Intestinal Microbiota Protects against MCD Diet-Induced Steatohepatitis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
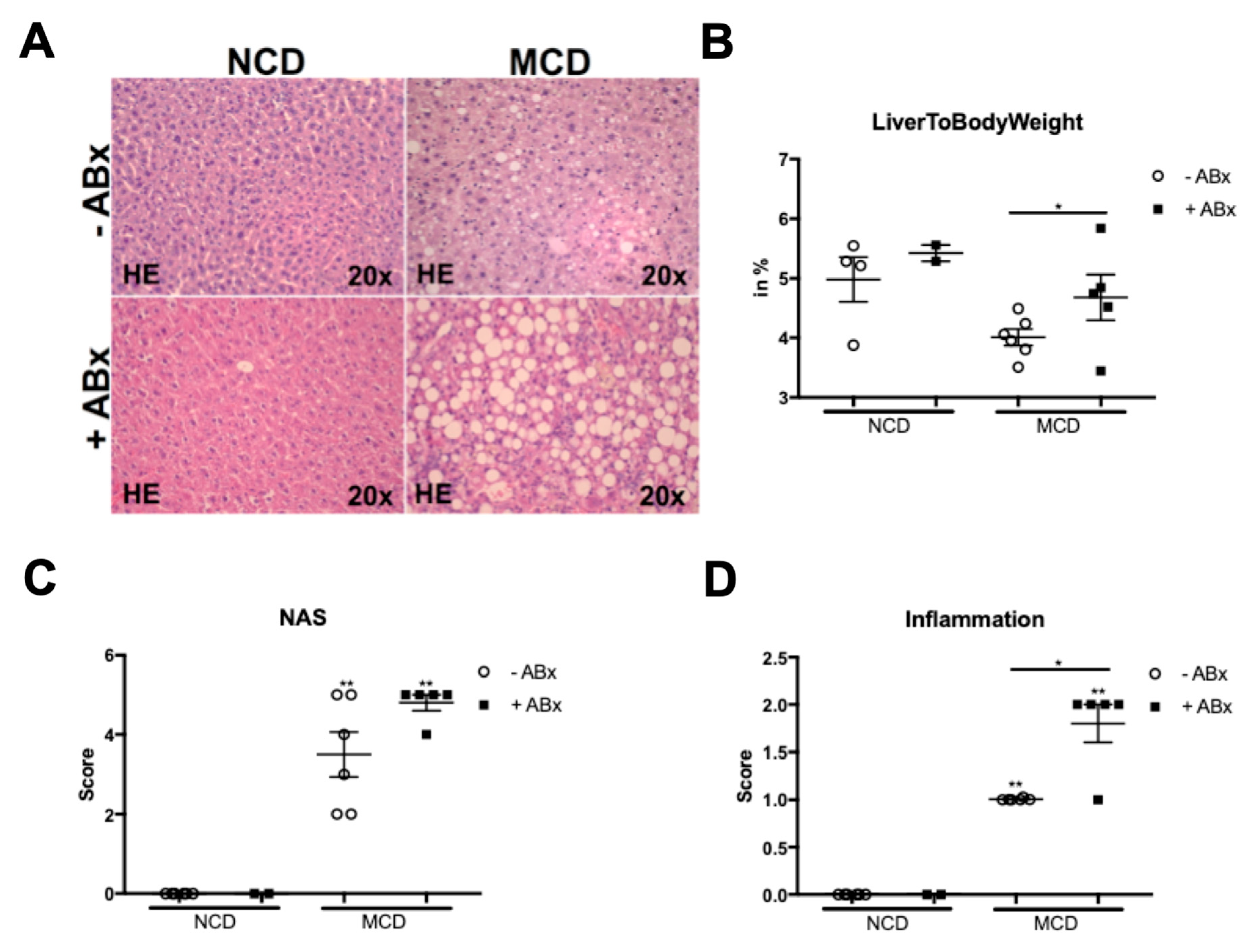
2.1. Microbiota Depletion Augments Steatohepatitis Development in the Murine MCD Model
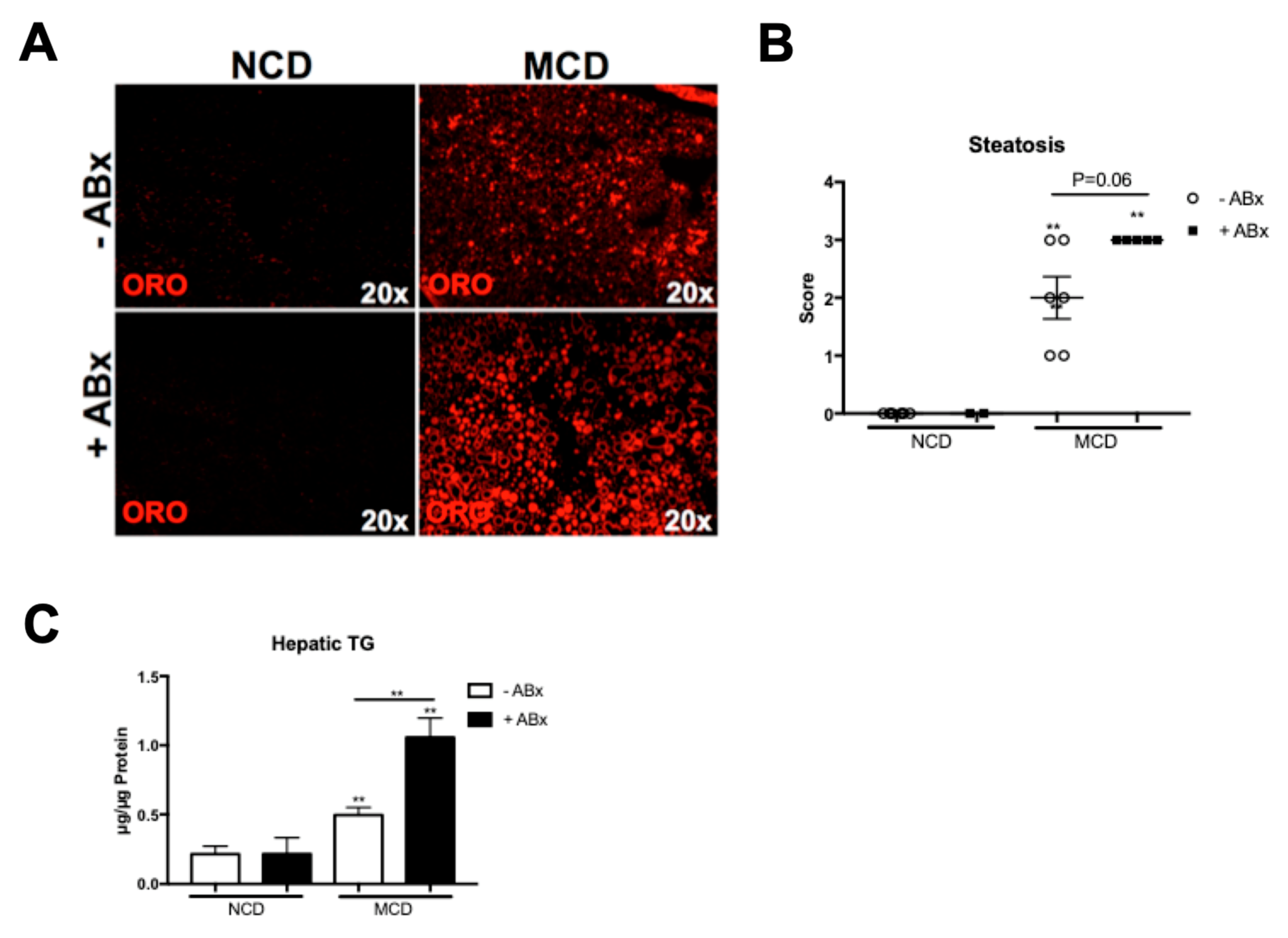
2.2. Antibiotic Treatment Increases Hepatic Fat Accumulation in the MCD-Fed Mice, But Is Not Associated with a Metabolic Phenotype Characteristic of the Human NASH
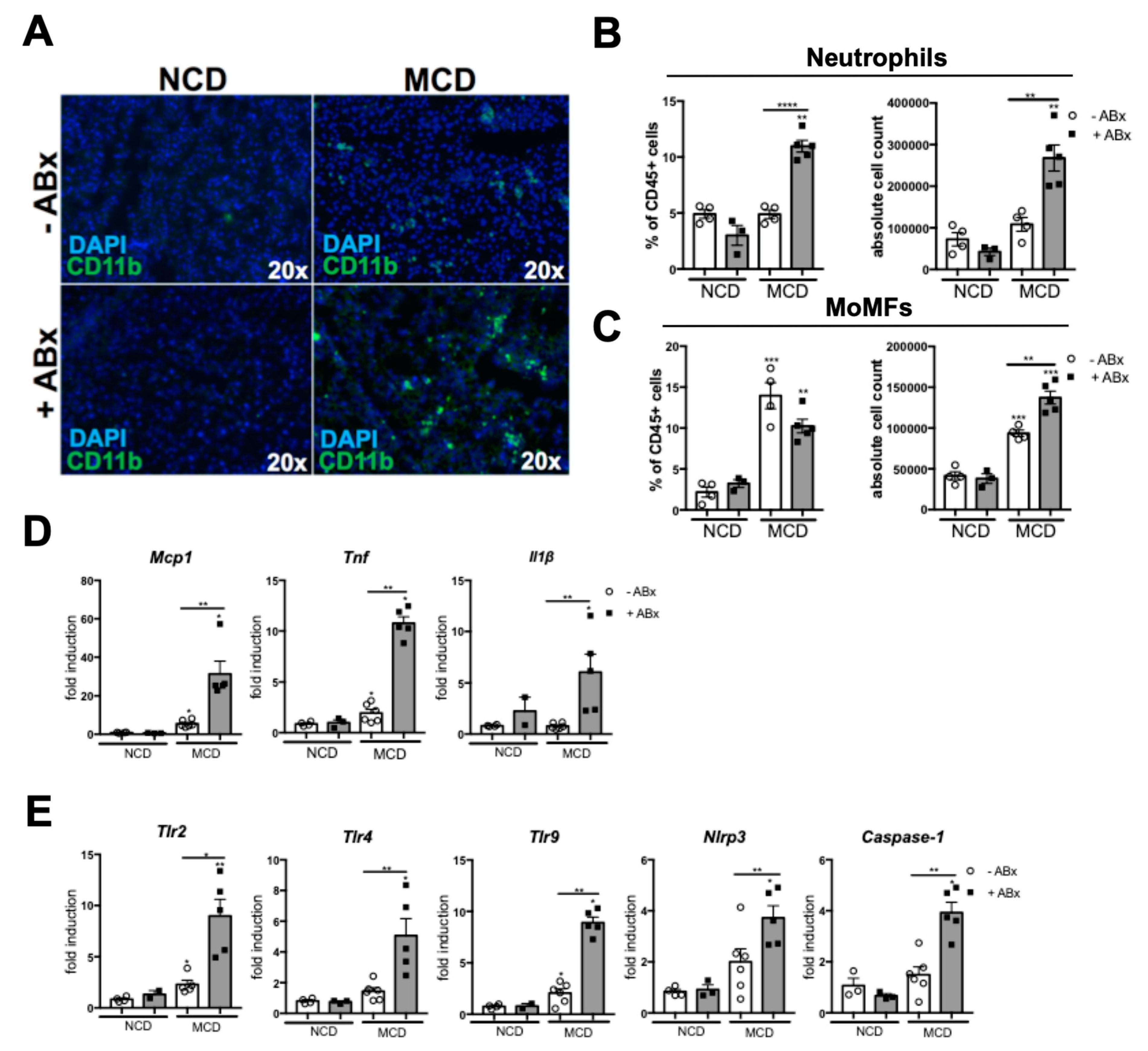
2.3. Microbiota Depletion Augments the Inflammatory Response During the MCD-Induced Steatohepatitis
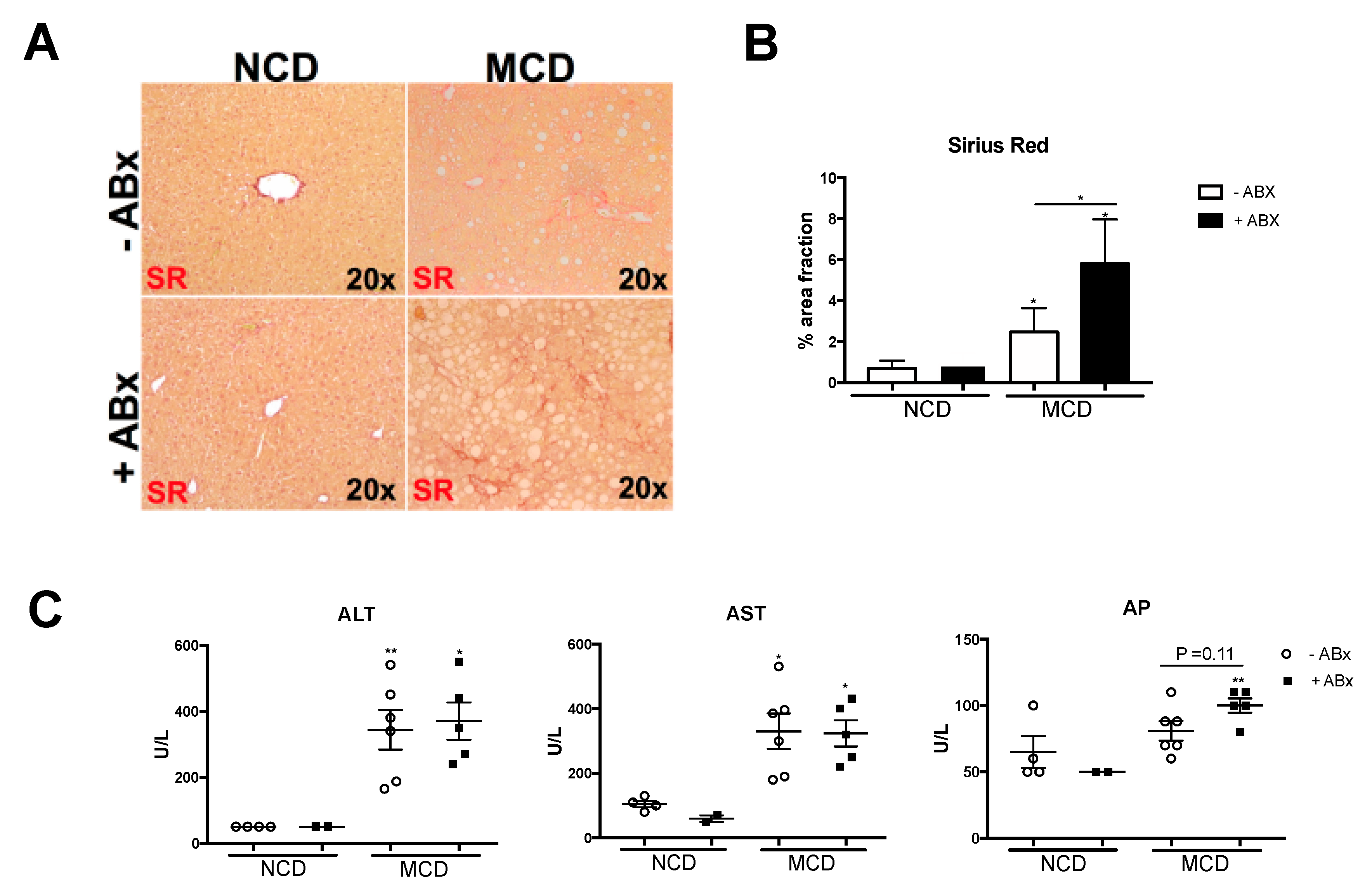
2.4. Intestinal Microbiota Protects against Excessive Liver Fibrosis
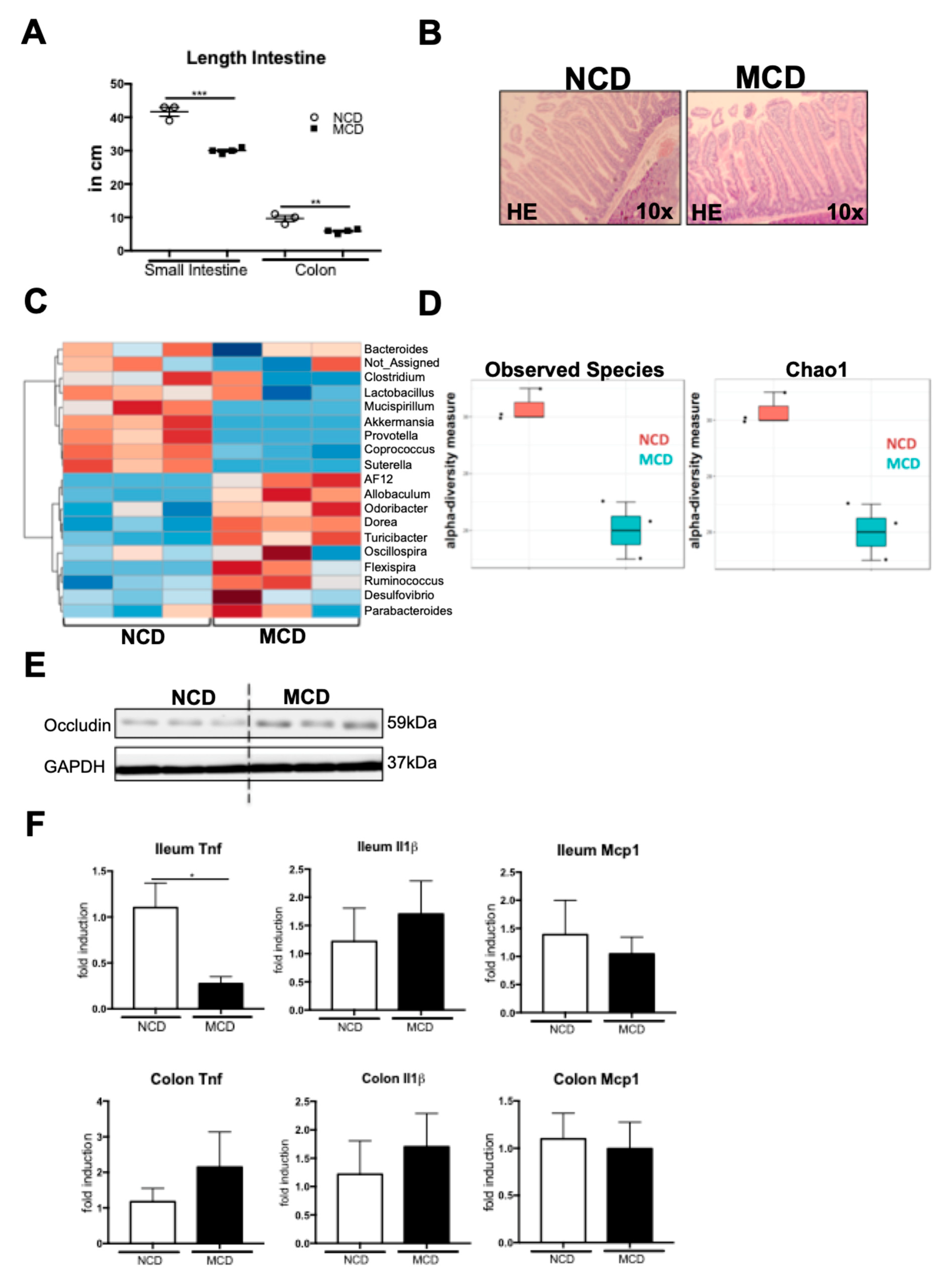
2.5. MCD Diet Impacts the Intestinal Homeostasis and Microbiota Composition
3. Discussion
4. Materials and Methods
4.1. Mice Experiments
4.2. Administration of the Broad-Spectrum Antibiotics
4.3. Immunofluorescence Analysis
4.4. Microbiota—16S rRNA V1-V3 Amplicon Library Preparation
4.5. Microbiota—Data Analysis
4.6. Histology
4.7. Flow Cytometry Analysis of the Intrahepatic and Intestinal Leukocytes
4.8. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Satapathy, S.K.; Sanyal, A.J. Epidemiology and Natural History of Nonalcoholic Fatty Liver Disease. Semin. Liver Dis. 2015, 35, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; Broderick, L.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 627–636. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Lotersztajn, S. Pathophysiology of NASH: Perspectives for a Targeted Treatment. Curr. Pharm. Des. 2013, 19, 5250–5269. [Google Scholar] [CrossRef] [PubMed]
- Dyson, J.K.; Anstee, Q.M.; McPherson, S. Non-alcoholic fatty liver disease: A practical approach to diagnosis and staging. Frontline Gastroenterol. 2014, 5, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Rensen, S.S.; Bieghs, V.; Xanthoulea, S.; Arfianti, E.; Bakker, J.A.; Shiri-Sverdlov, R.; Hofker, M.H.; Greve, J.W.; Buurman, W.A. Neutrophil-Derived Myeloperoxidase Aggravates Non-Alcoholic Steatohepatitis in Low-Density Lipoprotein Receptor-Deficient Mice. PLoS ONE 2012, 7, e52411. [Google Scholar] [CrossRef]
- Zhou, Z.; Xu, M.J.; Cai, Y.; Wang, W.; Jiang, J.X.; Varga, Z.V.; Feng, D.; Pacher, P.; Kunos, G.; Torok, N.J.; et al. Neutrophil–Hepatic Stellate Cell Interactions Promote Fibrosis in Experimental Steatohepatitis. CMGH 2018, 5, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Puengel, T.; Govaere, O.; Abdallah, A.T.; Mossanen, J.C.; Kohlhepp, M.; Liepelt, A.; Lefebvre, E.; Luedde, T.; Hellerbrand, C.; et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018, 67, 1270–1283. [Google Scholar] [CrossRef]
- Friedman, S.L.; Ratziu, V.; Harrison, S.A.; Abdelmalek, M.F.; Aithal, G.P.; Caballeria, J.; Francque, S.; Farrell, G.; Kowdley, K.V.; Craxi, A.; et al. A Randomized, Placebo-Controlled Trial of Cenicriviroc for Treatment of Nonalcoholic Steatohepatitis with Fibrosis. Hepatology 2017. [Google Scholar] [CrossRef]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Schnabl, B. Linking intestinal homeostasis and liver disease. Curr. Opin. Gastroenterol. 2013, 29, 264–270. [Google Scholar] [CrossRef]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Baothman, O.A.; Zamzami, M.A.; Taher, I.; Abubaker, J.; Abu-Farha, M. The role of Gut Microbiota in the development of obesity and Diabetes. Lipids Health Dis. 2016, 15, 108. [Google Scholar] [CrossRef] [PubMed]
- Wigg, A.J.; Roberts-Thomson, I.C.; Dymock, R.B.; McCarthy, P.J.; Grose, R.H.; Cummins, A.G. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut 2001, 48, 206–211. [Google Scholar] [CrossRef]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef]
- Volynets, V.; Küper, M.A.; Strahl, S.; Maier, I.B.; Spruss, A.; Wagnerberger, S.; Königsrainer, A.; Bischoff, S.C.; Bergheim, I. Nutrition, intestinal permeability, and blood ethanol levels are altered in patients with nonalcoholic fatty liver disease (NAFLD). Dig. Dis. Sci. 2012, 57, 1932–1941. [Google Scholar] [CrossRef]
- Schneider, K.M.; Bieghs, V.; Heymann, F.; Hu, W.; Dreymueller, D.; Liao, L.; Frissen, M.; Ludwig, A.; Gassler, N.; Pabst, O.; et al. CX3CR1 is a gatekeeper for intestinal barrier integrity in mice: Limiting steatohepatitis by maintaining intestinal homeostasis. Hepatology 2015, 62, 1405–1416. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Elias, M.S.; Smolak, R.R.; Fu, T.; Borensztajn, J.; Green, R.M. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J. Lipid Res. 2008, 49, 1068–1076. [Google Scholar] [CrossRef]
- Vance, D.E. Role of phosphatidylcholine biosynthesis in the regulation of lipoprotein homeostasis. Curr. Opin. Lipidol. 2008, 19, 229–234. [Google Scholar] [CrossRef]
- Caballero, F.; Fernández, A.; Matías, N.; Martínez, L.; Fucho, R.; Elena, M.; Caballeria, J.; Morales, A.; Fernández-Checa, J.C.; García-Ruiz, C. Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: Impact on mitochondrial S-adenosyl-L-methionine and glutathione. J. Biol. Chem. 2010, 285, 18528–18536. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Velayudham, A.; Romics, L.; Mandrekar, P. Modulation of non-alcoholic steatohepatitis by pattern recognition receptors in mice: The role of toll-like receptors 2 and 4. Alcohol. Clin. Exp. Res. 2005, 29, 140S–145S. [Google Scholar] [CrossRef] [PubMed]
- Csak, T.; Velayudham, A.; Hritz, I.; Petrasek, J.; Levin, I.; Lippai, D.; Catalano, D.; Mandrekar, P.; Dolganiuc, A.; Kurt-Jones, E.; et al. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. AJP Gastrointest. Liver Physiol. 2011, 300, G433–G441. [Google Scholar] [CrossRef] [PubMed]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef]
- Kumar, R.; Mohan, S. Non-alcoholic Fatty Liver Disease in Lean Subjects: Characteristics and Implications. J. Clin. Transl. Hepatol. 2017, 5, 1–8. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Arrese, M.; Cabrera, D.; Kalergis, A.M.; Feldstein, A.E. Innate Immunity and Inflammation in NAFLD/NASH. Dig. Dis. Sci. 2016, 61, 1294–1303. [Google Scholar] [CrossRef]
- Schnabl, B.; Brenner, D.A. Interactions between the intestinal microbiome and liver diseases. Gastroenterology 2014, 146, 1513–1524. [Google Scholar] [CrossRef]
- Schneider, K.M.; Albers, S.; Trautwein, C. Role of bile acids in the gut-liver axis. J. Hepatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Dapito, D.H.; Mencin, A.; Gwak, G.Y.; Pradere, J.P.; Jang, M.K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of Hepatocellular Carcinoma by the Intestinal Microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef]
- Brandl, K.; Schnabl, B. Intestinal microbiota and nonalcoholic steatohepatitis. Curr. Opin. Gastroenterol. 2017, 33, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Bergheim, I.; Weber, S.; Vos, M.; Krämer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992. [Google Scholar] [CrossRef]
- Brandt, A.; Jin, C.J.; Nolte, K.; Sellmann, C.; Engstler, A.J.; Bergheim, I. Short-term intake of a fructose-, fat- and cholesterol-rich diet causes hepatic steatosis in mice: Effect of antibiotic treatment. Nutrients 2017, 9, 1013. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Ramadori, P.; Weiskirchen, R.; Trebicka, J.; Streetz, K. Mouse models of metabolic liver injury. Lab. Anim. 2015, 49, 47–58. [Google Scholar] [CrossRef]
- Machado, M.V.; Michelotti, G.A.; Xie, G.; De Almeida, T.P.; Boursier, J.; Bohnic, B.; Guy, C.D.; Diehl, A.M. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS ONE 2015, 10, e0127991. [Google Scholar] [CrossRef]
- Rizki, G.; Arnaboldi, L.; Gabrielli, B.; Yan, J.; Lee, G.S.; Ng, R.K.; Turner, S.M.; Badger, T.M.; Pitas, R.E.; Maher, J.J. Mice fed a lipogenic methionine-choline-deficient diet develop hypermetabolism coincident with hepatic suppression of SCD-1. J. Lipid Res. 2006, 47, 2280–2290. [Google Scholar] [CrossRef]
- Velayudham, A.; Dolganiuc, A.; Ellis, M.; Petrasek, J.; Kodys, K.; Mandrekar, P.; Szabo, G. VSL#3 probiotic treatment attenuates fibrosis without changes in steatohepatitis in a diet-induced NASH model in mice. Hepatology 2009, 49, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Lendrum, J.; Seebach, B.; Liu, S. Depletion of murine microbiota with broad-spectrum antibiotics alters intestinal motility, secretion, and mucosal barrier function. FASEB J. 2016, 30, 1027-6. [Google Scholar]
- Josefsdottir, K.S.; Baldridge, M.T.; Kadmon, C.S.; King, K.Y. Antibiotics impair murine hematopoiesis by depleting the intestinal microbiota. Blood 2017, 129, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Zarrinpar, A.; Chaix, A.; Xu, Z.Z.; Chang, M.W.; Marotz, C.A.; Saghatelian, A.; Knight, R.; Panda, S. Antibiotic-induced microbiome depletion alters metabolic homeostasis by affecting gut signaling and colonic metabolism. Nat. Commun. 2018, 9, 2872. [Google Scholar] [CrossRef]
- Mazagova, M.; Wang, L.; Anfora, A.T.; Wissmueller, M.; Lesley, S.A.; Miyamoto, Y.; Eckmann, L.; Dhungana, S.; Pathmasiri, W.; Sumner, S.; et al. Commensal microbiota is hepatoprotective and prevents liver fibrosis in mice. FASEB J. 2015, 29, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Rivera, C.A.; Gaskin, L.; Allman, M.; Pang, J.; Brady, K.; Adegboyega, P.; Pruitt, K. Toll-like receptor-2 deficiency enhances non-alcoholic steatohepatitis. BMC Gastroenterol. 2010, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Mesmin, L.; Vijay-Kumar, M.; Gewirtz, A.T.; Chassaing, B. Hepatocyte Toll-Like Receptor 5 Promotes Bacterial Clearance and Protects Mice Against High-Fat Diet–Induced Liver Disease. CMGH 2016, 2, 584–604. [Google Scholar] [CrossRef]
- Ehses, J.A.; Meier, D.T.; Wueest, S.; Rytka, J.; Boller, S.; Wielinga, P.Y.; Schraenen, A.; Lemaire, K.; Debray, S.; Van Lommel, L.; et al. Toll-like receptor 2-deficient mice are protected from insulin resistance and beta cell dysfunction induced by a high-fat diet. Diabetologia 2010, 53, 1795–1806. [Google Scholar] [CrossRef]
- Himes, R.W.; Smith, C.W. Tlr2 is critical for diet-induced metabolic syndrome in a murine model. FASEB J. 2010, 24, 731–739. [Google Scholar] [CrossRef]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-Like Receptor 9 Promotes Steatohepatitis by Induction of Interleukin-1β in Mice. Gastroenterology 2010, 139, 323–334. [Google Scholar] [CrossRef]
- Poggi, M.; Bastelica, D.; Gual, P.; Iglesias, M.A.; Gremeaux, T.; Knauf, C.; Peiretti, F.; Verdier, M.; Juhan-Vague, I.; Tanti, J.F.; et al. C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia 2007, 50, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Ohnishi, H. Role of gut microbiota and Toll-like receptors in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 7381–7391. [Google Scholar] [CrossRef] [PubMed]
- Hatting, M.; Zhao, G.; Schumacher, F.; Sellge, G.; Al Masaoudi, M.; Gaßler, N.; Boekschoten, M.; Müller, M.; Liedtke, C.; Cubero, F.J.; Trautwein, C. Hepatocyte caspase-8 is an essential modulator of steatohepatitis in rodents. Hepatology 2013, 57, 2189–2201. [Google Scholar] [CrossRef] [PubMed]
- Kroy, D.C.; Schumacher, F.; Ramadori, P.; Hatting, M.; Bergheim, I.; Gassler, N.; Boekschoten, M.V.; Müller, M.; Streetz, K.L.; Trautwein, C. Hepatocyte specific deletion of c-Met leads to the development of severe non-alcoholic steatohepatitis in mice. J. Hepatol. 2014, 61, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pẽa, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010. [Google Scholar] [CrossRef]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; Desantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610. [Google Scholar] [CrossRef]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Belt, P.; Neuschwander-Tetri, B.A.; NASH Clinical Research Network. Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: Distinct clinicopathologic meanings. Hepatology 2011, 53, 810–820. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schneider, K.M.; Mohs, A.; Kilic, K.; Candels, L.S.; Elfers, C.; Bennek, E.; Schneider, L.B.; Heymann, F.; Gassler, N.; Penders, J.; et al. Intestinal Microbiota Protects against MCD Diet-Induced Steatohepatitis. Int. J. Mol. Sci. 2019, 20, 308. https://doi.org/10.3390/ijms20020308
Schneider KM, Mohs A, Kilic K, Candels LS, Elfers C, Bennek E, Schneider LB, Heymann F, Gassler N, Penders J, et al. Intestinal Microbiota Protects against MCD Diet-Induced Steatohepatitis. International Journal of Molecular Sciences. 2019; 20(2):308. https://doi.org/10.3390/ijms20020308
Chicago/Turabian StyleSchneider, Kai Markus, Antje Mohs, Konrad Kilic, Lena Susanna Candels, Carsten Elfers, Eveline Bennek, Lukas Ben Schneider, Felix Heymann, Nikolaus Gassler, John Penders, and et al. 2019. "Intestinal Microbiota Protects against MCD Diet-Induced Steatohepatitis" International Journal of Molecular Sciences 20, no. 2: 308. https://doi.org/10.3390/ijms20020308
APA StyleSchneider, K. M., Mohs, A., Kilic, K., Candels, L. S., Elfers, C., Bennek, E., Schneider, L. B., Heymann, F., Gassler, N., Penders, J., & Trautwein, C. (2019). Intestinal Microbiota Protects against MCD Diet-Induced Steatohepatitis. International Journal of Molecular Sciences, 20(2), 308. https://doi.org/10.3390/ijms20020308