Thrombin Upregulates PAI-1 and Mesothelial–Mesenchymal Transition Through PAR-1 and Contributes to Tuberculous Pleural Fibrosis


 ,
,
Abstract
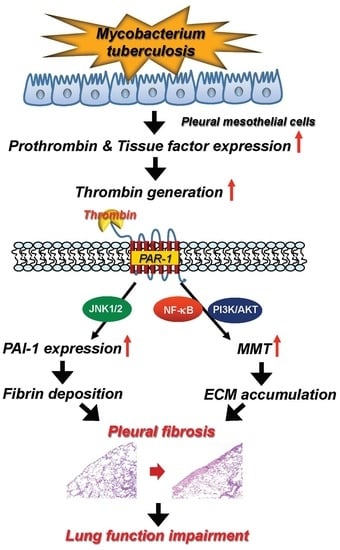
1. Introduction
2. Results
2.1. Thrombin Levels Between Transudative Pleural Effusion and Tuberculous Pleural Effusion
2.2. Cytokines and Fibrinolytic Factors between TBPE Patients with Residual Pleural Thickening (RPT) ≤ 10 mm and RPT > 10 mm
2.3. Correlation Between Thrombin and Inflammatory Parameters, Fibrinolytic Factors and Cytokines in TBPE
2.4. Multivariate Logistic Regression Analysis
2.5. Optimal Sensitivity, Specificity, and Cutoff Value of Variables to Predict RPT > 10 mm
2.6. Correlation Between Effusion Thrombin or Plasminogen Activator Inhibitor-1 (PAI-1) and Residual Pleural Fibrosis
2.7. Protease-Activated Receptor (PAR)-1 Silencing Attenuates Pleural Fibrosis in Vivo
2.8. Thrombin Induces Mesothelial–Mesenchymal Transition in Vivo and in Vitro Through PAR-1, PI3K/AKT and NF-κB Signalings
2.9. Thrombin Induces PAI-1 Expression in Human Pleural Mesothelial Cells Through PAR-1/JNK Signaling
2.10. Heat-Killed Mycobacterium Tuberculosis H37Ra Increased Thrombin Production in Human PMCs
2.11. MTBRa Induced mRNA and Protein Expression of Tissue Factor and Prothrombin in Human PMCs
2.12. MTBRa Upregulates PAI-1, α-SMA and Fibronectin Expression Through PAR-1 in Human PMCs
2.13. Expression of PAR-1, Prothrombin, PAI-1, and α-SMA in the Pleural Mesothelium of Patients with TBPE
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Patient Selection
4.3. Thoracentesis and Pleural Fluid Analysis
4.4. Chest Radiographs and Pulmonary Function
4.5. Measurement of Cytokines and Fibrinolytic Factors
4.6. Carbon Black/Bleomycin Pleural Fibrosis Mouse Model
4.7. Cell Cultivation of Pleural Mesothelial Cells
4.8. Protease-Activated Receptor-1 Silencing
4.9. Western Blotting Assay
4.10. PAI-1 Luciferase Activity Assay
4.11. Reverse Transcription–Polymerase Chain Reaction
4.12. Immunofluorescence Staining Assay
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| α-SMA | α-smooth muscle actin |
| CBB | Carbon black/bleomycin |
| ECM | Extracelluar matrix |
| MMT | Mesothelial–mesenchymal transition |
| MTBRa | Mycobacterium tuberculosis H37Ra |
| PAI-1 | Plasminogen activator inhibitor-1 |
| PAR-1 | Protease-activated receptor-1 |
| PMC | Pleural mesothelial cell |
| RPT | Residual pleural thickening |
| TBPE | Tuberculous pleural effusion |
| TF | Tissue factor |
| TPE | Transudative pleural effusion |
References
- GBD Tuberculosis Collaborators. Global, regional, and national burden of tuberculosis, 1990–2016: Results from the Global Burden of Diseases, Injuries, and Risk Factors 2016 Study. Lancet Infect. Dis. 2018, 18, 1329–1349. [Google Scholar] [CrossRef]
- Jantz, M.A.; Antony, V.B. Pleural fibrosis. Clin. Chest. Med. 2006, 27, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Antony, V.B. Immunological mechanisms in pleural disease. Eur. Respir. J. 2003, 21, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Nahm, C.H.; Choi, J.W. Thrombin-antithrombin III complex, proinflammatory cytokines, and fibrinolytic indices for assessing the severity of inflammation in pleural effusions. Ann. Clin. Lab. Sci. 2010, 40, 342–347. [Google Scholar] [PubMed]
- Wu, Z.H.; Tsai, J.H.; Hsieh, C.Y.; Chen, W.L.; Chung, C.L. Endothelin-1 induces mesothelial mesenchymal transition and correlates with pleural fibrosis in tuberculous pleural effusions. J. Clin. Med. 2019, 8, E426. [Google Scholar] [CrossRef] [PubMed]
- Komissarov, A.A.; Rahman, N.; Lee, Y.C.G.; Florova, G.; Shetty, S.; Idell, R.; Ikebe, M.; Das, K.; Tucker, T.A.; Idell, S. Fibrin turnover and pleural organization: Bench to bedside. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L757–L768. [Google Scholar] [CrossRef] [PubMed]
- Mutsaers, S.E. Mesothelial cells: Their structure, function and role in serosal repair. Respirology 2002, 7, 171–191. [Google Scholar] [CrossRef]
- Mutsaers, S.E.; Prele, C.M.; Brody, A.R.; Idell, S. Pathogenesis of pleural fibrosis. Respirology 2004, 9, 428–440. [Google Scholar] [CrossRef]
- Owens, S.; Jeffers, A.; Boren, J.; Tsukasaki, Y.; Koenig, K.; Ikebe, M.; Idell, S.; Tucker, T.A. Mesomesenchymal transition of pleural mesothelial cells is PI3K and NF-κB dependent. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L1265–L1273. [Google Scholar] [CrossRef]
- Decologne, N.; Wettstein, G.; Kolb, M.; Margetts, P.; Garrido, C.; Camus, P.; Bonniaud, P. Bleomycin induces pleural and subpleural fibrosis in the presence of carbon particles. Eur. Respir. J. 2010, 35, 176–185. [Google Scholar] [CrossRef]
- Tucker, T.A.; Jeffers, A.; Alvarez, A.; Owens, S.; Koenig, K.; Quaid, B.; Komissarov, A.A.; Florova, G.; Kothari, H.; Pendurthi, U.; et al. Plasminogen activator inhibitor-1 deficiency augments visceral mesothelial organization, intrapleural coagulation, and lung restriction in mice with carbon black/bleomycin-induced pleural injury. Am. J. Respir. Cell Mol. Biol. 2014, 50, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Rodríguez, N.A.; Cambrey, A.D.; Harrison, N.K.; Chambers, R.C.; Gray, A.J.; Southcott, A.M.; duBois, R.M.; Black, C.M.; Scully, M.F.; McAnulty, R.J. Role of thrombin in pulmonary fibrosis. Lancet 1995, 346, 1071–1073. [Google Scholar] [CrossRef]
- Mercer, P.F.; Chambers, R.C. Coagulation and coagulation signalling in fibrosis. Biochim. Biophys. Acta 2013, 1832, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Kothari, H.; Kaur, G.; Sahoo, S.; Idell, S.; Rao, L.V.; Pendurthi, U. Plasmin enhances cell surface tissue factor activity in mesothelial and endothelial cells. J. Thromb. Haemost. 2009, 7, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Jeffers, A.; Owens, S.; Koenig, K.; Quaid, B.; Pendurthi, U.R.; Rao, V.M.; Idell, S.; Tucker, T.A. Thrombin down-regulates tissue factor pathway inhibitor expression in a PI3K/nuclear factor-κB-dependent manner in human pleural mesothelial cells. Am. J. Respir. Cell Mol. Biol. 2015, 52, 674–682. [Google Scholar] [CrossRef] [PubMed]
- José, R.J.; Williams, A.E.; Chambers, R.C. Proteinase-activated receptors in fibroproliferative lung disease. Thorax 2014, 69, 190–192. [Google Scholar] [CrossRef] [PubMed]
- Iakhiaev, A.V.; Nalian, A.; Koenig, K.; Idell, S. Thrombin-thrombomodulin inhibits prourokinase-mediated pleural mesothelial cell-dependent fibrinolysis. Thromb. Res. 2007, 120, 715–725. [Google Scholar] [CrossRef][Green Version]
- Howell, D.C.; Johns, R.H.; Lasky, J.A.; Shan, B.; Scotton, C.J.; Laurent, G.J.; Chambers, R.C. Absence of proteinase-activated receptor-1 signaling affords protection from bleomycin-induced lung inflammation and fibrosis. Am. J. Pathol. 2005, 166, 1353–1365. [Google Scholar] [CrossRef]
- Shea, B.S.; Probst, C.K.; Brazee, P.L.; Rotile, N.J.; Blasi, F.; Weinreb, P.H.; Black, K.E.; Sosnovik, D.E.; Van Cott, E.M.; Violette, S.M.; et al. Uncoupling of the profibrotic and hemostatic effects of thrombin in lung fibrosis. JCI Insight 2017, 2, 86608. [Google Scholar] [CrossRef]
- Chen, W.L.; Sheu, J.R.; Chen, R.J.; Hsiao, S.H.; Hsiao, C.J.; Chou, Y.C.; Chung, C.L.; Hsiao, G. Mycobacterium tuberculosis upregulates TNF-α expression via TLR2/ERK signaling and induces MMP-1 and MMP-9 production in human pleural mesothelial cells. PLoS ONE 2015, 10, e0137979. [Google Scholar] [CrossRef]
- Siller-Matula, J.M.; Schwameis, M.; Blann, A.; Mannhalter, C.; Jilma, B. Thrombin as a multi-functional enzyme. Focus on in vitro and in vivo effects. Thromb. Haemost. 2011, 106, 1020–1033. [Google Scholar] [PubMed]
- Bogatkevich, G.S.; Tourkina, E.; Silver, R.M.; Ludwicka-Bradley, A. Thrombin differentiates normal lung fibroblasts to a myofibroblast phenotype via the proteolytically activated receptor-1 and a protein kinase C-dependent pathway. J. Biol. Chem. 2001, 276, 45184–45192. [Google Scholar] [CrossRef]
- Archiniegas, E.; Neves, C.Y.; Candelle, D.; Cardier, J.E. Thrombin and its protease-activated receptor-1 (PAR1) participate in the endothelial-mesenchymal transdifferentiation process. DNA Cell Biol. 2004, 23, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Song, J.S.; Kang, C.M.; Park, C.K.; Yoon, H.K. Thrombin induces epithelial-mesenchymal transition via PAR-1, PKC, and ERK1/2 pathways in A549 cells. Exp. Lung Res. 2013, 39, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Chambers, R.C.; Scotton, C.J. Coagulation cascade proteinases in lung injury and fibrosis. Proc. Am. Thorac. Soc. 2012, 9, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Dichek, D.; Quertermous, T. Thrombin regulation of mRNA levels of tissue plasminogen activator and plasminogen activator inhibitor-1 in cultured human umbilical vein endothelial cells. Blood 1989, 74, 222–228. [Google Scholar] [CrossRef]
- Bogatkevich, G.S.; Ludwicka-Bradley, A.; Silver, R.M. Dabigatran, a direct thrombin inhibitor, demonstrates antifibrotic effects on lung fibroblasts. Arthritis Rheum. 2009, 60, 3455–3464. [Google Scholar] [CrossRef]
- Howell, D.C.; Goldsack, N.R.; Marshall, R.P.; McAnulty, R.J.; Starke, R.; Purdy, G.; Laurent, G.J.; Chambers, R.C. Direct thrombin inhibition reduces lung collagen, accumulation, and connective tissue growth factor mRNA levels in bleomycin-induced pulmonary fibrosis. Am. J. Pathol. 2001, 159, 1383–1395. [Google Scholar] [CrossRef]
- Bogatkevich, G.S.; Ludwicka-Bradley, A.; Nietert, P.J.; Akter, T.; van Ryn, J.; Silver, R.M. Antiinflammatory and antifibrotic effects of the oral direct thrombin inhibitor dabigatran etexilate in a murine model of interstitial lung disease. Arthritis Rheum. 2011, 63, 1416–1425. [Google Scholar] [CrossRef]
- Silver, R.; Atanelishvili, I.; Akter, T.; Kajdasz, K.; Wilson, D.; Nietert, P.; Huggins, J.T.; Highland, K.B.; Bogatkevich, G. Safety and suitability of a direct thrombin inhibitor, Dabigatran etexilate, in scleroderma-associated interstitial lung disease (SSC-ILD) patients. Am. J. Respir. Crit. Care Med. 2018, 197, A1055. [Google Scholar]
- De Pablo, A.; Villena, V.; Echave-Sustaeta, J.; Encuentra, A.L. Are pleural fluid parameters related to the development of residual pleural thickening in tuberculosis? Chest 1997, 112, 1293–1297. [Google Scholar] [CrossRef] [PubMed]
- Duecker, R.; Baer, P.; Eickmeier, O.; Strecker, M.; Kurz, J.; Schaible, A.; Henrich, D.; Zielen, S.; Schubert, R. Oxidative stress-driven pulmonary inflammation and fibrosis in a mouse model of human ataxia-telangiectasia. Redox Biol. 2018, 14, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Rajput, P.S.; Lyden, P.D.; Chen, B.; Lamb, J.A.; Pereira, B.; Lamb, A.; Zhao, L.; Lei, I.F.; Bai, J. Protease activated receptor-1 mediates cytotoxicity during ischemia using in vivo and in vitro models. Neuroscience 2014, 281, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.L.; Sheu, J.R.; Chen, W.L.; Chou, Y.C.; Hsiao, C.J.; Hsiao, S.H.; Hsu, M.J.; Cheng, Y.W.; Hsiao, G. Histone deacetylase inhibitor m-carboxycinnamic acid bis-hydroxamide attenuates plasminogen activator inhibitor-1 expression in human pleural mesothelial cells. Am. J. Respir. Cell Mol. Biol. 2012, 46, 437–445. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TPE | TBPE | p-Value * | ||
|---|---|---|---|---|
| RPT ≤ 10 mm | RPT > 10 mm | |||
| Subject, n | 22 | 14 | 10 | |
| Age, years | 80 (65–88) | 64 (20–87) | 79 (34–91) | 0.186 |
| Males, n | 14 | 9 | 6 | 0.830 |
| Symptom onset to enrollment, days | 7 (5–10) | 14 (6–20) | 16 (7–25) | 0.304 |
| Pleural fluid | ||||
| pH value | 7.39 (7.30–7.46) | 7.37 (7.25–7.46) | 7.30 (7.23–7.37) | 0.010 |
| Glucose, mg/dL | 141 (102–193) | 126 (68–175) | 88 (25–181) | 0.105 |
| Protein, g/L | 2.3 (1.1–3.6) | 4.7 (3.3–5.8) | 4.6 (3.6–6.5) | 0.829 |
| LDH, IU/dL | 83 (22–153) | 274 (121–1370) | 495 (215–2001) | 0.064 |
| Leukocyte count, cells/μL | 313 (108–1080) | 959 (235–5840) | 1531 (137–6872) | 0.667 |
| ADA, IU/L | 13 (6–36) | 112 (59–262) | 180 (68–220) | 0.045 |
| Thrombin, pg/mL | 0.7 (0.1–1.1) | 5.0 (3.9–5.4) | 5.7 (5.4–6.7) | <0.0001 |
| PAI-1, ng/mL | 36.6 (3.1–140.5) | 107.2 (40.4–150.1) | 137.5 (100.9–225.3) | 0.003 |
| tPA, ng/mL | 12.1 (6.9–35.0) | 24.1 (1.0–56.9) | 14.1 (1.8–41.5) | 0.186 |
| TNF-α, pg/mL | 17.0 (4.5–109.8) | 37.7 (15.0–184.4) | 88.8 (35.8–287.2) | 0.025 |
| IL-1β, pg/mL | 1.7 (1.4–3.7) | 5.9 (4.3–34.3) | 17.6 (5.8–63.1) | 0.021 |
| Initial pleural effusion CXR score, % | 49 (25–65) | 43 (29–59) | 61 (42–93) | 0.003 |
| Residual pleural shadowing CXR score, % | 0 (0–1) | 2.0 (0.6–4.0) | 4.5 (2.5–5.9) | <0.001 |
| FVC at 12 months, % predicted | 79 (77–82) | 80 (78–81) | 74 (71–76) | <0.001 |
| Variables | Coefficient * | p-Value |
|---|---|---|
| pH value | −0.46 | 0.051 |
| Glucose, mg/dL | −0.16 | 0.460 |
| LDH, IU/dL | 0.35 | 0.070 |
| ADA, IU/L | 0.40 | 0.063 |
| PAI-1, ng/mL | 0.65 | <0.0001 |
| tPA, ng/mL | −0.04 | 0.862 |
| TNF-α, pg/mL | 0.38 | 0.066 |
| IL-1β, pg/mL | 0.41 | 0.058 |
| Variables | Thrombin Excluded | PAI-1 Excluded | ||||
|---|---|---|---|---|---|---|
| OR | 95% CI | p-Value | OR | 95% CI | p-Value | |
| Thrombin, U/mL | 9.05 | 3.09–28.42 | 0.007 | |||
| pH value | 0.92 | 0.83–1.02 | 0.135 | 0.25 | 0.01–1.181 | 0.623 |
| ADA, IU/L | 1.29 | 0.77–1.83 | 0.609 | 1.12 | 0.99–1.26 | 0.721 |
| TNF-α, pg/mL | 1.33 | 0.91–1.95 | 0.147 | 1.04 | 0.95–1.09 | 0.925 |
| IL-1β, pg/mL | 1.02 | 0.98–1.07 | 0.272 | 1.08 | 0.95–1.18 | 0.182 |
| PAI-1, ng/mL | 2.26 | 1.05–4.87 | 0.037 | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, C.-Y.; Sheu, J.-R.; Yang, C.-H.; Chen, W.-L.; Tsai, J.-H.; Chung, C.-L. Thrombin Upregulates PAI-1 and Mesothelial–Mesenchymal Transition Through PAR-1 and Contributes to Tuberculous Pleural Fibrosis. Int. J. Mol. Sci. 2019, 20, 5076. https://doi.org/10.3390/ijms20205076
Hsieh C-Y, Sheu J-R, Yang C-H, Chen W-L, Tsai J-H, Chung C-L. Thrombin Upregulates PAI-1 and Mesothelial–Mesenchymal Transition Through PAR-1 and Contributes to Tuberculous Pleural Fibrosis. International Journal of Molecular Sciences. 2019; 20(20):5076. https://doi.org/10.3390/ijms20205076
Chicago/Turabian StyleHsieh, Cheng-Ying, Joen-Rong Sheu, Chih-Hao Yang, Wei-Lin Chen, Jie-Heng Tsai, and Chi-Li Chung. 2019. "Thrombin Upregulates PAI-1 and Mesothelial–Mesenchymal Transition Through PAR-1 and Contributes to Tuberculous Pleural Fibrosis" International Journal of Molecular Sciences 20, no. 20: 5076. https://doi.org/10.3390/ijms20205076
APA StyleHsieh, C.-Y., Sheu, J.-R., Yang, C.-H., Chen, W.-L., Tsai, J.-H., & Chung, C.-L. (2019). Thrombin Upregulates PAI-1 and Mesothelial–Mesenchymal Transition Through PAR-1 and Contributes to Tuberculous Pleural Fibrosis. International Journal of Molecular Sciences, 20(20), 5076. https://doi.org/10.3390/ijms20205076