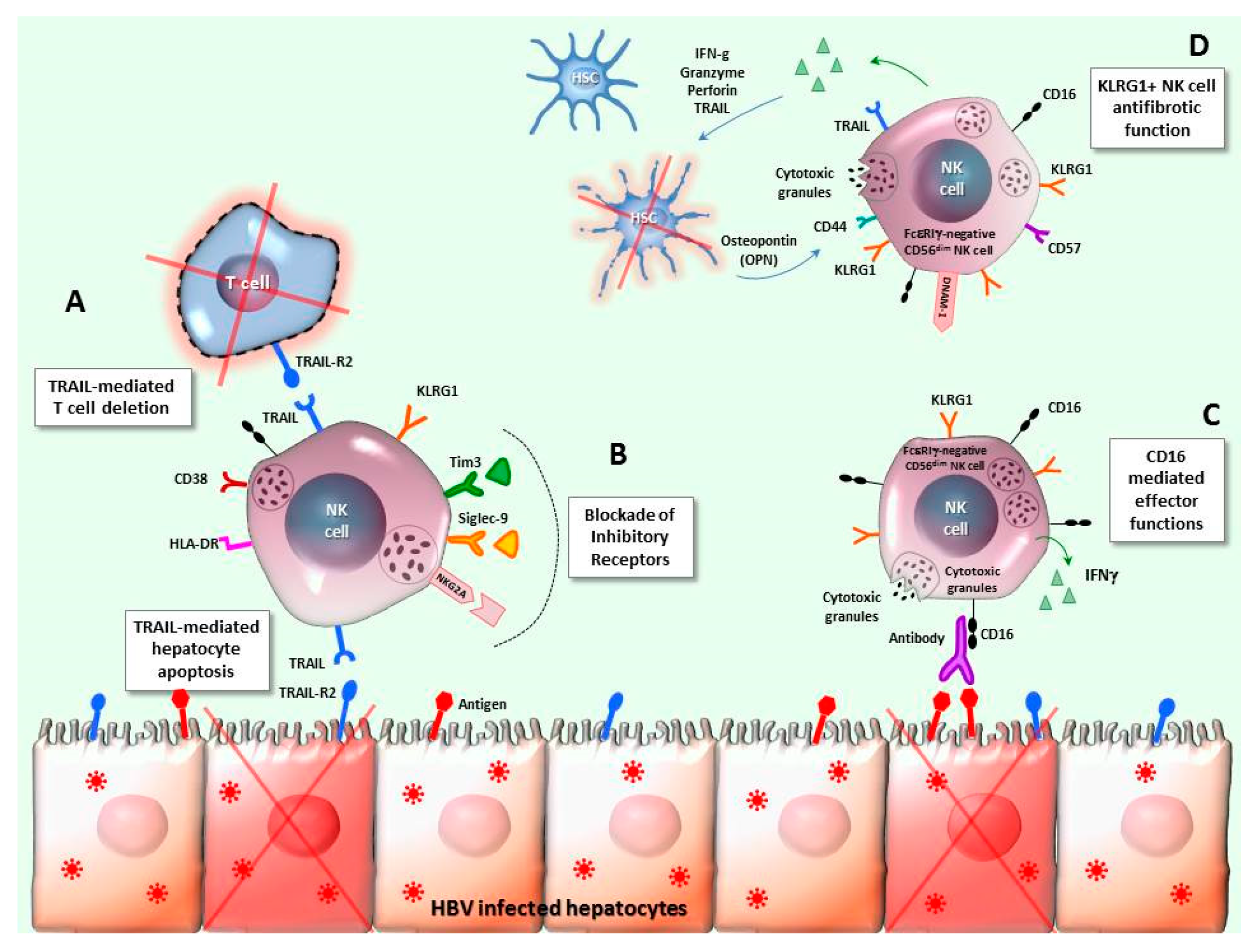
6. NK-T Cell Interplay in Chronic HBV Infection
7. Final Remarks and Potential Clinical Applications
Acknowledgments
Conflicts of Interest
References
- Schweitzer, A.; Horn, J.; Mikolajczyk, R.T.; Krause, G.; Ott, J.J. Estimations of worldwide prevalence of chronic hepatitis B virus infection: A systematic review of data published between 1965 and 2013. Lancet 2015, 386, 1546–1555. [Google Scholar] [CrossRef]
- Zoulim, F.; Lebossé, F.; Levrero, M. Current treatments for chronic hepatitis B virus infections. Curr. Opin. Virol. 2016, 18, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Lampertico, P.; Agarwal, K.; Berg, T.; Buti, M.; Janssen, H.L.A.; Papatheodoridis, G.; Zoulim, F.; Tacke, F. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef] [PubMed]
- Bertoletti, A.; Ferrari, C. Adaptive immunity in HBV infection. J. Hepatol. 2016, 64, S71–S83. [Google Scholar] [CrossRef]
- Maini, M.K.; Gehring, A.J. The role of innate immunity in the immunopathology and treatment of HBV infection. J. Hepatol. 2016, 64, S60–S70. [Google Scholar] [CrossRef]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef]
- Kiessling, R.; Klein, E.; Wigzell, H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur. J. Immunol. 1975, 5, 112–117. [Google Scholar] [CrossRef]
- Kiessling, R.; Klein, E.; Pross, H.; Wigzell, H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur. J. Immunol. 1975, 5, 117–121. [Google Scholar] [CrossRef]
- Herberman, R.B.; Nunn, M.E.; Lavrin, D.H. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. I. Distribution of reactivity and specificity. Int. J. Cancer 1975, 16, 216–229. [Google Scholar] [CrossRef]
- Herberman, R.B.; Nunn, M.E.; Holden, H.T.; Lavrin, D.H. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int. J. Cancer 1975, 16, 230–239. [Google Scholar] [CrossRef]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or Adaptive Immunity? The Example of Natural Killer Cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Sivori, S.; Vacca, P.; Del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK cells: Surface receptors, inhibitory checkpoints, and translational applications. Cell. Mol. Immunol. 2019, 16, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Del Zotto, G.; Marcenaro, E.; Vacca, P.; Sivori, S.; Pende, D.; Della Chiesa, M.; Moretta, F.; Ingegnere, T.; Mingari, M.C.; Moretta, A.; et al. Markers and function of human NK cells in normal and pathological conditions. Cytom. Part B Clin. Cytom. 2017, 92, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Adib-Conquy, M.; Scott-Algara, D.; Cavaillon, J.-M.; Souza-Fonseca-Guimaraes, F. TLR-mediated activation of NK cells and their role in bacterial/viral immune responses in mammals. Immunol. Cell Biol. 2014, 92, 256–262. [Google Scholar] [CrossRef]
- Bi, J.; Tian, Z. NK Cell Exhaustion. Front. Immunol. 2017, 8, 760. [Google Scholar] [CrossRef]
- O’Brien, K.L.; Finlay, D.K. Immunometabolism and natural killer cell responses. Nat. Rev. Immunol. 2019, 19, 282–290. [Google Scholar] [CrossRef]
- Stabile, H.; Nisti, P.; Morrone, S.; Pagliara, D.; Bertaina, A.; Locatelli, F.; Santoni, A.; Gismondi, A. Multifunctional human CD56low CD16low natural killer cells are the prominent subset in bone marrow of both healthy pediatric donors and leukemic patients. Haematologica 2015, 100, 489–498. [Google Scholar] [CrossRef]
- Roberto, A.; Di Vito, C.; Zaghi, E.; Mazza, E.M.C.; Capucetti, A.; Calvi, M.; Tentorio, P.; Zanon, V.; Sarina, B.; Mariotti, J.; et al. The early expansion of anergic NKG2A pos /CD56 dim /CD16 neg natural killer represents a therapeutic target in haploidentical hematopoietic stem cell transplantation. Haematologica 2018, 103, 1390–1402. [Google Scholar] [CrossRef]
- Lugli, E.; Marcenaro, E.; Mavilio, D. NK Cell Subset Redistribution during the Course of Viral Infections. Front. Immunol. 2014, 5, 390. [Google Scholar] [CrossRef]
- Müller-Durovic, B.; Grählert, J.; Devine, O.P.; Akbar, A.N.; Hess, C. CD56-negative NK cells with impaired effector function expand in CMV and EBV co-infected healthy donors with age. Aging 2019, 11, 724–740. [Google Scholar] [CrossRef]
- Zhou, J.; Peng, H.; Li, K.; Qu, K.; Wang, B.; Wu, Y.; Ye, L.; Dong, Z.; Wei, H.; Sun, R.; et al. Liver-Resident NK Cells Control Antiviral Activity of Hepatic T Cells via the PD-1-PD-L1 Axis. Immunity 2019, 50, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Hudspeth, K.; Donadon, M.; Cimino, M.; Pontarini, E.; Tentorio, P.; Preti, M.; Hong, M.; Bertoletti, A.; Bicciato, S.; Invernizzi, P.; et al. Human liver-resident CD56bright/CD16neg NK cells are retained within hepatic sinusoids via the engagement of CCR5 and CXCR6 pathways. J. Autoimmun. 2016, 66, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Mikulak, J.; Bruni, E.; Oriolo, F.; Di Vito, C.; Mavilio, D. Hepatic Natural Killer Cells: Organ-Specific Sentinels of Liver Immune Homeostasis and Physiopathology. Front. Immunol. 2019, 10, 946. [Google Scholar] [CrossRef] [PubMed]
- Della Chiesa, M.; Pesce, S.; Muccio, L.; Carlomagno, S.; Sivori, S.; Moretta, A.; Marcenaro, E. Features of Memory-Like and PD-1+ Human NK Cell Subsets. Front. Immunol. 2016, 7, 351. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Tian, Z. Natural Killer Cell Memory: Progress and Implications. Front. Immunol. 2017, 8, 1143. [Google Scholar] [CrossRef]
- Mariotti, F.R.; Quatrini, L.; Munari, E.; Vacca, P.; Moretta, L. Innate Lymphoid Cells: Expression of PD-1 and Other Checkpoints in Normal and Pathological Conditions. Front. Immunol. 2019, 10, 910. [Google Scholar] [CrossRef]
- Mariotti, F.R.; Petrini, S.; Ingegnere, T.; Tumino, N.; Besi, F.; Scordamaglia, F.; Munari, E.; Pesce, S.; Marcenaro, E.; Moretta, A.; et al. PD-1 in human NK cells: Evidence of cytoplasmic mRNA and protein expression. Oncoimmunology 2019, 8, 1557030. [Google Scholar] [CrossRef]
- Molgora, M.; Bonavita, E.; Ponzetta, A.; Riva, F.; Barbagallo, M.; Jaillon, S.; Popović, B.; Bernardini, G.; Magrini, E.; Gianni, F.; et al. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature 2017, 551, 110–114. [Google Scholar] [CrossRef]
- Georgiev, H.; Ravens, I.; Papadogianni, G.; Bernhardt, G. Coming of Age: CD96 Emerges as Modulator of Immune Responses. Front. Immunol. 2018, 9, 1072. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, X.; Li, S.; Twelkmeyer, T.; Wang, W.; Zhang, S.; Wang, S.; Chen, J.; Jin, X.; Wu, Y.; et al. NKG2A is a NK cell exhaustion checkpoint for HCV persistence. Nat. Commun. 2019, 10, 1507. [Google Scholar] [CrossRef]
- Zhou, X.; Yu, J.; Cheng, X.; Zhao, B.; Manyam, G.C.; Zhang, L.; Schluns, K.; Li, P.; Wang, J.; Sun, S.-C. The deubiquitinase Otub1 controls the activation of CD8+ T cells and NK cells by regulating IL-15-mediated priming. Nat. Immunol. 2019, 20, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Voigt, J.; Malone, D.F.G.; Dias, J.; Leeansyah, E.; Björkström, N.K.; Ljunggren, H.-G.; Gröbe, L.; Klawonn, F.; Heyner, M.; Sandberg, J.K.; et al. Proteome analysis of human CD56 neg NK cells reveals a homogeneous phenotype surprisingly similar to CD56 dim NK cells. Eur. J. Immunol. 2018, 48, 1456–1469. [Google Scholar] [CrossRef] [PubMed]
- Fasbender, F.; Widera, A.; Hengstler, J.G.; Watzl, C. Natural Killer Cells and Liver Fibrosis. Front. Immunol. 2016, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Ishiyama, K.; Ohdan, H.; Ohira, M.; Mitsuta, H.; Arihiro, K.; Asahara, T. Difference in cytotoxicity against hepatocellular carcinoma between liver and periphery natural killer cells in humans. Hepatology 2006, 43, 362–372. [Google Scholar] [CrossRef]
- Belkaya, S.; Koro, C.; Michailidis, E.; Kabbani, M.; Cobat, A.; Bastard, P.; Lee, Y.; Hernandez, N.; Drutman, S.; de Jong, Y.; et al. Inherited IL-18BP deficiency in human fulminant viral hepatitis. J. Exp. Med. 2019, 216, 1777–1790. [Google Scholar] [CrossRef]
- Nielsen, C.M.; Wolf, A.-S.; Goodier, M.R.; Riley, E.M. Synergy between Common γ Chain Family Cytokines and IL-18 Potentiates Innate and Adaptive Pathways of NK Cell Activation. Front. Immunol. 2016, 7, 101. [Google Scholar] [CrossRef]
- Stegmann, K.A.; Robertson, F.; Hansi, N.; Gill, U.; Pallant, C.; Christophides, T.; Pallett, L.J.; Peppa, D.; Dunn, C.; Fusai, G.; et al. CXCR6 marks a novel subset of T-bet(lo)Eomes(hi) natural killer cells residing in human liver. Sci. Rep. 2016, 6, 26157. [Google Scholar] [CrossRef]
- Cuff, A.O.; Robertson, F.P.; Stegmann, K.A.; Pallett, L.J.; Maini, M.K.; Davidson, B.R.; Male, V. Eomes hi NK Cells in Human Liver Are Long-Lived and Do Not Recirculate but Can Be Replenished from the Circulation. J. Immunol. 2016, 197, 4283–4291. [Google Scholar] [CrossRef]
- Wieland, S.; Thimme, R.; Purcell, R.H.; Chisari, F.V. Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 6669–6674. [Google Scholar] [CrossRef]
- Webster, G.; Reignat, S.; Maini, M.K.; Whalley, S.A.; Ogg, G.S.; King, A.; Brown, D.; Amlot, P.L.; Williams, R.; Vergani, D.; et al. Incubation Phase of Acute Hepatitis B in Man: Dynamic of Cellular Immune Mechanisms. Hepatology 2000, 32, 1117–1124. [Google Scholar] [CrossRef]
- Fisicaro, P.; Valdatta, C.; Boni, C.; Massari, M.; Mori, C.; Zerbini, A.; Orlandini, A.; Sacchelli, L.; Missale, G.; Ferrari, C. Early kinetics of innate and adaptive immune responses during hepatitis B virus infection. Gut 2009, 58, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Lunemann, S.; Malone, D.F.G.; Hengst, J.; Port, K.; Grabowski, J.; Deterding, K.; Markova, A.; Bremer, B.; Schlaphoff, V.; Cornberg, M.; et al. Compromised Function of Natural Killer Cells in Acute and Chronic Viral Hepatitis. J. Infect. Dis. 2014, 209, 1362–1373. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Li, Y.; Jin, L.; Zhang, S.; Fan, R.; Sun, Y.; Zhou, C.; Shang, Q.; Li, W.; Zhang, Z.; et al. Natural Killer Cells Are Characterized by the Concomitantly Increased Interferon-γ and Cytotoxicity in Acute Resolved Hepatitis B Patients. PLoS ONE 2012, 7, e49135. [Google Scholar] [CrossRef] [PubMed]
- Stelma, F.; Willemse, S.B.; Erken, R.; de Niet, A.; Sinnige, M.J.; van Dort, K.; Zaaijer, H.L.; van Leeuwen, E.M.M.; Kootstra, N.A.; Reesink, H.W. Dynamics of the Immune Response in Acute Hepatitis B Infection. Open Forum Infect. Dis. 2017, 4, ofx231. [Google Scholar] [CrossRef]
- Dunn, C.; Peppa, D.; Khanna, P.; Nebbia, G.; Jones, M.; Brendish, N.; Lascar, R.M.; Brown, D.; Gilson, R.J.; Tedder, R.J.; et al. Temporal Analysis of Early Immune Responses in Patients With Acute Hepatitis B Virus Infection. Gastroenterology 2009, 137, 1289–1300. [Google Scholar] [CrossRef]
- Yu, W.-H.; Cosgrove, C.; Berger, C.T.; Cheney, P.C.; Krykbaeva, M.; Kim, A.Y.; Lewis-Ximenez, L.; Lauer, G.M.; Alter, G. ADCC-Mediated CD56dim NK Cell Responses Are Associated with Early HBsAg Clearance in Acute HBV Infection. Pathog. Immun. 2018, 3, 2–18. [Google Scholar] [CrossRef]
- Oliviero, B.; Varchetta, S.; Paudice, E.; Michelone, G.; Zaramella, M.; Mavilio, D.; De Filippi, F.; Bruno, S.; Mondelli, M.U. Natural Killer Cell Functional Dichotomy in Chronic Hepatitis B and Chronic Hepatitis C Virus Infections. Gastroenterology 2009, 137, 1151–1160. [Google Scholar] [CrossRef]
- Boni, C.; Lampertico, P.; Talamona, L.; Giuberti, T.; Invernizzi, F.; Barili, V.; Fisicaro, P.; Rossi, M.; Cavallo, M.C.; Vecchi, A.; et al. Natural killer cell phenotype modulation and natural killer/T-cell interplay in nucleos(t)ide analogue-treated hepatitis e antigen-negative patients with chronic hepatitis B. Hepatology 2015, 62, 1697–1709. [Google Scholar] [CrossRef]
- Schuch, A.; Zecher, B.F.; Müller, P.A.; Correia, M.P.; Daul, F.; Rennert, C.; Tauber, C.; Schlitt, K.; Boettler, T.; Neumann-Haefelin, C.; et al. NK-cell responses are biased towards CD16-mediated effector functions in chronic hepatitis B virus infection. J. Hepatol. 2019, 70, 351–360. [Google Scholar] [CrossRef]
- Peppa, D.; Micco, L.; Javaid, A.; Kennedy, P.T.F.; Schurich, A.; Dunn, C.; Pallant, C.; Ellis, G.; Khanna, P.; Dusheiko, G.; et al. Blockade of Immunosuppressive Cytokines Restores NK Cell Antiviral Function in Chronic Hepatitis B Virus Infection. PLoS Pathog. 2010, 6, e1001227. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, S.; Zou, Z.; Shi, J.; Zhao, J.; Fan, R.; Qin, E.; Li, B.; Li, Z.; Xu, X.; et al. Hypercytolytic activity of hepatic natural killer cells correlates with liver injury in chronic hepatitis B patients. Hepatology 2011, 53, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, T.; Tatsumi, T.; Miyagi, T.; Mukai, K.; Nishio, K.; Nishio, A.; Yokoyama, Y.; Suda, T.; Kegasawa, T.; Shigekawa, M.; et al. Frequency and role of NKp46 and NKG2A in hepatitis B virus infection. PLoS ONE 2017, 12, e0174103. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Nandi, M.; Pal, S.; Mukhopadhyay, D.; Chakraborty, B.C.; Khatun, M.; Bhowmick, D.; Mondal, R.K.; Das, S.; Das, K.; et al. Natural killer cells contribute to hepatic injury and help in viral persistence during progression of hepatitis B e-antigen-negative chronic hepatitis B virus infection. Clin. Microbiol. Infect. 2016, 22, 733.e9–e19. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Zhu, Y.Y.; Chen, J.; Ye, Y.B.; Li, J.Y.; Liu, Y.R.; Hu, M.L.; Zheng, Y.C.; Jiang, J.J. Activated natural killer cells accelerate liver damage in patients with chronic hepatitis B virus infection. Clin. Exp. Immunol. 2015, 180, 499–508. [Google Scholar] [CrossRef]
- Li, W.; Jiang, Y.; Wang, X.; Jin, J.; Qi, Y.; Chi, X.; Zhang, H.; Feng, X.; Niu, J. Natural Killer p46 Controls Hepatitis B Virus Replication and Modulates Liver Inflammation. PLoS ONE 2015, 10, e0135874. [Google Scholar] [CrossRef]
- Wang, W.-T.; Zhao, X.-Q.; Li, G.-P.; Chen, Y.-Z.; Wang, L.; Han, M.-F.; Li, W.-N.; Chen, T.; Chen, G.; Xu, D.; et al. Immune response pattern varies with the natural history of chronic hepatitis B. World J. Gastroenterol. 2019, 25, 1950–1963. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, W.; Shen, C.; Wang, Y.; Jiao, M.; Yu, W.; Yin, H.; Shang, X.; Liang, Q.; Zhao, C. NKG2D modulates aggravation of liver inflammation by activating NK cells in HBV infection. Sci. Rep. 2017, 7, 88. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.-J.; Gao, S.; Liu, Q.; Bai, J.; Zhao, X.-Q.; Hao, Y.-H.; Ding, H.-H.; Zhu, F.; Yang, D.-L.; et al. Decreased Peripheral Natural Killer Cells Activity in the Immune Activated Stage of Chronic Hepatitis B. PLoS ONE 2014, 9, e86927. [Google Scholar] [CrossRef]
- De Groen, R.A.; Hou, J.; van Oord, G.W.; Groothuismink, Z.M.A.; van der Heide, M.; de Knegt, R.J.; Boonstra, A. NK cell phenotypic and functional shifts coincide with specific clinical phases in the natural history of chronic HBV infection. Antivir. Res. 2017, 140, 18–24. [Google Scholar] [CrossRef]
- Han, W.; Ni, Q.; Liu, K.; Yao, Y.; Zhao, D.; Liu, X.; Chen, Y. Decreased CD122 on CD56 dim NK associated with its impairment in asymptomatic chronic HBV carriers with high levels of HBV DNA, HBsAg and HBeAg. Life Sci. 2018, 195, 53–60. [Google Scholar] [CrossRef]
- Zheng, B.; Yang, Y.; Han, Q.; Yin, C.; Pan, Z.; Zhang, J. STAT3 directly regulates NKp46 transcription in NK cells of HBeAg-negative CHB patients. J. Leukoc. Biol. 2019, 106, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, C.L.; Rinker, F.; Höner zu Siederdissen, C.; Manns, M.P.; Wedemeyer, H.; Cornberg, M.; Björkström, N.K. Increased NK Cell Function After Cessation of Long-Term Nucleos(t)ide Analogue Treatment in Chronic Hepatitis B Is Associated With Liver Damage and HBsAg Loss. J. Infect. Dis. 2018, 217, 1656–1666. [Google Scholar] [CrossRef] [PubMed]
- Micco, L.; Peppa, D.; Loggi, E.; Schurich, A.; Jefferson, L.; Cursaro, C.; Panno, A.M.; Bernardi, M.; Brander, C.; Bihl, F.; et al. Differential boosting of innate and adaptive antiviral responses during pegylated-interferon-alpha therapy of chronic hepatitis B. J. Hepatol. 2013, 58, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Bruder Costa, J.; Dufeu-Duchesne, T.; Leroy, V.; Bertucci, I.; Bouvier-Alias, M.; Pouget, N.; Brevot-Lutton, O.; Bourliere, M.; Zoulim, F.; Plumas, J.; et al. Pegylated Interferon α-2a Triggers NK-Cell Functionality and Specific T-Cell Responses in Patients with Chronic HBV Infection without HBsAg Seroconversion. PLoS ONE 2016, 11, e0158297. [Google Scholar] [CrossRef]
- Tan, A.T.; Hoang, L.T.; Chin, D.; Rasmussen, E.; Lopatin, U.; Hart, S.; Bitter, H.; Chu, T.; Gruenbaum, L.; Ravindran, P.; et al. Reduction of HBV replication prolongs the early immunological response to IFNα therapy. J. Hepatol. 2014, 60, 54–61. [Google Scholar] [CrossRef]
- Gill, U.S.; Peppa, D.; Micco, L.; Singh, H.D.; Carey, I.; Foster, G.R.; Maini, M.K.; Kennedy, P.T.F. Interferon Alpha Induces Sustained Changes in NK Cell Responsiveness to Hepatitis B Viral Load Suppression In Vivo. PLOS Pathog. 2016, 12, e1005788. [Google Scholar] [CrossRef]
- Ju, Y.; Hou, N.; Meng, J.; Wang, X.; Zhang, X.; Zhao, D.; Liu, Y.; Zhu, F.; Zhang, L.; Sun, W.; et al. T cell immunoglobulin- and mucin-domain-containing molecule-3 (Tim-3) mediates natural killer cell suppression in chronic hepatitis B. J. Hepatol. 2010, 52, 322–329. [Google Scholar] [CrossRef]
- Li, F.; Wei, H.; Wei, H.; Gao, Y.; Xu, L.; Yin, W.; Sun, R.; Tian, Z. Blocking the Natural Killer Cell Inhibitory Receptor NKG2A Increases Activity of Human Natural Killer Cells and Clears Hepatitis B Virus Infection in Mice. Gastroenterology 2013, 144, 392–401. [Google Scholar] [CrossRef]
- Zhao, D.; Jiang, X.; Xu, Y.; Yang, H.; Gao, D.; Li, X.; Gao, L.; Ma, C.; Liang, X. Decreased Siglec-9 Expression on Natural Killer Cell Subset Associated With Persistent HBV Replication. Front. Immunol. 2018, 9, 1124. [Google Scholar] [CrossRef]
- Sun, C.; Fu, B.; Gao, Y.; Liao, X.; Sun, R.; Tian, Z.; Wei, H. TGF-β1 Down-Regulation of NKG2D/DAP10 and 2B4/SAP Expression on Human NK Cells Contributes to HBV Persistence. PLoS Pathog. 2012, 8, e1002594. [Google Scholar] [CrossRef]
- Yang, Y.; Han, Q.; Hou, Z.; Zhang, C.; Tian, Z.; Zhang, J. Exosomes mediate hepatitis B virus (HBV) transmission and NK-cell dysfunction. Cell. Mol. Immunol. 2017, 14, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Béziat, V.; Dalgard, O.; Asselah, T.; Halfon, P.; Bedossa, P.; Boudifa, A.; Hervier, B.; Theodorou, I.; Martinot, M.; Debré, P.; et al. CMV drives clonal expansion of NKG2C + NK cells expressing self-specific KIRs in chronic hepatitis patients. Eur. J. Immunol. 2012, 42, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Malone, D.F.G.; Lunemann, S.; Hengst, J.; Ljunggren, H.-G.; Manns, M.P.; Sandberg, J.K.; Cornberg, M.; Wedemeyer, H.; Björkström, N.K. Cytomegalovirus-Driven Adaptive-Like Natural Killer Cell Expansions Are Unaffected by Concurrent Chronic Hepatitis Virus Infections. Front. Immunol. 2017, 8, 525. [Google Scholar] [CrossRef] [PubMed]
- Paust, S.; Blish, C.A.; Reeves, R.K. Redefining Memory: Building the Case for Adaptive NK Cells. J. Virol. 2017, 91, e00169–e00174. [Google Scholar] [CrossRef] [PubMed]
- Schlums, H.; Cichocki, F.; Tesi, B.; Theorell, J.; Beziat, V.; Holmes, T.D.; Han, H.; Chiang, S.C.C.; Foley, B.; Mattsson, K.; et al. Cytomegalovirus Infection Drives Adaptive Epigenetic Diversification of NK Cells with Altered Signaling and Effector Function. Immunity 2015, 42, 443–456. [Google Scholar] [CrossRef]
- Wijaya, R.S.; Read, S.A.; Schibeci, S.; Eslam, M.; Azardaryany, M.K.; El-Khobar, K.; van der Poorten, D.; Lin, R.; Yuen, L.; Lam, V.; et al. KLRG1+ natural killer cells exert a novel antifibrotic function in chronic hepatitis B. J. Hepatol. 2019, 71, 252–264. [Google Scholar] [CrossRef]
- Pallmer, K.; Oxenius, A. Recognition and Regulation of T Cells by NK Cells. Front. Immunol. 2016, 7, 251. [Google Scholar] [CrossRef]
- Crouse, J.; Xu, H.C.; Lang, P.A.; Oxenius, A. NK cells regulating T cell responses: Mechanisms and outcome. Trends Immunol. 2015, 36, 49–58. [Google Scholar] [CrossRef]
- Cook, K.D.; Waggoner, S.N.; Whitmire, J.K. NK cells and their ability to modulate T cells during virus infections. Crit. Rev. Immunol. 2014, 34, 359–388. [Google Scholar] [CrossRef]
- Waggoner, S.N.; Cornberg, M.; Selin, L.K.; Welsh, R.M. Natural killer cells act as rheostats modulating antiviral T cells. Nature 2012, 481, 394–398. [Google Scholar] [CrossRef]
- Lang, P.A.; Lang, K.S.; Xu, H.C.; Grusdat, M.; Parish, I.A.; Recher, M.; Elford, A.R.; Dhanji, S.; Shaabani, N.; Tran, C.W.; et al. Natural killer cell activation enhances immune pathology and promotes chronic infection by limiting CD8+ T-cell immunity. Proc. Natl. Acad. Sci. USA 2012, 109, 1210–1215. [Google Scholar] [CrossRef] [PubMed]
- Gerosa, F.; Baldani-Guerra, B.; Nisii, C.; Marchesini, V.; Carra, G.; Trinchieri, G. Reciprocal activating interaction between natural killer cells and dendritic cells. J. Exp. Med. 2002, 195, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Piccioli, D.; Sbrana, S.; Melandri, E.; Valiante, N.M. Contact-dependent stimulation and inhibition of dendritic cells by natural killer cells. J. Exp. Med. 2002, 195, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Chijioke, O.; Münz, C. Dendritic Cell Derived Cytokines in Human Natural Killer Cell Differentiation and Activation. Front. Immunol. 2013, 4, 365. [Google Scholar] [CrossRef]
- Ge, M.Q.; Ho, A.W.S.; Tang, Y.; Wong, K.H.S.; Chua, B.Y.L.; Gasser, S.; Kemeny, D.M. NK Cells Regulate CD8+ T Cell Priming and Dendritic Cell Migration during Influenza A Infection by IFN-γ and Perforin-Dependent Mechanisms. J. Immunol. 2012, 189, 2099–2109. [Google Scholar] [CrossRef]
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat. Med. 2018, 24, 1178–1191. [Google Scholar] [CrossRef]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037. [Google Scholar] [CrossRef]
- Krebs, P.; Barnes, M.J.; Lampe, K.; Whitley, K.; Bahjat, K.S.; Beutler, B.; Janssen, E.; Hoebe, K. NK cell-mediated killing of target cells triggers robust antigen-specific T cell-mediated and humoral responses. Blood 2009, 113, 6593–6602. [Google Scholar] [CrossRef]
- Deauvieau, F.; Ollion, V.; Doffin, A.-C.; Achard, C.; Fonteneau, J.-F.; Verronese, E.; Durand, I.; Ghittoni, R.; Marvel, J.; Dezutter-Dambuyant, C.; et al. Human natural killer cells promote cross-presentation of tumor cell-derived antigens by dendritic cells. Int. J. Cancer 2015, 136, 1085–1094. [Google Scholar] [CrossRef]
- Srivastava, R.M.; Trivedi, S.; Concha-Benavente, F.; Gibson, S.P.; Reeder, C.; Ferrone, S.; Ferris, R.L. CD137 Stimulation Enhances Cetuximab-Induced Natural Killer: Dendritic Cell Priming of Antitumor T-Cell Immunity in Patients with Head and Neck Cancer. Clin. Cancer Res. 2017, 23, 707–716. [Google Scholar] [CrossRef]
- Cook, K.D.; Whitmire, J.K. The Depletion of NK Cells Prevents T Cell Exhaustion to Efficiently Control Disseminating Virus Infection. J. Immunol. 2013, 190, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Barber, M.A.; Zhang, T.; Gagne, B.A.; Sentman, C.L. NK Cells Negatively Regulate Antigen Presentation and Tumor-Specific CTLs in a Syngeneic Lymphoma Model. J. Immunol. 2007, 178, 6140–6147. [Google Scholar] [CrossRef] [PubMed]
- Mandaric, S.; Walton, S.M.; Rülicke, T.; Richter, K.; Girard-Madoux, M.J.H.; Clausen, B.E.; Zurunic, A.; Kamanaka, M.; Flavell, R.A.; Jonjic, S.; et al. IL-10 Suppression of NK/DC Crosstalk Leads to Poor Priming of MCMV-Specific CD4 T Cells and Prolonged MCMV Persistence. PLoS Pathog. 2012, 8, e1002846. [Google Scholar] [CrossRef] [PubMed]
- Ferlazzo, G.; Tsang, M.L.; Moretta, L.; Melioli, G.; Steinman, R.M.; Münz, C. Human dendritic cells activate resting natural killer (NK) cells and are recognized via the NKp30 receptor by activated NK cells. J. Exp. Med. 2002, 195, 343–351. [Google Scholar] [CrossRef]
- Mitrovic, M.; Arapovic, J.; Jordan, S.; Fodil-Cornu, N.; Ebert, S.; Vidal, S.M.; Krmpotic, A.; Reddehase, M.J.; Jonjic, S. The NK Cell Response to Mouse Cytomegalovirus Infection Affects the Level and Kinetics of the Early CD8+ T-Cell Response. J. Virol. 2012, 86, 2165–2175. [Google Scholar] [CrossRef]
- Ali, A.; Gyurova, I.E.; Waggoner, S.N. Mutually assured destruction: The cold war between viruses and natural killer cells. Curr. Opin. Virol. 2019, 34, 130–139. [Google Scholar] [CrossRef]
- Boni, C.; Vecchi, A.; Rossi, M.; Laccabue, D.; Giuberti, T.; Alfieri, A.; Lampertico, P.; Grossi, G.; Facchetti, F.; Brunetto, M.R.; et al. TLR7 Agonist Increases Responses of Hepatitis B Virus–Specific T Cells and Natural Killer Cells in Patients With Chronic Hepatitis B Treated With Nucleos(T)Ide Analogues. Gastroenterology 2018, 154, 1764–1777. [Google Scholar] [CrossRef]
- Waggoner, S.N.; Taniguchi, R.T.; Mathew, P.A.; Kumar, V.; Welsh, R.M. Absence of mouse 2B4 promotes NK cell–mediated killing of activated CD8+ T cells, leading to prolonged viral persistence and altered pathogenesis. J. Clin. Investig. 2010, 120, 1925–1938. [Google Scholar] [CrossRef]
- Guo, H.; Cranert, S.A.; Lu, Y.; Zhong, M.-C.; Zhang, S.; Chen, J.; Li, R.; Mahl, S.E.; Wu, N.; Davidson, D.; et al. Deletion of Slam locus in mice reveals inhibitory role of SLAM family in NK cell responses regulated by cytokines and LFA-1. J. Exp. Med. 2016, 213, 2187–2207. [Google Scholar] [CrossRef]
- Xu, H.C.; Huang, J.; Pandyra, A.A.; Lang, E.; Zhuang, Y.; Thöns, C.; Timm, J.; Häussinger, D.; Colonna, M.; Cantor, H.; et al. Lymphocytes Negatively Regulate NK Cell Activity via Qa-1b following Viral Infection. Cell Rep. 2017, 21, 2528–2540. [Google Scholar] [CrossRef]
- Cerboni, C.; Zingoni, A.; Cippitelli, M.; Piccoli, M.; Frati, L.; Santoni, A. Antigen-activated human T lymphocytes express cell-surface NKG2D ligands via an ATM/ATR-dependent mechanism and become susceptible to autologous NK- cell lysis. Blood 2007, 110, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-C.; Easom, N.J.; Tang, X.-Z.; Gill, U.S.; Singh, H.; Robertson, F.; Chang, C.; Trowsdale, J.; Davidson, B.R.; Rosenberg, W.M.; et al. T Cells Infiltrating Diseased Liver Express Ligands for the NKG2D Stress Surveillance System. J. Immunol. 2017, 198, 1172–1182. [Google Scholar] [CrossRef] [PubMed]
- Ardolino, M.; Zingoni, A.; Cerboni, C.; Cecere, F.; Soriani, A.; Iannitto, M.L.; Santoni, A. DNAM-1 ligand expression on Ag-stimulated T lymphocytes is mediated by ROS-dependent activation of DNA-damage response: Relevance for NK-T cell interaction. Blood 2011, 117, 4778–4786. [Google Scholar] [CrossRef] [PubMed]
- Schuster, I.S.; Wikstrom, M.E.; Brizard, G.; Coudert, J.D.; Estcourt, M.J.; Manzur, M.; O’Reilly, L.A.; Smyth, M.J.; Trapani, J.A.; Hill, G.R.; et al. TRAIL+ NK Cells Control CD4+ T Cell Responses during Chronic Viral Infection to Limit Autoimmunity. Immunity 2014, 41, 646–656. [Google Scholar] [CrossRef]
- Peppa, D.; Gill, U.S.; Reynolds, G.; Easom, N.J.W.; Pallett, L.J.; Schurich, A.; Micco, L.; Nebbia, G.; Singh, H.D.; Adams, D.H.; et al. Up-regulation of a death receptor renders antiviral T cells susceptible to NK cell-mediated deletion. J. Exp. Med. 2013, 210, 99–114. [Google Scholar] [CrossRef]
- Crouse, J.; Bedenikovic, G.; Wiesel, M.; Ibberson, M.; Xenarios, I.; Von Laer, D.; Kalinke, U.; Vivier, E.; Jonjic, S.; Oxenius, A. Type I Interferons Protect T Cells against NK Cell Attack Mediated by the Activating Receptor NCR1. Immunity 2014, 40, 961–973. [Google Scholar] [CrossRef]
- Pallmer, K.; Barnstorf, I.; Baumann, N.S.; Borsa, M.; Jonjic, S.; Oxenius, A. NK cells negatively regulate CD8 T cells via natural cytotoxicity receptor (NCR) 1 during LCMV infection. PLOS Pathog. 2019, 15, e1007725. [Google Scholar] [CrossRef]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743. [Google Scholar] [CrossRef]
- Ramsuran, V.; Naranbhai, V.; Horowitz, A.; Qi, Y.; Martin, M.P.; Yuki, Y.; Gao, X.; Walker-Sperling, V.; Del Prete, G.Q.; Schneider, D.K.; et al. Elevated HLA-A expression impairs HIV control through inhibition of NKG2A-expressing cells. Science 2018, 359, 86–90. [Google Scholar] [CrossRef]
- Li, H.; Zhai, N.; Wang, Z.; Song, H.; Yang, Y.; Cui, A.; Li, T.; Wang, G.; Niu, J.; Crispe, I.N.; et al. Regulatory NK cells mediated between immunosuppressive monocytes and dysfunctional T cells in chronic HBV infection. Gut 2018, 67, 2035–2044. [Google Scholar] [CrossRef]
- Lam, V.C.; Lanier, L.L. NK cells in host responses to viral infections. Curr. Opin. Immunol. 2017, 44, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Martinet, J.; Dufeu–Duchesne, T.; Bruder Costa, J.; Larrat, S.; Marlu, A.; Leroy, V.; Plumas, J.; Aspord, C. Altered Functions of Plasmacytoid Dendritic Cells and Reduced Cytolytic Activity of Natural Killer Cells in Patients With Chronic HBV Infection. Gastroenterology 2012, 143, 1586–1596. [Google Scholar] [CrossRef] [PubMed]
- Tjwa, E.T.T.L.; van Oord, G.W.; Biesta, P.J.; Boonstra, A.; Janssen, H.L.A.; Woltman, A.M. Restoration of TLR3-Activated Myeloid Dendritic Cell Activity Leads to Improved Natural Killer Cell Function in Chronic Hepatitis B Virus Infection. J. Virol. 2012, 86, 4102–4109. [Google Scholar] [CrossRef] [PubMed]
- Costa-García, M.; Ataya, M.; Moraru, M.; Vilches, C.; López-Botet, M.; Muntasell, A. Human Cytomegalovirus Antigen Presentation by HLA-DR+ NKG2C+ Adaptive NK Cells Specifically Activates Polyfunctional Effector Memory CD4+ T Lymphocytes. Front. Immunol. 2019, 10, 687. [Google Scholar] [CrossRef]
- Zamora, A.E.; Aguilar, E.G.; Sungur, C.M.; Khuat, L.T.; Dunai, C.; Lochhead, G.R.; Du, J.; Pomeroy, C.; Blazar, B.R.; Longo, D.L.; et al. Licensing delineates helper and effector NK cell subsets during viral infection. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Zingoni, A.; Ardolino, M.; Santoni, A.; Cerboni, C. NKG2D and DNAM-1 activating receptors and their ligands in NK-T cell interactions: Role in the NK cell-mediated negative regulation of T cell responses. Front. Immunol. 2013, 3, 408. [Google Scholar] [CrossRef]
- Zingoni, A.; Molfetta, R.; Fionda, C.; Soriani, A.; Paolini, R.; Cippitelli, M.; Cerboni, C.; Santoni, A. NKG2D and Its Ligands: “One for All, All for One. ” Front. Immunol. 2018, 9, 476. [Google Scholar] [CrossRef]
- Kearney, C.J.; Ramsbottom, K.M.; Voskoboinik, I.; Darcy, P.K.; Oliaro, J. Loss of DNAM-1 ligand expression by acute myeloid leukemia cells renders them resistant to NK cell killing. Oncoimmunology 2016, 5, e1196308. [Google Scholar] [CrossRef]
- Veneziani, I.; Fruci, D.; Compagnone, M.; Pistoia, V.; Rossi, P.; Cifaldi, L. The BET-bromodomain inhibitor JQ1 renders neuroblastoma cells more resistant to NK cell-mediated recognition and killing by downregulating ligands for NKG2D and DNAM-1 receptors. Oncotarget 2019, 10, 2151–2160. [Google Scholar] [CrossRef][Green Version]
- Turchinovich, G.; Ganter, S.; Bärenwaldt, A.; Finke, D. NKp46 Calibrates Tumoricidal Potential of Type 1 Innate Lymphocytes by Regulating TRAIL Expression. J. Immunol. 2018, 200, 3762–3768. [Google Scholar] [CrossRef]
- Sheppard, S.; Schuster, I.S.; Andoniou, C.E.; Cocita, C.; Adejumo, T.; Kung, S.K.P.; Sun, J.C.; Degli-Esposti, M.A.; Guerra, N. The Murine Natural Cytotoxic Receptor NKp46/NCR1 Controls TRAIL Protein Expression in NK Cells and ILC1s. Cell Rep. 2018, 22, 3385–3392. [Google Scholar] [CrossRef] [PubMed]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Souza-Fonseca-Guimaraes, F.; Cursons, J.; Huntington, N.D. The Emergence of Natural Killer Cells as a Major Target in Cancer Immunotherapy. Trends Immunol. 2019, 40, 142–158. [Google Scholar] [CrossRef]
- Fang, F.; Xiao, W.; Tian, Z. NK cell-based immunotherapy for cancer. Semin. Immunol. 2017, 31, 37–54. [Google Scholar] [CrossRef]
- Ishikawa, T.; Okayama, T.; Sakamoto, N.; Ideno, M.; Oka, K.; Enoki, T.; Mineno, J.; Yoshida, N.; Katada, K.; Kamada, K.; et al. Phase I clinical trial of adoptive transfer of expanded natural killer cells in combination with IgG1 antibody in patients with gastric or colorectal cancer. Int. J. Cancer 2018, 142, 2599–2609. [Google Scholar] [CrossRef]
- Adotevi, O.; Godet, Y.; Galaine, J.; Lakkis, Z.; Idirene, I.; Certoux, J.M.; Jary, M.; Loyon, R.; Laheurte, C.; Kim, S.; et al. In situ delivery of allogeneic natural killer cell (NK) combined with Cetuximab in liver metastases of gastrointestinal carcinoma: A phase I clinical trial. Oncoimmunology 2018, 7, e1424673. [Google Scholar] [CrossRef]
- Romee, R.; Rosario, M.; Berrien-Elliott, M.M.; Wagner, J.A.; Jewell, B.A.; Schappe, T.; Leong, J.W.; Abdel-Latif, S.; Schneider, S.E.; Willey, S.; et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 2016, 8, 357ra123. [Google Scholar] [CrossRef]
- Hu, Y.; Tian, Z.; Zhang, C. Natural Killer Cell-Based Immunotherapy for Cancer: Advances and Prospects. Engineering 2019, 31, 37–54. [Google Scholar] [CrossRef]
- Romee, R.; Cooley, S.; Berrien-Elliott, M.M.; Westervelt, P.; Verneris, M.R.; Wagner, J.E.; Weisdorf, D.J.; Blazar, B.R.; Ustun, C.; DeFor, T.E.; et al. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood 2018, 131, 2515–2527. [Google Scholar] [CrossRef] [PubMed]
- Felices, M.; Chu, S.; Kodal, B.; Bendzick, L.; Ryan, C.; Lenvik, A.J.; Boylan, K.L.M.; Wong, H.C.; Skubitz, A.P.N.; Miller, J.S.; et al. IL-15 super-agonist (ALT-803) enhances natural killer (NK) cell function against ovarian cancer. Gynecol. Oncol. 2017, 145, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Daher, M.; Rezvani, K. Next generation natural killer cells for cancer immunotherapy: The promise of genetic engineering. Curr. Opin. Immunol. 2018, 51, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Revill, P.; Testoni, B.; Locarnini, S.; Zoulim, F. Global strategies are required to cure and eliminate HBV infection. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Durantel, D.; Zoulim, F. New antiviral targets for innovative treatment concepts for hepatitis B virus and hepatitis delta virus. J. Hepatol. 2016, 64, S117–S131. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| NK Cell Subset | Phenotypic Characteristics | Functional Activity | Comments |
|---|---|---|---|
| CD56bright NK circulating cells | CD56bright CD16neg/low | High cytokine production Low cytolytic activity | Considered as precursors of the more mature CD56 dim NK cells [13] |
| CD56dim NK circulating cells | CD56dim CD16bright | Cytolytic activity Low cytokine production | Main circulating NK subset Terminally differentiated NK cells [13] |
| CD56dim CD16neg | High cytolytic activity High cytokine production in healthy individuals | Not fully functional in malignancies [17,18] | |
| CD56neg NK circulating cells | CD56neg CD16bright | Low cytolytic activity Low cytokine production | Minor subset in healthy donors Significantly expanded in chronic HIV and HCV chronic infections [19] and in CMV/EBV co-infected older healthy donors [20] |
| Hepatic conventional NK cells | CD56dim CD16bright CCR5neg CXCR6neg | High cytolytic activity [21] | Similar to peripheral CD56dim [22] Promote T cell function [21] |
| Liver-resident NK cells | CD56bright CD16low CD69+ Tbetlow EomeshiCCR5+ CXCR6+ | low levels of perforin and granzyme B Low cytokine production Low cytolytic activity [23] | Regulatory role through the PD1/PD-L1 signaling [21] Can acquire memory to haptens and viral antigens [24,25] |
| Mechanisms of NK/T Cell Interplay | Animal Studies | Human Studies | HBV Studies (human) | ||
|---|---|---|---|---|---|
| Indirect mechanisms | enhancement | DC maturation and IL-12 production | [77,82,83,84] | ||
| DC recruitment | [87] | [86] | |||
| Promoting Ag cross-presentation by DC | [88] | [89,90] | |||
| inhibition | APC capacity reduction | [91] | |||
| DC killing | [92,93] | [94] | |||
| Ag availability modulation | [95] | ||||
| Direct mechanisms | enhancement | a.Cytokine-mediated interaction Anti-viral/pro-inflammatory cytokine secretion | [96] | [96] | [97] |
b.Receptor/Ligand NK-T cell cross-talk
| |||||
| [98,99] | ||||
| [100] | ||||
| inhibition | a.Cytokine-mediated interaction
| [79] | [79] | ||
b.Receptor/Ligand NK-T cell cross-talk
| |||||
| [80,81] | [101] | [102] | ||
| [103] | ||||
| [104] | [48,105] | |||
| [106,107] | ||||
| c.Checkpoint inhibitory pathways | |||||
| [108] | [108] | |||
| [109,110] | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fisicaro, P.; Rossi, M.; Vecchi, A.; Acerbi, G.; Barili, V.; Laccabue, D.; Montali, I.; Zecca, A.; Penna, A.; Missale, G.; et al. The Good and the Bad of Natural Killer Cells in Virus Control: Perspective for Anti-HBV Therapy. Int. J. Mol. Sci. 2019, 20, 5080. https://doi.org/10.3390/ijms20205080
Fisicaro P, Rossi M, Vecchi A, Acerbi G, Barili V, Laccabue D, Montali I, Zecca A, Penna A, Missale G, et al. The Good and the Bad of Natural Killer Cells in Virus Control: Perspective for Anti-HBV Therapy. International Journal of Molecular Sciences. 2019; 20(20):5080. https://doi.org/10.3390/ijms20205080
Chicago/Turabian StyleFisicaro, Paola, Marzia Rossi, Andrea Vecchi, Greta Acerbi, Valeria Barili, Diletta Laccabue, Ilaria Montali, Alessandra Zecca, Amalia Penna, Gabriele Missale, and et al. 2019. "The Good and the Bad of Natural Killer Cells in Virus Control: Perspective for Anti-HBV Therapy" International Journal of Molecular Sciences 20, no. 20: 5080. https://doi.org/10.3390/ijms20205080
APA StyleFisicaro, P., Rossi, M., Vecchi, A., Acerbi, G., Barili, V., Laccabue, D., Montali, I., Zecca, A., Penna, A., Missale, G., Ferrari, C., & Boni, C. (2019). The Good and the Bad of Natural Killer Cells in Virus Control: Perspective for Anti-HBV Therapy. International Journal of Molecular Sciences, 20(20), 5080. https://doi.org/10.3390/ijms20205080