The Immune Escape Mechanisms of Mycobacterium Tuberculosis


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. M. tuberculosis Inhibits the Maturation and Acidification of Phagolysosomes
2.1. M. tuberculosis Inhibits the Maturation of Phagolysosomes
2.2. M. tuberculosis Inhibits the Acidification of Phagolysosomes
3. M. tuberculosis Inhibits Oxidative Stress and the Function of Reactive Oxygen and Reactive Nitrogen Intermediates
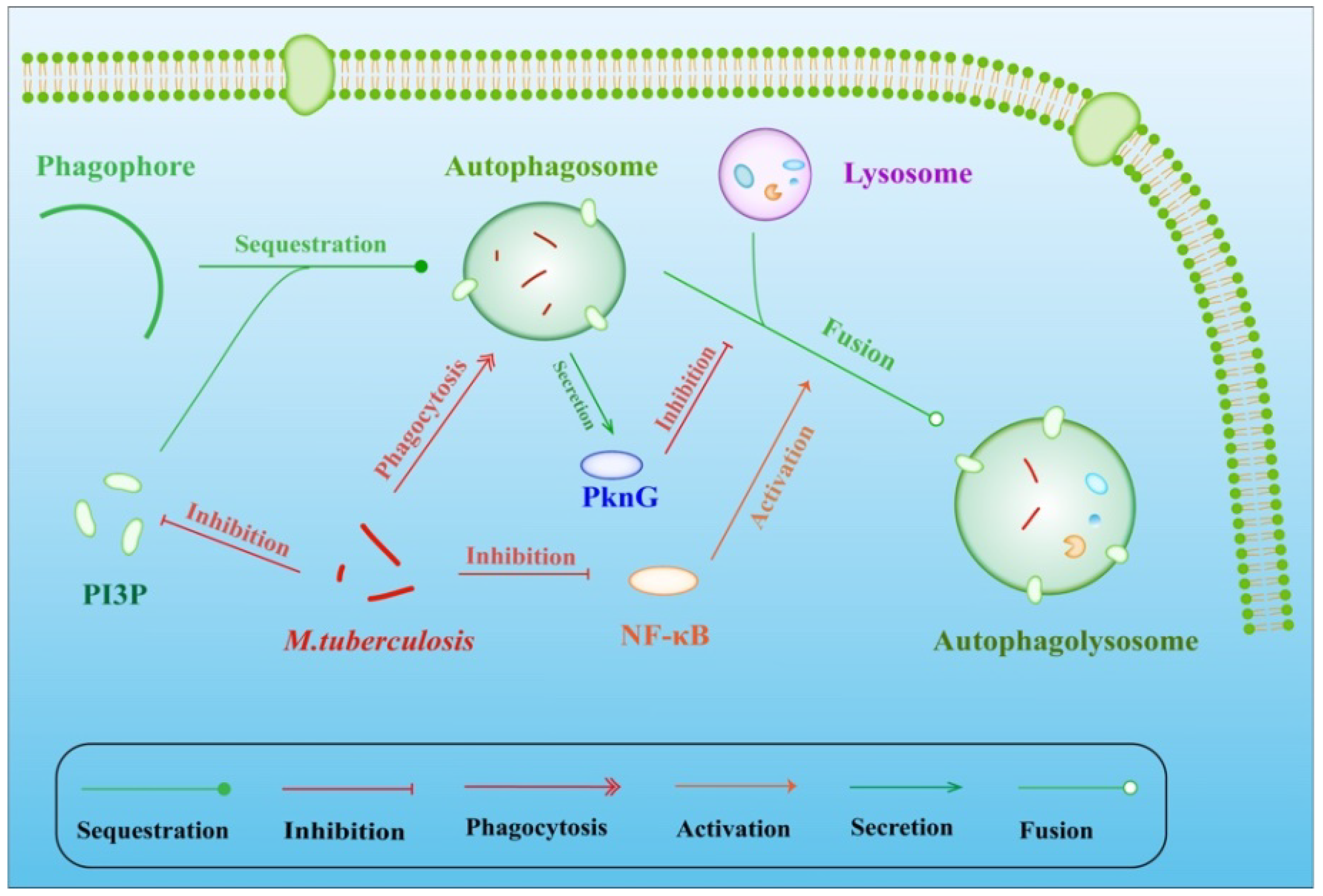
4. M. tuberculosis Inhibits Apoptosis and Autophagy
4.1. Weakly Virulent M. tuberculosis Promotes Apoptosis
4.2. Strongly Virulent M. tuberculosis Inhibits the Apoptosis of Macrophages
5. The Effects of Iron, Hydrogen and Calcium Ions in Macrophages
5.1. Iron Ions Inhibit Lysosome Formation
5.2. Hydrogen Ions Inhibit Lysosome Formation
5.3. Calcium Ions Promote Lysosome Formation
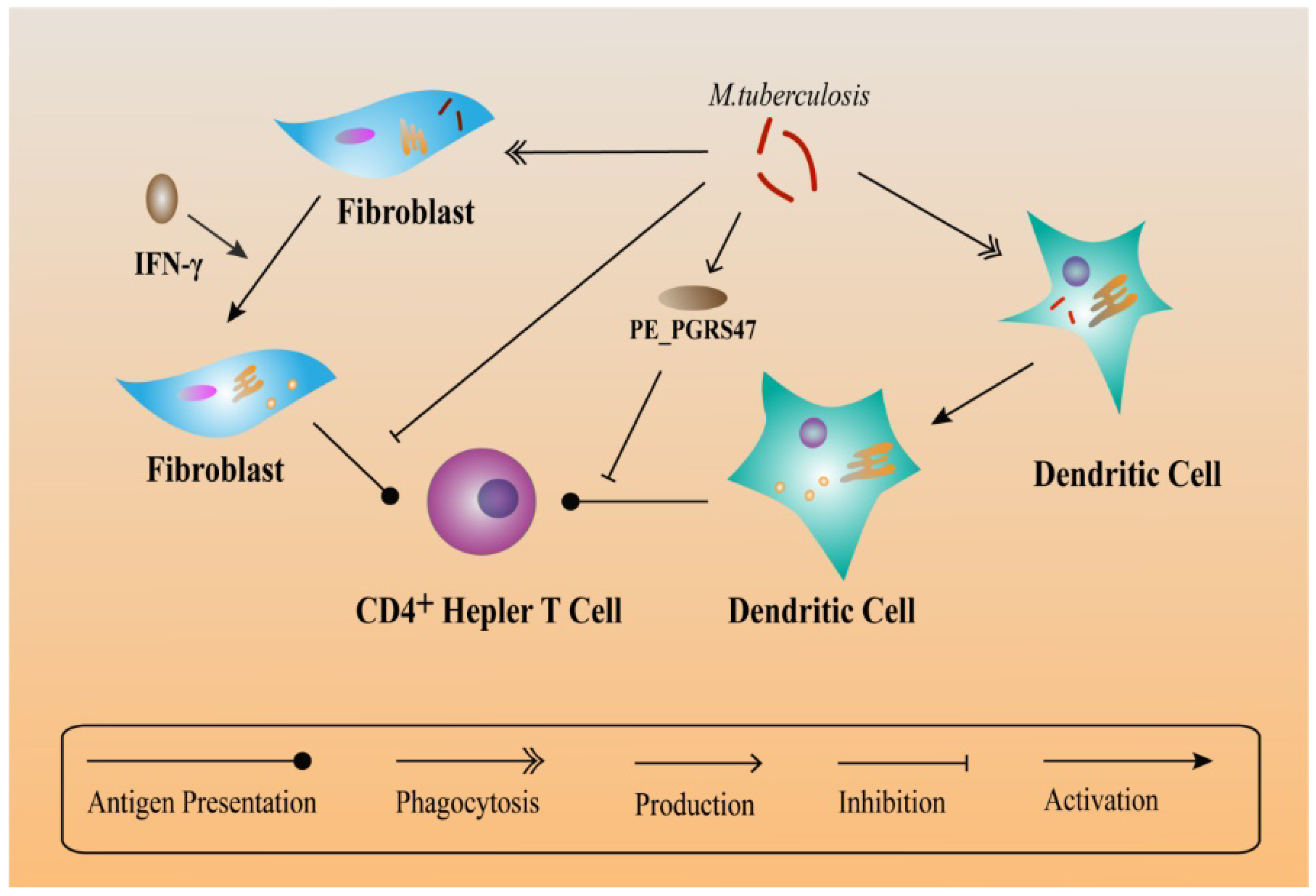
6. Other Mechanisms Helping M. tuberculosis to Escape Immune Responses
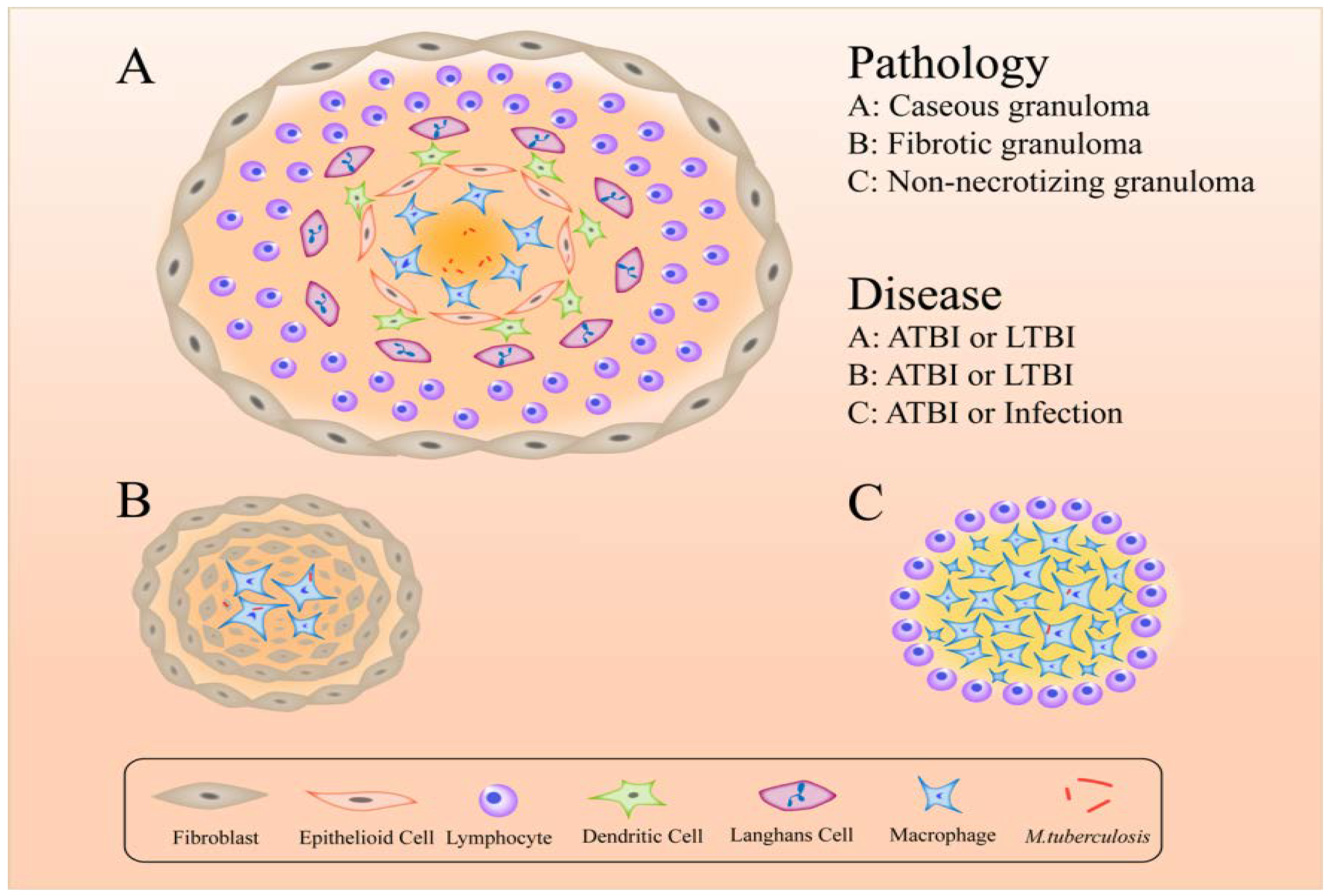
7. Formation of Granulomas Help M. tuberculosis to Escape Immune Responses
8. Pre-clinical Animal Models
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MTB | Mycobacterium tuberculosis |
| TLRs | Toll-like receptors |
| NF-κB | nuclear factor kappa B |
| PI3P | Phosphatidylinositol 3-phosphate |
| LAM | lipoarabinomannan |
| DC-SIGN | DC-specific intercellular adhesion molecule-3 grabbing nonintegrin |
| PtkA | protein tyrosine kinase A |
| V-ATPase | Vacuolar H+-ATPase |
| CISH | Cytokine-inducible SH2-containing protein |
| Rv0014c-Rv0019c | [PknA (encoded by Rv0014c) and FtsZ-interacting protein A (FipA) (encoded by Rv0019c)] |
| FipA | FtsZ-interacting protein A |
| MSH | Mycothiol; H2O2: Hydrogen peroxide |
| PknG | Protein kinase G |
| PMNs | Polymorphonuclear neutrophils |
| ROS | Reactive oxygen species |
| LAM | Latin America and the Mediterranean |
| CFP-10 | Culture filtrate protein-10 kDa |
| ESAT-6 | Early secreted antigenic target-6 kDa |
| ERG | Ergothioneine |
| GGC | Gamma-glutamylcysteine |
| RNS | Reactive nitrogen species |
| DCs | Dendritic cells |
| Nramp1 | Membrane protein 1 |
| P2Y2 | Purinergic receptor P2Y, G-protein coupled, 2, P2RY2 |
| P2Y7 | Purinergic receptor P2Y, G-protein coupled, 7, P2RY7 |
| GTP | Guanosine triphosphate |
| Th17 | T helper 17 |
| Treg | T regulatory |
| Glu MODs | Cell-wall-related alpha-glucan can induce mononuclear cells to differentiate into DCS |
| Mt-MoDC | MTB-infected monocytes |
| BMDCs | Bone marrow-derived DCs |
| PPARγ | Proliferator-activated receptor γ |
| PPE | Pro-Pro-Glu |
| PPE38 | Pro-Pro-Glu 38 |
References
- Ahmad, S. New approaches in the diagnosis and treatment of latent tuberculosis infection. Respir. Res. 2010, 11, 169. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, E.S.; Early, J.; Parish, T. The future for early-stage tuberculosis drug discovery. Future Microbiol. 2015, 10, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.L.; Chan, J. Immunology of tuberculosis. Annu. Rev. Immunol. 2001, 19, 93–129. [Google Scholar] [CrossRef] [PubMed]
- Rohini, K.; Srikumar, P.S. Insights from the docking and molecular dynamics simulation of the Phosphopantetheinyl transferase (PptT) structural model from Mycobacterium tuberculosis. Bioinformation 2013, 9, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, A.; Dietzold, J.; Verma, S.; Bhagavathula, M.; Salgame, P. Toll-like receptor 2 prevents neutrophil-driven immunopathology during infection with Mycobacterium tuberculosis by curtailing CXCL5 production. Infect. Immun. 2018. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, L.S.; Hull, S.R.; Kaufman, T.M. Binding of the terminal mannosyl units of lipoarabinomannan from a virulent strain of Mycobacterium tuberculosis to human macrophages. J. Immunol. 1994, 152, 4070–4079. [Google Scholar] [PubMed]
- Gutierrez, M.G.; Mishra, B.B.; Jordao, L.; Elliott, E.; Anes, E.; Griffiths, G. NF-kappa B activation controls phagolysosome fusion-mediated killing of mycobacteria by macrophages. J. Immunol. 2008, 181, 2651–2663. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, G. Mycobacterium tuberculosis response to stress from reactive oxygen and nitrogen species. Front. Microbiol. 2011, 2, 176. [Google Scholar] [CrossRef] [PubMed]
- Raja, A. Immunology of tuberculosis. Indian J. Med. Res. 2004, 120, 213–232. [Google Scholar]
- Bulut, Y.; Michelsen, K.S.; Hayrapetian, L.; Naiki, Y.; Spallek, R.; Singh, M.; Arditi, M. Mycobacterium tuberculosis heat shock proteins use diverse Toll-like receptor pathways to activate pro-inflammatory signals. J. Biol. Chem. 2005, 280, 20961–20967. [Google Scholar] [CrossRef]
- Davila, S.; Hibberd, M.L.; Hari Dass, R.; Wong, H.E.; Sahiratmadja, E.; Bonnard, C.; Alisjahbana, B.; Szeszko, J.S.; Balabanova, Y.; Drobniewski, F.; et al. Genetic association and expression studies indicate a role of toll-like receptor 8 in pulmonary tuberculosis. PLoS Genet. 2008, 4, e1000218. [Google Scholar] [CrossRef] [PubMed]
- Master, S.S.; Rampini, S.K.; Davis, A.S.; Keller, C.; Ehlers, S.; Springer, B.; Timmins, G.S.; Sander, P.; Deretic, V. Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe 2008, 3, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Inohara, N.; Nunez, G. The NOD: A signaling module that regulates apoptosis and host defense against pathogens. Oncogene 2001, 20, 6473–6481. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.D.; Lamkanfi, M.; Nunez, G. Intracellular NOD-like receptors in host defense and disease. Immunity 2007, 27, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Rothfuchs, A.G.; Bafica, A.; Feng, C.G.; Egen, J.G.; Williams, D.L.; Brown, G.D.; Sher, A. Dectin-1 interaction with Mycobacterium tuberculosis leads to enhanced IL-12p40 production by splenic dendritic cells. J. Immunol. 2007, 179, 3463–3471. [Google Scholar] [CrossRef]
- Arora, G.; Misra, R.; Sajid, A. Model Systems for Pulmonary Infectious Diseases: Paradigms of Anthrax and Tuberculosis. Curr Top. Med. Chem. 2017, 17, 2077–2099. [Google Scholar] [CrossRef]
- Domingo-Gonzalez, R.; Prince, O.; Cooper, A.; Khader, S.A. Cytokines and Chemokines in Mycobacterium tuberculosis Infection. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Rohde, K.; Yates, R.M.; Purdy, G.E.; Russell, D.G. Mycobacterium tuberculosis and the environment within the phagosome. Immunol. Rev. 2007, 219, 37–54. [Google Scholar] [CrossRef]
- Schuller, S.; Neefjes, J.; Ottenhoff, T.; Thole, J.; Young, D. Coronin is involved in uptake of Mycobacterium bovis BCG in human macrophages but not in phagosome maintenance. Cell Microbiol. 2001, 3, 785–793. [Google Scholar] [CrossRef]
- Flynn, J.L.; Chan, J. Immune evasion by Mycobacterium tuberculosis: Living with the enemy. Curr. Opin. Immunol. 2003, 15, 450–455. [Google Scholar] [CrossRef]
- Ma, J.; Yang, B.; Yu, S.; Zhang, Y.; Zhang, X.; Lao, S.; Chen, X.; Li, B.; Wu, C. Tuberculosis antigen-induced expression of IFN-alpha in tuberculosis patients inhibits production of IL-1beta. FASEB J. 2014, 28, 3238–3248. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.; Chao, J.D.; Av-Gay, Y. Mycobacterium tuberculosis-secreted phosphatases: From pathogenesis to targets for TB drug development. Trends Microbiol. 2013, 21, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Sajid, A.; Arora, G.; Singhal, A.; Kalia, V.C.; Singh, Y. Protein Phosphatases of Pathogenic Bacteria: Role in Physiology and Virulence. Annu. Rev. Microbiol. 2015, 69, 527–547. [Google Scholar] [CrossRef] [PubMed]
- Walburger, A.; Koul, A.; Ferrari, G.; Nguyen, L.; Prescianotto-Baschong, C.; Huygen, K.; Klebl, B.; Thompson, C.; Bacher, G.; Pieters, J. Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science 2004, 304, 1800–1804. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.K.; Sassetti, C.M. Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl. Acad. Sci. USA 2008, 105, 4376–4380. [Google Scholar] [CrossRef] [PubMed]
- Vergne, I.; Chua, J.; Deretic, V. Tuberculosis toxin blocking phagosome maturation inhibits a novel Ca2+/calmodulin-PI3K hVPS34 cascade. J. Exp. Med. 2003, 198, 653–659. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.; Van Vliet, S.J.; Koppel, E.A.; Sanchez-Hernandez, M.; Vandenbroucke-Grauls, C.M.; Appelmelk, B.; Van Kooyk, Y. Mycobacteria target DC-SIGN to suppress dendritic cell function. J. Exp. Med. 2003, 197, 7–17. [Google Scholar] [CrossRef]
- Tailleux, L.; Schwartz, O.; Herrmann, J.L.; Pivert, E.; Jackson, M.; Amara, A.; Legres, L.; Dreher, D.; Nicod, L.P.; Gluckman, J.C.; et al. DC-SIGN is the major Mycobacterium tuberculosis receptor on human dendritic cells. J. Exp. Med. 2003, 197, 121–127. [Google Scholar] [CrossRef]
- Jamwal, S.V.; Mehrotra, P.; Singh, A.; Siddiqui, Z.; Basu, A.; Rao, K.V. Mycobacterial escape from macrophage phagosomes to the cytoplasm represents an alternate adaptation mechanism. Sci. Rep. 2016, 6, 23089. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, T.; Liu, Z.; Zhang, G.; Wang, J.; Feng, S.; Liang, J. Inhibition of Autophagy by MiR-30A Induced by Mycobacteria tuberculosis as a Possible Mechanism of Immune Escape in Human Macrophages. Jpn. J. Infect. Dis. 2015, 68, 420–424. [Google Scholar] [CrossRef]
- Wong, D.; Li, W.; Chao, J.D.; Zhou, P.; Narula, G.; Tsui, C.; Ko, M.; Xie, J.; Martinez-Frailes, C.; Av-Gay, Y. Protein tyrosine kinase, PtkA, is required for Mycobacterium tuberculosis growth in macrophages. Sci. Rep. 2018, 8, 155. [Google Scholar] [CrossRef] [PubMed]
- Queval, C.J.; Song, O.R.; Carralot, J.P.; Saliou, J.M.; Bongiovanni, A.; Deloison, G.; Deboosere, N.; Jouny, S.; Iantomasi, R.; Delorme, V.; et al. Mycobacterium tuberculosis Controls Phagosomal Acidification by Targeting CISH-Mediated Signaling. Cell Rep. 2017, 20, 3188–3198. [Google Scholar] [CrossRef] [PubMed]
- MacMicking, J.D.; Taylor, G.A.; McKinney, J.D. Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science 2003, 302, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Sharp, J.D.; Singh, A.K.; Park, S.T.; Lyubetskaya, A.; Peterson, M.W.; Gomes, A.L.; Potluri, L.P.; Raman, S.; Galagan, J.E.; Husson, R.N. Comprehensive Definition of the SigH Regulon of Mycobacterium tuberculosis Reveals Transcriptional Control of Diverse Stress Responses. PLoS ONE 2016, 11, e0152145. [Google Scholar] [CrossRef]
- Dutta, N.K.; Mehra, S.; Martinez, A.N.; Alvarez, X.; Renner, N.A.; Morici, L.A.; Pahar, B.; Maclean, A.G.; Lackner, A.A.; Kaushal, D. The stress-response factor SigH modulates the interaction between Mycobacterium tuberculosis and host phagocytes. PLoS ONE 2012, 7, e28958. [Google Scholar] [CrossRef]
- Sureka, K.; Hossain, T.; Mukherjee, P.; Chatterjee, P.; Datta, P.; Kundu, M.; Basu, J. Novel role of phosphorylation-dependent interaction between FtsZ and FipA in mycobacterial cell division. PLoS ONE 2010, 5, e8590. [Google Scholar] [CrossRef]
- Buchmeier, N.A.; Newton, G.L.; Fahey, R.C. A mycothiol synthase mutant of Mycobacterium tuberculosis has an altered thiol-disulfide content and limited tolerance to stress. J. Bacteriol. 2006, 188, 6245–6252. [Google Scholar] [CrossRef]
- Mukherjee, P.; Sureka, K.; Datta, P.; Hossain, T.; Barik, S.; Das, K.P.; Kundu, M.; Basu, J. Novel role of Wag31 in protection of mycobacteria under oxidative stress. Mol. Microbiol. 2009, 73, 103–119. [Google Scholar] [CrossRef]
- Arora, G.; Sajid, A.; Singhal, A.; Joshi, J.; Virmani, R.; Gupta, M.; Verma, N.; Maji, A.; Misra, R.; Baronian, G.; et al. Identification of Ser/Thr kinase and forkhead associated domains in Mycobacterium ulcerans: Characterization of novel association between protein kinase Q and MupFHA. PLoS Negl. Trop. Dis. 2014, 8, e3315. [Google Scholar] [CrossRef]
- Romero, M.M.; Balboa, L.; Basile, J.I.; Lopez, B.; Ritacco, V.; de la Barrera, S.S.; Sasiain, M.C.; Barrera, L.; Aleman, M. Clinical isolates of Mycobacterium tuberculosis differ in their ability to induce respiratory burst and apoptosis in neutrophils as a possible mechanism of immune escape. Clin. Dev. Immunol. 2012, 2012, 152546. [Google Scholar] [CrossRef]
- Imai, K.; Kurita-Ochiai, T.; Ochiai, K. Mycobacterium bovis bacillus Calmette-Guerin infection promotes SOCS induction and inhibits IFN-gamma-stimulated JAK/STAT signaling in J774 macrophages. FEMS Immunol. Med. Microbiol. 2003, 39, 173–180. [Google Scholar] [CrossRef]
- Ganguly, N.; Giang, P.H.; Gupta, C.; Basu, S.K.; Siddiqui, I.; Salunke, D.M.; Sharma, P. Mycobacterium tuberculosis secretory proteins CFP-10, ESAT-6 and the CFP10:ESAT6 complex inhibit lipopolysaccharide-induced NF-kappaB transactivation by downregulation of reactive oxidative species (ROS) production. Immunol. Cell Biol. 2008, 86, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Colangeli, R.; Haq, A.; Arcus, V.L.; Summers, E.; Magliozzo, R.S.; McBride, A.; Mitra, A.K.; Radjainia, M.; Khajo, A.; Jacobs, W.R., Jr.; et al. The multifunctional histone-like protein Lsr2 protects mycobacteria against reactive oxygen intermediates. Proc. Natl. Acad. Sci. USA 2009, 106, 4414–4418. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.M.; Jeon, B.Y.; Lee, H.M.; Jin, H.S.; Yuk, J.M.; Song, C.H.; Lee, S.H.; Lee, Z.W.; Cho, S.N.; Kim, J.M.; et al. Mycobacterium tuberculosis eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PLoS Pathog. 2010, 6, e1001230. [Google Scholar] [CrossRef] [PubMed]
- Sao Emani, C.; Williams, M.J.; Van Helden, P.D.; Taylor, M.J.C.; Wiid, I.J.; Baker, B. Gamma-glutamylcysteine protects ergothioneine-deficient Mycobacterium tuberculosis mutants against oxidative and nitrosative stress. Biochem. Biophys. Res. Commun. 2018, 495, 174–178. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Framework for Conducting Reviews of Tuberculosis Programmes; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Riendeau, C.J.; Kornfeld, H. THP-1 cell apoptosis in response to Mycobacterial infection. Infect. Immun. 2003, 71, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Gajer, P.; Brotman, R.M.; Bai, G.; Sakamoto, J.; Schutte, U.M.; Zhong, X.; Koenig, S.S.; Fu, L.; Ma, Z.S.; Zhou, X.; et al. Temporal dynamics of the human vaginal microbiota. Sci. Transl. Med. 2012, 4, 132ra52. [Google Scholar] [CrossRef]
- Liu, M.; Li, W.; Xiang, X.; Xie, J. Mycobacterium tuberculosis effectors interfering host apoptosis signaling. Apoptosis 2015, 20, 883–891. [Google Scholar] [CrossRef]
- Flynn, J.L.; Chan, J. What’s good for the host is good for the bug. Trends Microbiol. 2005, 13, 98–102. [Google Scholar] [CrossRef]
- Dieli, F.; Taniguchi, M.; Kronenberg, M.; Sidobre, S.; Ivanyi, J.; Fattorini, L.; Iona, E.; Orefici, G.; De Leo, G.; Russo, D.; et al. An anti-inflammatory role for V alpha 14 NK T cells in Mycobacterium bovis bacillus Calmette-Guerin-infected mice. J. Immunol. 2003, 171, 1961–1968. [Google Scholar] [CrossRef]
- Ulrichs, T.; Kaufmann, S.H. New insights into the function of granulomas in human tuberculosis. J. Pathol. 2006, 208, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.G. Who puts the tubercle in tuberculosis? Nat. Rev. Microbiol. 2007, 5, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Slight, S.R.; Khader, S.A. Chemokines shape the immune responses to tuberculosis. Cytokine Growth Factor Rev. 2013, 24, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Lowe, D.M.; Redford, P.S.; Wilkinson, R.J.; O’Garra, A.; Martineau, A.R. Neutrophils in tuberculosis: Friend or foe? Trends Immunol. 2012, 33, 14–25. [Google Scholar] [CrossRef]
- Srivastava, S.; Ernst, J.D.; Desvignes, L. Beyond macrophages: The diversity of mononuclear cells in tuberculosis. Immunol. Rev. 2014, 262, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Dorhoi, A.; Kaufmann, S.H. Versatile myeloid cell subsets contribute to tuberculosis-associated inflammation. Eur J. Immunol. 2015, 45, 2191–2202. [Google Scholar] [CrossRef] [PubMed]
- Zielske, S.P.; Stevenson, M. Importin 7 may be dispensable for human immunodeficiency virus type 1 and simian immunodeficiency virus infection of primary macrophages. J. Virol. 2005, 79, 11541–11546. [Google Scholar] [CrossRef]
- Schaller, T.; Ocwieja, K.E.; Rasaiyaah, J.; Price, A.J.; Brady, T.L.; Roth, S.L.; Hue, S.; Fletcher, A.J.; Lee, K.; KewalRamani, V.N.; et al. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 2011, 7, e1002439. [Google Scholar] [CrossRef]
- Torrado, E.; Robinson, R.T.; Cooper, A.M. Cellular response to mycobacteria: Balancing protection and pathology. Trends Immunol. 2011, 32, 66–72. [Google Scholar] [CrossRef]
- Price, A.J.; Jacques, D.A.; McEwan, W.A.; Fletcher, A.J.; Essig, S.; Chin, J.W.; Halambage, U.D.; Aiken, C.; James, L.C. Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog. 2014, 10, e1004459. [Google Scholar] [CrossRef]
- Swaminathan, S.; Nandini, K.S.; Hanna, L.E.; Somu, N.; Narayanan, P.R.; Barnes, P.F. T-lymphocyte subpopulations in tuberculosis. Indian Pediatr. 2000, 37, 489–495. [Google Scholar] [PubMed]
- Apte, R.N.; Voronov, E. Is interleukin-1 a good or bad ‘guy’ in tumor immunobiology and immunotherapy? Immunol. Rev. 2008, 222, 222–241. [Google Scholar] [CrossRef] [PubMed]
- Aliprantis, A.O.; Yang, R.B.; Weiss, D.S.; Godowski, P.; Zychlinsky, A. The apoptotic signaling pathway activated by Toll-like receptor-2. EMBO J. 2000, 19, 3325–3336. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V. Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol. Rev. 2011, 240, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, I.; Amano, A.; Mizushima, N.; Yamamoto, A.; Yamaguchi, H.; Kamimoto, T.; Nara, A.; Funao, J.; Nakata, M.; Tsuda, K.; et al. Autophagy defends cells against invading group A Streptococcus. Science 2004, 306, 1037–1040. [Google Scholar] [CrossRef]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef]
- Deretic, V. Multiple regulatory and effector roles of autophagy in immunity. Curr. Opin. Immunol. 2009, 21, 53–62. [Google Scholar] [CrossRef]
- Loeuillet, C.; Martinon, F.; Perez, C.; Munoz, M.; Thome, M.; Meylan, P.R. Mycobacterium tuberculosis subverts innate immunity to evade specific effectors. J. Immunol. 2006, 177, 6245–6255. [Google Scholar] [CrossRef]
- Oddo, M.; Renno, T.; Attinger, A.; Bakker, T.; MacDonald, H.R.; Meylan, P.R. Fas ligand-induced apoptosis of infected human macrophages reduces the viability of intracellular Mycobacterium tuberculosis. J. Immunol. 1998, 160, 5448–5454. [Google Scholar]
- Vergne, I.; Singh, S.; Roberts, E.; Kyei, G.; Master, S.; Harris, J.; de Haro, S.; Naylor, J.; Davis, A.; Delgado, M.; et al. Autophagy in immune defense against Mycobacterium tuberculosis. Autophagy 2006, 2, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Altare, F.; Jouanguy, E.; Lamhamedi, S.; Doffinger, R.; Fischer, A.; Casanova, J.L. Mendelian susceptibility to mycobacterial infection in man. Curr. Opin. Immunol. 1998, 10, 413–417. [Google Scholar] [CrossRef]
- Fieschi, C.; Dupuis, S.; Catherinot, E.; Feinberg, J.; Bustamante, J.; Breiman, A.; Altare, F.; Baretto, R.; Le Deist, F.; Kayal, S.; et al. Low penetrance, broad resistance, and favorable outcome of interleukin 12 receptor beta1 deficiency: Medical and immunological implications. J. Exp. Med. 2003, 197, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Cruz, A.; Fraga, A.G.; Fountain, J.J.; Rangel-Moreno, J.; Torrado, E.; Saraiva, M.; Pereira, D.R.; Randall, T.D.; Pedrosa, J.; Cooper, A.M.; et al. Pathological role of interleukin 17 in mice subjected to repeated BCG vaccination after infection with Mycobacterium tuberculosis. J. Exp. Med. 2010, 207, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Behar, S.M.; Martin, C.J.; Booty, M.G.; Nishimura, T.; Zhao, X.; Gan, H.X.; Divangahi, M.; Remold, H.G. Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal Immunol. 2011, 4, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Gehring, A.J.; Rojas, R.E.; Canaday, D.H.; Lakey, D.L.; Harding, C.V.; Boom, W.H. The Mycobacterium tuberculosis 19-kilodalton lipoprotein inhibits gamma interferon-regulated HLA-DR and Fc gamma R1 on human macrophages through Toll-like receptor 2. Infect. Immun 2003, 71, 4487–4497. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.; Sly, L.M.; Luu, Y.; Young, D.; Cooper, H.; Reiner, N.E. The 19-kDa Mycobacterium tuberculosis protein induces macrophage apoptosis through Toll-like receptor-2. J. Immunol. 2003, 170, 2409–2416. [Google Scholar] [CrossRef]
- Diaz-Silvestre, H.; Espinosa-Cueto, P.; Sanchez-Gonzalez, A.; Esparza-Ceron, M.A.; Pereira-Suarez, A.L.; Bernal-Fernandez, G.; Espitia, C.; Mancilla, R. The 19-kDa antigen of Mycobacterium tuberculosis is a major adhesin that binds the mannose receptor of THP-1 monocytic cells and promotes phagocytosis of mycobacteria. Microb. Pathog. 2005, 39, 97–107. [Google Scholar] [CrossRef]
- Danelishvili, L.; Yamazaki, Y.; Selker, J.; Bermudez, L.E. Secreted Mycobacterium tuberculosis Rv3654c and Rv3655c proteins participate in the suppression of macrophage apoptosis. PLoS ONE 2010, 5, e10474. [Google Scholar] [CrossRef]
- Miller, J.L.; Velmurugan, K.; Cowan, M.J.; Briken, V. The type I NADH dehydrogenase of Mycobacterium tuberculosis counters phagosomal NOX2 activity to inhibit TNF-alpha-mediated host cell apoptosis. PLoS Pathog. 2010, 6, e1000864. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Olivier, M.; Garcia, L.F. Activation of JAK2/STAT1-alpha-dependent signaling events during Mycobacterium tuberculosis-induced macrophage apoptosis. Cell Immunol. 2002, 217, 58–66. [Google Scholar] [CrossRef]
- Clay, H.; Volkman, H.E.; Ramakrishnan, L. Tumor necrosis factor signaling mediates resistance to mycobacteria by inhibiting bacterial growth and macrophage death. Immunity 2008, 29, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Feng, Y.; Chatterjee, S.; Tuske, S.; Ho, M.X.; Arnold, E.; Ebright, R.H. Structural basis of transcription initiation. Science 2012, 338, 1076–1080. [Google Scholar] [CrossRef]
- Axelrod, S.; Oschkinat, H.; Enders, J.; Schlegel, B.; Brinkmann, V.; Kaufmann, S.H.; Haas, A.; Schaible, U.E. Delay of phagosome maturation by a mycobacterial lipid is reversed by nitric oxide. Cell Microbiol. 2008, 10, 1530–1545. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.; Espinosa, P.; Garcia, T.; Mancilla, R. The 19 kDa Mycobacterium tuberculosis lipoprotein (LpqH) induces macrophage apoptosis through extrinsic and intrinsic pathways: A role for the mitochondrial apoptosis-inducing factor. Clin. Dev. Immunol. 2012, 2012, 950503. [Google Scholar] [CrossRef] [PubMed]
- Pante, N.; Kann, M. Nuclear pore complex is able to transport macromolecules with diameters of about 39 nm. Mol. Biol. Cell 2002, 13, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Allam, A.M.; Hassan, M.M.; Elzainy, T.A. Formation and cleavage of 2-keto-3-deoxygluconate by 2-keto-3-deoxygluconate aldolase of Aspergillus niger. J. Bacteriol. 1975, 124, 1128–1131. [Google Scholar]
- Ball, J.R.; Ullman, K.S. Versatility at the nuclear pore complex: Lessons learned from the nucleoporin Nup153. Chromosoma 2005, 114, 319–330. [Google Scholar] [CrossRef]
- Xu, X.; Liu, T.; Zhang, A.; Huo, X.; Luo, Q.; Chen, Z.; Yu, L.; Li, Q.; Liu, L.; Lun, Z.; et al. Reactive oxygen species-triggered trophoblast apoptosis is initiated by endoplasmic reticulum stress via activation of caspase-12, CHOP, and the JNK pathway in Toxoplasma gondii infection in mice. Infect. Immun. 2015, 83, 1735. [Google Scholar] [CrossRef]
- Singh, V.; Donini, S.; Pacitto, A.; Sala, C.; Hartkoorn, R.C.; Dhar, N.; Keri, G.; Ascher, D.B.; Mondesert, G.; Vocat, A.; et al. The Inosine Monophosphate Dehydrogenase, GuaB2, Is a Vulnerable New Bactericidal Drug Target for Tuberculosis. ACS Infect. Dis. 2017, 3, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Zuber, P.; Healy, J.; Carter, H.L., 3rd; Cutting, S.; Moran, C.P., Jr.; Losick, R. Mutation changing the specificity of an RNA polymerase sigma factor. J. Mol. Biol. 1989, 206, 605–614. [Google Scholar] [CrossRef]
- Iwabuchi, K. [Lactosylceramide-enriched Lipid Raft-mediated Infection Immunity]. Med. Mycol. J. 2018, 59, J51–J61. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M. Molecular mechanism of the nuclear protein import cycle. Nat. Rev. Mol. Cell Biol. 2007, 8, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.H.; Shin, D.M.; Kang, G.; Kim, K.H.; Park, J.B.; Hur, G.M.; Lee, H.M.; Lim, Y.J.; Park, J.K.; Jo, E.K.; et al. Endoplasmic reticulum stress response is involved in Mycobacterium tuberculosis protein ESAT-6-mediated apoptosis. FEBS Lett. 2010, 584, 2445–2454. [Google Scholar] [CrossRef] [PubMed]
- McMillan, A.; Rulisa, S.; Sumarah, M.; Macklaim, J.M.; Renaud, J.; Bisanz, J.E.; Gloor, G.B.; Reid, G. A multi-platform metabolomics approach identifies highly specific biomarkers of bacterial diversity in the vagina of pregnant and non-pregnant women. Sci. Rep. 2015, 5, 14174. [Google Scholar] [CrossRef]
- Macklaim, J.M.; Clemente, J.C.; Knight, R.; Gloor, G.B.; Reid, G. Changes in vaginal microbiota following antimicrobial and probiotic therapy. Microb. Ecol. Health Dis. 2015, 26, 27799. [Google Scholar] [CrossRef]
- Macklaim, J.M.; Fernandes, A.D.; Di Bella, J.M.; Hammond, J.A.; Reid, G.; Gloor, G.B. Comparative meta-RNA-seq of the vaginal microbiota and differential expression by Lactobacillus iners in health and dysbiosis. Microbiome 2013, 1, 12. [Google Scholar] [CrossRef]
- Kusner, D.J.; Adams, J. ATP-induced killing of virulent Mycobacterium tuberculosis within human macrophages requires phospholipase D. J. Immunol. 2000, 164, 379–388. [Google Scholar] [CrossRef]
- Bouchonnet, F.; Boechat, N.; Bonay, M.; Hance, A.J. Alpha/beta interferon impairs the ability of human macrophages to control growth of Mycobacterium bovis BCG. Infect. Immun. 2002, 70, 3020–3025. [Google Scholar] [CrossRef]
- Seimon, T.A.; Kim, M.J.; Blumenthal, A.; Koo, J.; Ehrt, S.; Wainwright, H.; Bekker, L.G.; Kaplan, G.; Nathan, C.; Tabas, I.; et al. Induction of ER stress in macrophages of tuberculosis granulomas. PLoS ONE 2010, 5, e12772. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, K.A.; Serbina, N.V.; Flynn, J.L. Fate of Mycobacterium tuberculosis within murine dendritic cells. Infect. Immun. 2001, 69, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Dove, S.L.; Darst, S.A.; Hochschild, A. Region 4 of sigma as a target for transcription regulation. Mol. Microbiol. 2003, 48, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Niedergang, F.; Didierlaurent, A.; Kraehenbuhl, J.P.; Sirard, J.C. Dendritic cells: The host Achille’s heel for mucosal pathogens? Trends Microbiol. 2004, 12, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Reid, G. Cervicovaginal Microbiomes-Threats and Possibilities. Trends Endocrinol. Metab. 2016, 27, 446–454. [Google Scholar] [CrossRef]
- Tapping, R.I.; Tobias, P.S. Mycobacterial lipoarabinomannan mediates physical interactions between TLR1 and TLR2 to induce signaling. J. Endotoxin Res. 2003, 9, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, B.; Ramakrishnan, U.M.; Raghunand, T.R. The Mycobacterium tuberculosis protein pair PE9 (Rv1088)-PE10 (Rv1089) forms heterodimers and induces macrophage apoptosis through Toll-like receptor 4. Cell Microbiol. 2015, 17, 1653–1669. [Google Scholar] [CrossRef] [PubMed]
- Francez-Charlot, A.; Frunzke, J.; Reichen, C.; Ebneter, J.Z.; Gourion, B.; Vorholt, J.A. Sigma factor mimicry involved in regulation of general stress response. Proc. Natl. Acad. Sci. USA 2009, 106, 3467–3472. [Google Scholar] [CrossRef]
- Bafica, A.; Scanga, C.A.; Feng, C.G.; Leifer, C.; Cheever, A.; Sher, A. TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J. Exp. Med. 2005, 202, 1715–1724. [Google Scholar] [CrossRef]
- Shiratsuchi, A.; Watanabe, I.; Takeuchi, O.; Akira, S.; Nakanishi, Y. Inhibitory effect of Toll-like receptor 4 on fusion between phagosomes and endosomes/lysosomes in macrophages. J. Immunol. 2004, 172, 2039–2047. [Google Scholar] [CrossRef]
- Guenin-Mace, L.; Simeone, R.; Demangel, C. Lipids of pathogenic Mycobacteria: Contributions to virulence and host immune suppression. Transbound. Emerg. Dis. 2009, 56, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.D. Antigenic Variation and Immune Escape in the MTBC. Adv. Exp. Med. Biol 2017, 1019, 171–190. [Google Scholar] [PubMed]
- Mai, J.; Wang, H.; Yang, X.F. Th 17 cells interplay with Foxp3+ Tregs in regulation of inflammation and autoimmunity. Front. Biosci. (Landmark Ed.) 2010, 15, 986–1006. [Google Scholar] [CrossRef]
- Caccamo, N.; Pietra, G.; Sullivan, L.C.; Brooks, A.G.; Prezzemolo, T.; La Manna, M.P.; Di Liberto, D.; Joosten, S.A.; van Meijgaarden, K.E.; Di Carlo, P.; et al. Human CD8 T lymphocytes recognize Mycobacterium tuberculosis antigens presented by HLA-E during active tuberculosis and express type 2 cytokines. Eur. J. Immunol. 2015, 45, 1069–1081. [Google Scholar] [CrossRef]
- Li, Y.; Sun, W. Effects of Th17/Treg cell imbalance on HIV replication in patients with AIDS complicated with tuberculosis. Exp. Ther. Med. 2018, 15, 2879–2883. [Google Scholar] [CrossRef] [PubMed]
- Korb, V.C.; Phulukdaree, A.; Lalloo, U.G.; Chuturgoon, A.A.; Moodley, D. TB/HIV pleurisy reduces Th17 lymphocyte proportion independent of the cytokine microenvironment. Tuberculosis (Edinb) 2016, 99, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Geijtenbeek, T.B.; van Kooyk, Y. Pathogens target DC-SIGN to influence their fate DC-SIGN functions as a pathogen receptor with broad specificity. APMIS 2003, 111, 698–714. [Google Scholar] [CrossRef]
- Gagliardi, M.C.; Lemassu, A.; Teloni, R.; Mariotti, S.; Sargentini, V.; Pardini, M.; Daffe, M.; Nisini, R. Cell wall-associated alpha-glucan is instrumental for Mycobacterium tuberculosis to block CD1 molecule expression and disable the function of dendritic cell derived from infected monocyte. Cell Microbiol. 2007, 9, 2081–2092. [Google Scholar] [CrossRef]
- Haugen, S.P.; Berkmen, M.B.; Ross, W.; Gaal, T.; Ward, C.; Gourse, R.L. rRNA promoter regulation by nonoptimal binding of sigma region 1.2: An additional recognition element for RNA polymerase. Cell 2006, 125, 1069–1082. [Google Scholar] [CrossRef]
- Kusner, D.J.; Barton, J.A. ATP stimulates human macrophages to kill intracellular virulent Mycobacterium tuberculosis via calcium-dependent phagosome-lysosome fusion. J. Immunol. 2001, 167, 3308–3315. [Google Scholar] [CrossRef]
- Almeida, P.E.; Carneiro, A.B.; Silva, A.R.; Bozza, P.T. PPARgamma Expression and Function in Mycobacterial Infection: Roles in Lipid Metabolism, Immunity, and Bacterial Killing. PPAR Res. 2012, 2012, 383829. [Google Scholar] [CrossRef] [PubMed]
- Sha, S.; Shi, X.; Deng, G.; Chen, L.; Xin, Y.; Ma, Y. Mycobacterium tuberculosis Rv1987 induces Th2 immune responses and enhances Mycobacterium smegmatis survival in mice. Microbiol. Res. 2017, 197, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Xie, J. Ins and outs of Mycobacterium tuberculosis PPE family in pathogenesis and implications for novel measures against tuberculosis. J. Cell Biochem. 2012, 113, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Rath, P.; Huang, C.; Wang, T.; Wang, T.; Li, H.; Prados-Rosales, R.; Elemento, O.; Casadevall, A.; Nathan, C.F. Genetic regulation of vesiculogenesis and immunomodulation in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2013, 110, E4790–E4797. [Google Scholar] [CrossRef] [PubMed]
- Dorhoi, A.; Kaufmann, S.H. Pathology and immune reactivity: Understanding multidimensionality in pulmonary tuberculosis. Semin. Immunopathol. 2016, 38, 153–166. [Google Scholar] [CrossRef]
- Silva Miranda, M.; Breiman, A.; Allain, S.; Deknuydt, F.; Altare, F. The tuberculous granuloma: An unsuccessful host defence mechanism providing a safety shelter for the bacteria? Clin. Dev. Immunol. 2012, 2012, 139127. [Google Scholar] [CrossRef] [PubMed]
- Guirado, E.; Schlesinger, L.S. Modeling the Mycobacterium tuberculosis Granuloma—the Critical Battlefield in Host Immunity and Disease. Front. Immunol. 2013, 4, 98. [Google Scholar] [CrossRef]
- Barry, C.E., 3rd; Boshoff, H.I.; Dartois, V.; Dick, T.; Ehrt, S.; Flynn, J.; Schnappinger, D.; Wilkinson, R.J.; Young, D. The spectrum of latent tuberculosis: Rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 2009, 7, 845–855. [Google Scholar] [CrossRef]
- Flynn, J.L.; Chan, J.; Lin, P.L. Macrophages and control of granulomatous inflammation in tuberculosis. Mucosal Immunol. 2011, 4, 271–278. [Google Scholar] [CrossRef]
- Huynh, K.K.; Joshi, S.A.; Brown, E.J. A delicate dance: Host response to mycobacteria. Curr. Opin. Immunol. 2011, 23, 464–472. [Google Scholar] [CrossRef]
- Yoshida, A.; Inagawa, H.; Kohchi, C.; Nishizawa, T.; Soma, G. The role of toll-like receptor 2 in survival strategies of Mycobacterium tuberculosis in macrophage phagosomes. Anticancer Res. 2009, 29, 907–910. [Google Scholar] [PubMed]
- Ehlers, S.; Schaible, U.E. The granuloma in tuberculosis: Dynamics of a host-pathogen collusion. Front. Immunol. 2012, 3, 411. [Google Scholar] [CrossRef] [PubMed]
- Egen, J.G.; Rothfuchs, A.G.; Feng, C.G.; Winter, N.; Sher, A.; Germain, R.N. Macrophage and T cell dynamics during the development and disintegration of mycobacterial granulomas. Immunity 2008, 28, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Pieters, J. Mycobacterium tuberculosis and the macrophage: Maintaining a balance. Cell Host Microbe 2008, 3, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, S.; Sargentini, V.; Pardini, M.; Giannoni, F.; De Spirito, M.; Gagliardi, M.C.; Greco, E.; Teloni, R.; Fraziano, M.; Nisini, R. Mycobacterium tuberculosis may escape helper T cell recognition by infecting human fibroblasts. Hum. Immunol. 2013, 74, 722–729. [Google Scholar] [CrossRef]
- Saini, N.K.; Baena, A.; Ng, T.W.; Venkataswamy, M.M.; Kennedy, S.C.; Kunnath-Velayudhan, S.; Carreno, L.J.; Xu, J.; Chan, J.; Larsen, M.H.; et al. Suppression of autophagy and antigen presentation by Mycobacterium tuberculosis PE_PGRS47. Nat. Microbiol. 2016, 1, 16133. [Google Scholar] [CrossRef]
- Rhoades, E.R.; Frank, A.A.; Orme, I.M. Progression of chronic pulmonary tuberculosis in mice aerogenically infected with virulent Mycobacterium tuberculosis. Tuber. Lung Dis. 1997, 78, 57–66. [Google Scholar] [CrossRef]
- Manabe, Y.C.; Kesavan, A.K.; Lopez-Molina, J.; Hatem, C.L.; Brooks, M.; Fujiwara, R.; Hochstein, K.; Pitt, M.L.; Tufariello, J.; Chan, J.; et al. The aerosol rabbit model of TB latency, reactivation and immune reconstitution inflammatory syndrome. Tuberculosis (Edinb) 2008, 88, 187–196. [Google Scholar] [CrossRef]
- Kesavan, A.K.; Brooks, M.; Tufariello, J.; Chan, J.; Manabe, Y.C. Tuberculosis genes expressed during persistence and reactivation in the resistant rabbit model. Tuberculosis (Edinb) 2009, 89, 17–21. [Google Scholar] [CrossRef]
- Capuano, S.V., 3rd; Croix, D.A.; Pawar, S.; Zinovik, A.; Myers, A.; Lin, P.L.; Bissel, S.; Fuhrman, C.; Klein, E.; Flynn, J.L. Experimental Mycobacterium tuberculosis infection of cynomolgus macaques closely resembles the various manifestations of human M. tuberculosis infection. Infect. Immun. 2003, 71, 5831–5844. [Google Scholar] [CrossRef]
- Gandotra, S.; Schnappinger, D.; Monteleone, M.; Hillen, W.; Ehrt, S. In vivo gene silencing identifies the Mycobacterium tuberculosis proteasome as essential for the bacteria to persist in mice. Nat. Med. 2007, 13, 1515–1520. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhai, W.; Wu, F.; Zhang, Y.; Fu, Y.; Liu, Z. The Immune Escape Mechanisms of Mycobacterium Tuberculosis. Int. J. Mol. Sci. 2019, 20, 340. https://doi.org/10.3390/ijms20020340
Zhai W, Wu F, Zhang Y, Fu Y, Liu Z. The Immune Escape Mechanisms of Mycobacterium Tuberculosis. International Journal of Molecular Sciences. 2019; 20(2):340. https://doi.org/10.3390/ijms20020340
Chicago/Turabian StyleZhai, Weijie, Fengjuan Wu, Yiyuan Zhang, Yurong Fu, and Zhijun Liu. 2019. "The Immune Escape Mechanisms of Mycobacterium Tuberculosis" International Journal of Molecular Sciences 20, no. 2: 340. https://doi.org/10.3390/ijms20020340
APA StyleZhai, W., Wu, F., Zhang, Y., Fu, Y., & Liu, Z. (2019). The Immune Escape Mechanisms of Mycobacterium Tuberculosis. International Journal of Molecular Sciences, 20(2), 340. https://doi.org/10.3390/ijms20020340