Prediction Model with High-Performance Constitutive Androstane Receptor (CAR) Using DeepSnap-Deep Learning Approach from the Tox21 10K Compound Library



Abstract
1. Introduction
2. Results and Discussion
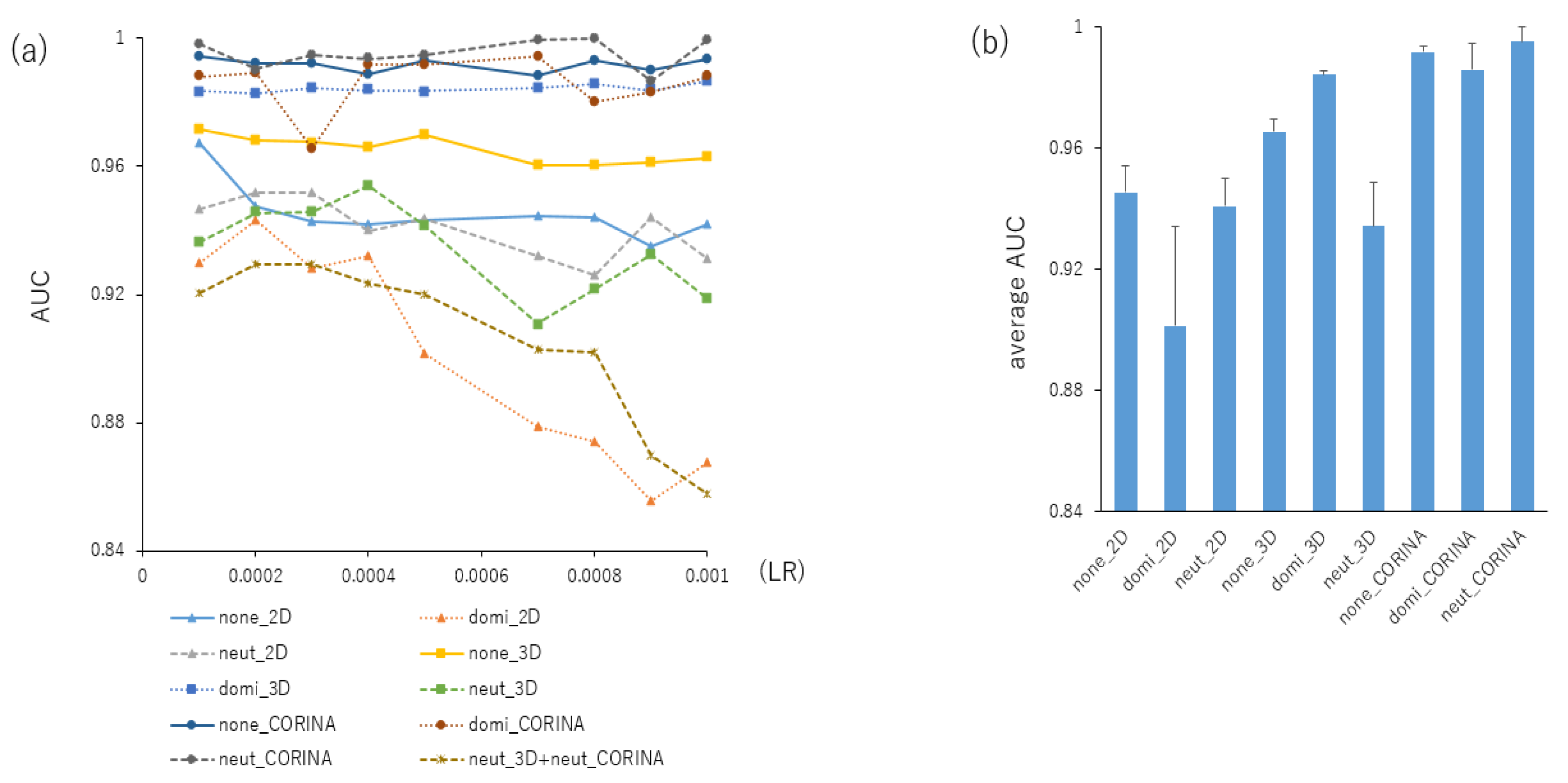
2.1. Contributions of Parameters for Prediction Performance in the DeepSnap-DL Approach
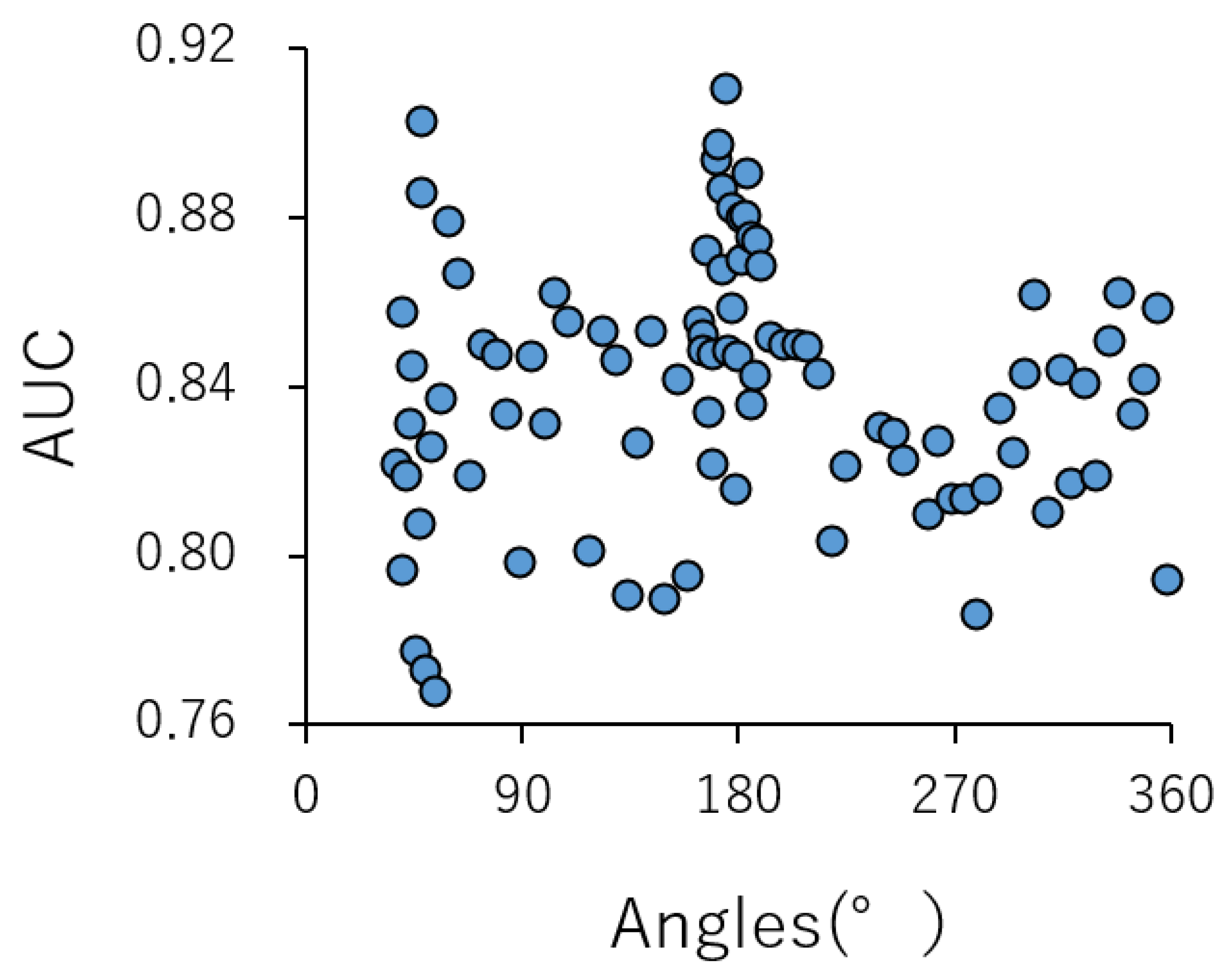
2.2. Contributions of Conformational Sampling of Chemical Compounds for Prediction Performance in the DeepSnap-DL Approach
2.3. The Prediction Performance of the DeepSnap-DL Approach Compared with the Conventional ML
3. Materials and Methods
3.1. Data
3.2. DeepSnap
3.3. ML Models
3.4. Evaluation of the Predictive Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2D | Two-dimensional |
| 3D | Three-dimensional |
| Acc | Accuracy in the test dataset |
| AO | Adverse outcome |
| AOP | Adverse outcome pathway |
| AUC | Area under the curve |
| AT | Atom size for van der Waals |
| AV | Accuracy in the validation dataset |
| BAC | Balanced accuracy |
| BMD | Bound minimum distance |
| BR | Bond radius |
| BSs | Batch sizes |
| BT | Bond tolerance |
| CAR | Constitutive androstane receptor |
| CNN | Convolutional neural network |
| DIGITS | Deep learning GPU training system |
| DL | Deep learning |
| DNNs | Deep neural networks |
| F | F value |
| KEs | Key events |
| KER | Key event relationship |
| LR | Learning rate |
| LV | Loss in the validation dataset |
| MBD | Minimum bond distance |
| MCC | Matthews correlation coefficient |
| MIE | Molecular initiating event |
| ML | Machine learning |
| MOE | Molecular operating environment |
| MPS | Number of molecules per SDF file to split into |
| NN | Neural network |
| RF | Random Forest |
| ROC | Receiver operating characteristic |
| SMILES | Simplified molecular input line entry system |
| SVM | Support vector machine |
| Tox21 | Toxicology in the 21st century |
| XGB | eXtreme Gradient Boosting |
| ZF | Zoom factor |
References
- Thomas, R.S.; Paules, R.S.; Simeonov, A.; Fitzpatrick, S.C.; Crofton, K.M.; Casey, W.M.; Mendrick, D.L. The US Federal Tox21 Program: A strategic and operational plan for continued leadership. ALTEX 2018, 35, 163–168. [Google Scholar] [CrossRef]
- Xia, M.; Huang, R.; Shi, Q.; Boyd, W.A.; Zhao, J.; Sun, N.; Rice, J.R.; Dunlap, P.E.; Hackstadt, A.J.; Bridge, M.F.; et al. Comprehensive Analyses and Prioritization of Tox21 10K Chemicals Affecting Mitochondrial Function by in-Depth Mechanistic Studies. Environ. Health Perspect. 2018, 126, 077010. [Google Scholar] [CrossRef] [PubMed]
- Sipes, N.S.; Wambaugh, J.F.; Pearce, R.; Auerbach, S.S.; Wetmore, B.A.; Hsieh, J.H.; Shapiro, A.J.; Svoboda, D.; DeVito, M.J.; Ferguson, S.S. An Intuitive Approach for Predicting Potential Human Health Risk with the Tox21 10k Library. Environ. Sci. Technol. 2017, 51, 10786–10796. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.; Mackowiak, B.; Huang, R.; Li, L.; Heyward, S.; Sakamuru, S.; Wang, H.; Xia, M. Identification of Modulators That Activate the Constitutive Androstane Receptor From the Tox21 10K Compound Library. Toxicol. Sci. 2019, 167, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Ankley, G.T.; Bennett, R.S.; Erickson, R.J.; Hoff, D.J.; Hornung, M.W.; Johnson, R.D.; Mount, D.R.; Nichols, J.W.; Russom, C.L.; Schmieder, P.K.; et al. Adverse outcome pathways: A conceptual framework to support ecotoxicology research and risk assessment. Environ. Toxicol. Chem. 2010, 29, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Vinken, M. The adverse outcome pathway concept: A pragmatic tool in toxicology. Toxicology 2013, 312, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, D.L.; Crump, D.; Garcia-Reyero, N.; Hecker, M.; Hutchinson, T.H.; LaLone, C.A.; Landesmann, B.; Lettieri, T.; Munn, S.; Nepelska, M.; et al. Adverse outcome pathway development II: Best practices. Toxicol. Sci. 2014, 142, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Bal-Price, A.; Crofton, K.M.; Sachana, M.; Shafer, T.J.; Behl, M.; Forsby, A.; Hargreaves, A.; Landesmann, B.; Lein, P.J.; Louisse, J.; et al. Putative adverse outcome pathways relevant to neurotoxicity. Crit. Rev. Toxicol. 2015, 45, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Bal-Price, A.; Lein, P.J.; Keil, K.P.; Sethi, S.; Shafer, T.; Barenys, M.; Fritsche, E.; Sachana, M.; Meek, M.E.B. Developing and applying the adverse outcome pathway concept for understanding and predicting neurotoxicity. Neurotoxicology 2017, 59, 240–255. [Google Scholar] [CrossRef]
- Perkins, E.J.; Antczak, P.; Burgoon, L.; Falciani, F.; Garcia-Reyero, N.; Gutsell, S.; Hodges, G.; Kienzler, A.; Knapen, D.; McBride, M.; et al. Adverse Outcome Pathways for Regulatory Applications: Examination of Four Case Studies With Different Degrees of Completeness and Scientific Confidence. Toxicol. Sci. 2015, 148, 14–25. [Google Scholar] [CrossRef]
- El-Masri, H.; Kleinstreuer, N.; Hines, R.N.; Adams, L.; Tal, T.; Isaacs, K.; Wetmore, B.A.; Tan, Y.M. Integration of Life-Stage Physiologically Based Pharmacokinetic Models with Adverse Outcome Pathways and Environmental Exposure Models to Screen for Environmental Hazards. Toxicol. Sci. 2016, 152, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Leist, M.; Ghallab, A.; Graepel, R.; Marchan, R.; Hassan, R.; Bennekou, S.H.; Limonciel, A.; Vinken, M.; Schildknecht, S.; Waldmann, T.; et al. Adverse outcome pathways: Opportunities, limitations and open questions. Arch. Toxicol. 2017, 91, 3477–3505. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.P.; Wiegers, T.C.; Wiegers, J.; Johnson, R.J.; Sciaky, D.; Grondin, C.J.; Mattingly, C.J. Chemical-Induced Phenotypes at CTD Help Inform the Predisease State and Construct Adverse Outcome Pathways. Toxicol. Sci. 2018, 165, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Terron, A.; Bal-Price, A.; Paini, A.; Monnet-Tschudi, F.; Bennekou, S.H.; EFSA WG EPI1 Members; Leist, M.; Schildknecht, S. An adverse outcome pathway for parkinsonian motor deficits associated with mitochondrial complex I inhibition. Arch. Toxicol. 2018, 92, 41–82. [Google Scholar] [CrossRef] [PubMed]
- Wang, D. Infer the in vivo point of departure with ToxCast in vitro assay data using a robust learning approach. Arch. Toxicol. 2018, 92, 2913–2922. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Charli, A.; Luo, J.; Riaz, Z.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Mechanistic Interplay between Autophagy and Apoptotic Signaling in Endosulfan-induced Dopaminergic Neurotoxicity: Relevance to the Adverse OutcomePathway in Pesticide Neurotoxicity. Toxicol. Sci. 2019, 169, 333–352. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Sakamuru, S.; Martin, M.T.; Reif, D.M.; Judson, R.S.; Houck, K.A.; Casey, W.; Hsieh, J.H.; Shockley, K.R.; Ceger, P.; et al. Profiling of the Tox21 10K compound library for agonists and antagonists of the estrogen receptor alpha signaling pathway. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Xia, M.; Sakamuru, S.; Zhao, J.; Shahane, S.A.; Attene-Ramos, M.; Zhao, T.; Austin, C.P.; Simeonov, A. Modelling the Tox21 10 K chemical profiles for in vivo toxicity prediction and mechanism characterization. Nat. Commun. 2016, 7, 10425. [Google Scholar] [CrossRef]
- Mahadevan, B.; Snyder, R.D.; Waters, M.D.; Benz, R.D.; Kemper, R.A.; Tice, R.R.; Richard, A.M. Genetic toxicology in the 21st century: Reflections and future directions. Environ. Mol. Mutagen. 2011, 52, 339–354. [Google Scholar] [CrossRef] [PubMed]
- Attene-Ramos, M.S.; Miller, N.; Huang, R.; Michael, S.; Itkin, M.; Kavlock, R.J.; Austin, C.P.; Shinn, P.; Simeonov, A.; Tice, R.R.; et al. The Tox21 robotic platform for the assessment of environmental chemicals--from vision to reality. Drug Discov. Today 2013, 18, 716–723. [Google Scholar] [CrossRef]
- Cherian, M.T.; Chai, S.C.; Chen, T. Small-molecule modulators of the constitutive androstane receptor. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1099–1114. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Hashimoto, M.; Honkakoski, P.; Negishi, M. Regulation of gene expression by CAR: An update. Arch. Toxicol. 2015, 89, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Mackowiak, B.; Wang, H. Mechanisms of xenobiotic receptor activation: Direct vs. indirect. Biochim. Biophys. Acta. 2016, 1859, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; You, H.; Choi, J.; No, K.T. Development of pharmacophore-based classification model for activators of constitutive androstane receptor. Drug Metab. Pharmacokinet. 2017, 32, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Yamaotsu, N.; Iwazaki, N.; Okamura, S.; Kume, T.; Hirono, S. Precise prediction of activators for the human constitutive androstane receptorusing structure-based three-dimensional quantitative structure-activity relationship methods. Drug Metab. Pharmacokinet. 2017, 32, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Freires, I.A.; Sardi, J.C.; de Castro, R.D.; Rosalen, P.L. Alternative Animal and Non-Animal Models for Drug Discovery and Development: Bonus or Burden? Pharm. Res. 2017, 34, 681–686. [Google Scholar] [CrossRef]
- Alves-Pimenta, S.; Colaço, B.; Oliveira, P.A.; Venâncio, C. Biological Concerns on the Selection of Animal Models for Teratogenic Testing. Methods Mol. Biol. 2018, 1797, 61–93. [Google Scholar] [PubMed]
- Ponzoni, I.; Sebastián-Pérez, V.; Requena-Triguero, C.; Roca, C.; Martínez, M.J.; Cravero, F.; Díaz, M.F.; Páez, J.A.; Arrayás, R.G.; Adrio, J.; et al. Hybridizing Feature Selection and Feature Learning Approaches in QSAR Modeling for Drug Discovery. Sci. Rep. 2017, 7, 2403. [Google Scholar] [CrossRef]
- Xia, L.Y.; Wang, Y.W.; Meng, D.Y.; Yao, X.J.; Chai, H.; Liang, Y. Descriptor Selection via Log-Sum Regularization for the Biological Activities of Chemical Structure. Int. J. Mol. Sci. 2017, 19, 30. [Google Scholar] [CrossRef]
- Khan, P.M.; Roy, K. Current approaches for choosing feature selection and learning algorithms in quantitative structure-activity relationships (QSAR). Expert Opin. Drug Discov. 2018, 13, 1075–1089. [Google Scholar] [CrossRef]
- Moriwaki, H.; Tian, Y.S.; Kawashita, N.; Takagi, T. Three-Dimensional Classification Structure-Activity RelationshipAnalysis Using Convolutional Neural Network. Chem. Pharm. Bull. 2019, 67, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Pei, J.; Lai, L. Deep Learning Based Regression and Multiclass Models for Acute Oral Toxicity Prediction with Automatic Chemical Feature Extraction. J. Chem. Inf. Model. 2017, 57, 2672–2685. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.; Ban, F.; Woo, G.; Hsing, M.; Yamazaki, T.; LeBlanc, E.; Rennie, P.S.; Welch, W.J.; Cherkasov, A. Toxic Colors: The Use of Deep Learning for Predicting Toxicity of Compounds Merely from Their Graphic Images. J. Chem. Inf. Model. 2018, 58, 1533–1543. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Chen, J.; Wang, Z.; Xie, H.; Hong, H. Deep learning for predicting toxicity of chemicals: A mini review. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2018, 36, 252–271. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Guo, P.; Zhou, Y.; Zhou, J.; Wang, Q.; Zhang, F.; Fang, J.; Cheng, F. Deep Learning-Based Prediction of Drug-Induced Cardiotoxicity. J. Chem. Inf. Model. 2019, 59, 1073–1084. [Google Scholar] [CrossRef]
- Koutsoukas, A.; Monaghan, K.J.; Li, X.; Huan, J. Deep-learning: Investigating deep neural networks hyper-parameters and comparison of performance to shallow methods for modeling bioactivity data. J. Cheminform. 2017, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Lenselink, E.B.; Ten Dijke, N.; Bongers, B.; Papadatos, G.; van Vlijmen, H.W.T.; Kowalczyk, W.; IJzerman, A.P.; van Westen, G.J.P. Beyond the hype: Deep neural networks outperform established methods using a ChEMBL bioactivity benchmark set. J. Cheminform. 2017, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Winkler, D.A.; Le, T.C. Performance of Deep and Shallow Neural Networks, the Universal Approximation Theorem, Activity Cliffs, and QSAR. Mol. Inform. 2017, 36. [Google Scholar]
- Liu, R.; Wang, H.; Glover, K.P.; Feasel, M.G.; Wallqvist, A. Dissecting Machine-Learning Prediction of Molecular Activity: Is an Applicability Domain Needed for Quantitative Structure-Activity Relationship Models Based on Deep Neural Networks? J. Chem. Inf. Model. 2019, 59, 117–126. [Google Scholar] [CrossRef]
- Simões, R.S.; Maltarollo, V.G.; Oliveira, P.R.; Honorio, K.M. Transfer and Multi-task Learning in QSAR Modeling: Advances and Challenges. Front. Pharm. 2018, 9, 74. [Google Scholar] [CrossRef]
- Park, J.G.; Jo, S. Bayesian Weight Decay on Bounded Approximation for Deep Convolutional Neural Networks. IEEE Trans. Neural Netw. Learn. Syst. 2019, 30, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Monteagudo, M.; Medina-Franco, J.L.; Pérez-Castillo, Y.; Nicolotti, O.; Cordeiro, M.N.; Borges, F. Activity cliffs in drug discovery: Dr Jekyll or Mr Hyde? Drug Discov. Today 2014, 19, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Horvath, D.; Marcou, G.; Varnek, A.; Kayastha, S.; de la Vega de León, A.; Bajorath, J. Prediction of Activity Cliffs Using Condensed Graphs of Reaction Representations, Descriptor Recombination, Support Vector Machine Classification, and Support Vector Regression. J. Chem. Inf. Model. 2016, 56, 1631–1640. [Google Scholar] [CrossRef] [PubMed]
- Uesawa, Y. Quantitative structure-activity relationship analysis using deep learning based on a novel molecular image input technique. Bioorg. Med. Chem. Lett. 2018, 28, 3400–3403. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaka, Y.; Uesawa, Y. Optimization of a Deep-Learning Method Based on the Classification of Images Generated by Parameterized Deep Snap a Novel Molecular-Image-Input Technique for Quantitative Structure-Activity Relationship (QSAR) Analysis. Front. Bioeng. Biotechnol. 2019, 7, 65. [Google Scholar] [CrossRef]
- Xu, M.; Papageorgiou, D.P.; Abidi, S.Z.; Dao, M.; Zhao, H.; Karniadakis, G.E. A deep convolutional neural network for classification of red blood cells in sickle cell anemia. PLoS Comput. Biol. 2017, 13, e1005746. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ye, Z.; Su, Y.; Zhao, Q.; Li, X.; Ouyang, D. Deep learning for in vitro prediction of pharmaceutical formulations. Acta. Pharm. Sin. B. 2019, 9, 177–185. [Google Scholar] [CrossRef]
- Cui, Y.; Dong, Q.; Hong, D.; Wang, X. Predicting protein-ligand binding residues with deep convolutional neural networks. BMC Bioinform. 2019, 20, 93. [Google Scholar] [CrossRef]
- Yang, Y.; Yan, L.F.; Zhang, X.; Han, Y.; Nan, H.Y.; Hu, Y.C.; Hu, B.; Yan, S.L.; Zhang, J.; Cheng, D.L.; et al. Glioma Grading on Conventional MR Images: A Deep Learning Study With Transfer Learning. Front. Neurosci. 2018, 12, 804. [Google Scholar] [CrossRef]
- Dybiec, K.; Molchanov, S.; Gryff-Keller, A. Structure of neutral molecules and monoanions of selected oxopurines in aqueous solutions as studied by NMR spectroscopy and theoretical calculations. J. Phys. Chem. A 2011, 115, 2057–2064. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.; Zhang, Y.; Yasin, A.; Zhang, L. Investigating Stability and Tautomerization of Gossypol-A Spectroscopy Study. Molecules 2019, 24, 1286. [Google Scholar] [CrossRef] [PubMed]
- Parthiban, V.; Gromiha, M.M.; Hoppe, C.; Schomburg, D. Structural analysis and prediction of protein mutant stability using distance and torsion potentials: Role of secondary structure and solvent accessibility. Proteins 2007, 66, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Mervin, L.H.; Afzal, A.M.; Drakakis, G.; Lewis, R.; Engkvist, O.; Bender, A. Target prediction utilising negative bioactivity data covering large chemicalspace. J. Cheminform. 2015, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Klingspohn, W.; Mathea, M.; Ter Laak, A.; Heinrich, N.; Baumann, K. Efficiency of different measures for defining the applicability domain of classification models. J. Cheminform. 2017, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Xia, M.; Sakamuru, S.; Zhao, J.; Lynch, C.; Zhao, T.; Zhu, H.; Austin, C.P.; Simeonov, A. Expanding biological space coverage enhances the prediction of drug adverse effects in human using in vitro activity profiles. Sci. Rep. 2018, 8, 3783. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Kim, S.; Kim, K.; Lee, D. Deep learning-based classification with improved time resolution for physical activities of children. Peer J. 2018, 6, e5764. [Google Scholar] [CrossRef] [PubMed]
- Low, Y.; Uehara, T.; Minowa, Y.; Yamada, H.; Ohno, Y.; Urushidani, T.; Sedykh, A.; Muratov, E.; Kuz’min, V.; Fourches, D.; et al. Predicting drug-induced hepatotoxicity using QSAR and toxicogenomics approaches. Chem. Res. Toxicol. 2011, 24, 1251–1262. [Google Scholar] [CrossRef]
- Marzo, M.; Kulkarni, S.; Manganaro, A.; Roncaglioni, A.; Wu, S.; Barton-Maclaren, T.S.; Lester, C.; Benfenati, E. Integrating in silico models to enhance predictivity for developmental toxicity. Toxicology. 2016, 370, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Maggiora, G.M. On outliers and activity cliffs--why QSAR often disappoints. J. Chem. Inf. Model. 2006, 46, 1535. [Google Scholar] [CrossRef]
- Stumpfe, D.; Bajorath, J. Exploring activity cliffs in medicinal chemistry. J. Med. Chem. 2012, 55, 2932–2942. [Google Scholar] [CrossRef]
- Stumpfe, D.; Hu, H.; Bajorath, J. Introducing a new category of activity cliffs with chemical modifications at multiple sites and rationalizing contributions of individual substitutions. Bioorg. Med. Chem. 2019, 27, 3605–3612. [Google Scholar] [CrossRef] [PubMed]
- Bajorath, J. Representation and identification of activity cliffs. Expert. Opin. Drug Discov. 2017, 12, 879–883. [Google Scholar] [CrossRef] [PubMed]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, H.; Tian, Y.S.; Kawashita, N.; Takagi, T. Mordred: A molecular descriptor calculator. J. Cheminform. 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Sakamuru, S.; Li, X.; Attene-Ramos, M.S.; Huang, R.; Lu, J.; Shou, L.; Shen, M.; Tice, R.R.; Austin, C.P.; Xia, M. Application of a homogenous membrane potential assay to assess mitochondrial function. Physiol. Genom. 2012, 44, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Inglese, J.; Auld, D.S.; Jadhav, A.; Johnson, R.L.; Simeonov, A.; Yasgar, A.; Zheng, W.; Austin, C.P. Quantitative high-throughput screening: A titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc. Natl. Acad. Sci. USA 2006, 103, 11473–11478. [Google Scholar] [CrossRef]
- Chen, I.J.; Foloppe, N. Conformational sampling of druglike molecules with MOE and catalyst: Implications for pharmacophore modeling and virtual screening. J. Chem. Inf. Model. 2008, 48, 1773–1791. [Google Scholar] [CrossRef]
- Agrafiotis, D.K.; Gibbs, A.C.; Zhu, F.; Izrailev, S.; Martin, E. Conformational sampling of bioactive molecules: A comparative study. J. Chem. Inf. Model. 2007, 47, 1067–1086. [Google Scholar] [CrossRef] [PubMed]
- Hanson, R.M. Jmol SMILES and Jmol SMARTS: Specifications and applications. J. Cheminform. 2016, 8, 50. [Google Scholar] [CrossRef]
- Scalfani, V.F.; Williams, A.J.; Tkachenko, V.; Karapetyan, K.; Pshenichnov, A.; Hanson, R.M.; Liddie, J.M.; Bara, J.E. Programmatic conversion of crystal structures into 3D printable files using Jmol. J. Cheminform. 2016, 8, 66. [Google Scholar] [CrossRef]
- Hanson, R.M.; Lu, X.J. DSSR-enhanced visualization of nucleic acid structures in Jmol. Nucleic Acids Res. 2017, 45, W528–W533. [Google Scholar] [CrossRef] [PubMed]
- Krizhevsky, A.; Sutskev, I.; Hinton, G.E. ImageNet classification with deep convolutional neural networks. Adv. Neural. Inf. Process. Syst. 2012, 1, 1097–1105. [Google Scholar] [CrossRef]
- Szegedy, C.; Liu, W.; Jia, Y.; Sermanet, P.; Reed, S.; Anguelov, D.; Erhan, D.; Vanhoucke, V.; Rabinovich, A. Going Deeper with Convolutions. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Boston, MA, USA, 7–12 June 2015; pp. 1–9. [Google Scholar]
- Rawat, W.; Wang, Z. Deep convolutional neural networks for image classification: A comprehensive review. Neural. Comput. 2017, 29, 2352–2449. [Google Scholar] [CrossRef] [PubMed]
- Karri, S.P.; Chakraborty, D.; Chatterjee, J. Transfer learning based classification of optical coherence tomography images with diabetic macular edema and dry age-related macular degeneration. Biomed. Op.T Express. 2017, 8, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Saubern, S.; Guha, R.; Baell, J.B. KNIME Workflow to Assess PAINS Filters in SMARTS Format. Comparison of RDKit and Indigo Cheminformatics Libraries. Mol. Inform. 2011, 30, 847–850. [Google Scholar] [CrossRef]
- Abraham, A.; Pedregosa, F.; Eickenberg, M.; Gervais, P.; Mueller, A.; Kossaifi, J.; Gramfort, A.; Thirion, B.; Varoquaux, G. Machine learning for neuroimaging with scikit-learn. Front. Neuroinform. 2014, 8, 14. [Google Scholar] [CrossRef]
- Kensert, A.; Alvarsson, J.; Norinder, U.; Spjuth, O. Evaluating parameters for ligand-based modeling with random forest on sparse data sets. J. Cheminform. 2018, 10, 49. [Google Scholar] [CrossRef]
- Sandino, J.; Gonzalez, F.; Mengersen, K.; Gaston, K.J. UAVs and Machine Learning Revolutionising Invasive Grass and Vegetation Surveys in Remote Arid Lands. Sensors 2018, 18, 605. [Google Scholar] [CrossRef]
- Hongjaisee, S.; Nantasenamat, C.; Carraway, T.S.; Shoombuatong, W. HIVCoR: A sequence-based tool for predicting HIV-1 CRF01_AE coreceptor usage. Comput. Biol. Chem. 2019, 80, 419–432. [Google Scholar] [CrossRef]
- Laengsri, V.; Nantasenamat, C.; Schaduangrat, N.; Nuchnoi, P.; Prachayasittikul, V.; Shoombuatong, W. TargetAntiAngio: A Sequence-Based Tool for the Prediction and Analysis of Anti-Angiogenic Peptides. Int. J. Mol. Sci. 2019, 20, 2950. [Google Scholar] [CrossRef]
- Shoombuatong, W.; Schaduangrat, N.; Pratiwi, R.; Nantasenamat, C. THPep: A machine learning-based approach for predicting tumor homing peptides. Comput. Biol. Chem. 2019, 80, 441–451. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| AUC | Acc | MCC | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Train:val:test | Protonation | Coordinate | Protonation | Coordinate | Average | SD | Average | SD | Average | SD |
| 1:1:1 | none | 2D | 0.930 | 0.006 | 0.967 | 0.007 | 0.821 | 0.035 | ||
| 1:1:1 | dominate | 2D | 0.904 | 0.011 | 0.926 | 0.048 | 0.668 | 0.131 | ||
| 1:1:1 | neutralize | 2D | 0.890 | 0.006 | 0.919 | 0.032 | 0.619 | 0.115 | ||
| 1:1:1 | none | 3D | 0.907 | 0.008 | 0.797 | 0.035 | 0.440 | 0.019 | ||
| 1:1:1 | dominate | 3D | 0.971 | 0.003 | 0.927 | 0.001 | 0.734 | 0.005 | ||
| 1:1:1 | neutralize | 3D | 0.924 | 0.007 | 0.969 | 0.003 | 0.827 | 0.017 | ||
| 1:1:1 | none | CORINA | 0.989 | 0.003 | 0.958 | 0.003 | 0.826 | 0.012 | ||
| 1:1:1 | dominate | CORINA | 0.996 | 0.002 | 0.982 | 0.005 | 0.914 | 0.021 | ||
| 1:1:1 | neutralize | CORINA | 0.998 | 0.002 | 0.991 | 0.006 | 0.954 | 0.026 | ||
| 1:1:1 | neutralize | 3D | neutralize | CORINA | 0.798 | 0.016 | 0.707 | 0.020 | 0.302 | 0.018 |
| 4:4:1 | none | 2D | 0.923 | 0.024 | 0.959 | 0.029 | 0.798 | 0.107 | ||
| 4:4:1 | dominate | 2D | 0.906 | 0.013 | 0.894 | 0.069 | 0.609 | 0.139 | ||
| 4:4:1 | neutralize | 2D | 0.898 | 0.019 | 0.903 | 0.059 | 0.621 | 0.125 | ||
| 4:4:1 | none | 3D | 0.911 | 0.009 | 0.801 | 0.043 | 0.458 | 0.033 | ||
| 4:4:1 | dominate | 3D | 0.972 | 0.003 | 0.928 | 0.012 | 0.739 | 0.030 | ||
| 4:4:1 | neutralize | 3D | 0.927 | 0.011 | 0.971 | 0.002 | 0.839 | 0.010 | ||
| 4:4:1 | none | CORINA | 0.990 | 0.003 | 0.957 | 0.009 | 0.821 | 0.029 | ||
| 4:4:1 | dominate | CORINA | 0.997 | 0.001 | 0.985 | 0.003 | 0.927 | 0.015 | ||
| 4:4:1 | neutralize | CORINA | 0.999 | 0.001 | 0.993 | 0.005 | 0.966 | 0.023 | ||
| 4:4:1 | neutralize | 3D | neutralize | CORINA | 0.802 | 0.014 | 0.684 | 0.043 | 0.311 | 0.021 |
| 176° | 280° | 360° | 280°PT | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Train:Val:Test | N | Average | SD | Average | SD | Average | SD | Average | SD | |
| AUC | 1:1:1 | 3 | 1.000 | 0.000 | 0.998 | 0.002 | 0.932 | 0.027 | 0.537 | 0.009 |
| 2:2:1 | 5 | 0.999 | 0.001 | 0.998 | 0.001 | 0.964 | 0.005 | 0.522 | 0.013 | |
| 3:3:1 | 6 | 0.999 | 0.000 | 0.998 | 0.001 | 0.972 | 0.009 | 0.544 | 0.019 | |
| 4:4:1 | 9 | 0.998 | 0.003 | 0.999 | 0.001 | 0.979 | 0.005 | 0.545 | 0.027 | |
| 5:5:1 | 11 | 0.998 | 0.003 | 0.998 | 0.002 | 0.983 | 0.005 | 0.534 | 0.016 | |
| 6:6:1 | 13 | 0.999 | 0.001 | 0.998 | 0.002 | 0.983 | 0.008 | 0.529 | 0.022 | |
| 7:7:1 | 15 | 0.998 | 0.002 | 0.998 | 0.002 | 0.982 | 0.007 | 0.555 | 0.043 | |
| 8:8:1 | 17 | 0.999 | 0.003 | 0.998 | 0.003 | 0.983 | 0.009 | 0.552 | 0.044 | |
| Acc | 1:1:1 | 3 | 0.997 | 0.001 | 0.991 | 0.006 | 0.851 | 0.037 | 0.422 | 0.009 |
| 2:2:1 | 5 | 0.995 | 0.002 | 0.993 | 0.005 | 0.898 | 0.005 | 0.554 | 0.013 | |
| 3:3:1 | 6 | 0.993 | 0.006 | 0.988 | 0.008 | 0.918 | 0.034 | 0.555 | 0.019 | |
| 4:4:1 | 9 | 0.995 | 0.003 | 0.993 | 0.005 | 0.925 | 0.020 | 0.449 | 0.027 | |
| 5:5:1 | 11 | 0.993 | 0.004 | 0.992 | 0.004 | 0.934 | 0.022 | 0.507 | 0.016 | |
| 6:6:1 | 13 | 0.995 | 0.002 | 0.993 | 0.007 | 0.942 | 0.022 | 0.498 | 0.022 | |
| 7:7:1 | 15 | 0.994 | 0.003 | 0.993 | 0.007 | 0.934 | 0.030 | 0.513 | 0.043 | |
| 8:8:1 | 17 | 0.996 | 0.003 | 0.992 | 0.009 | 0.931 | 0.049 | 0.527 | 0.044 | |
| MCC | 1:1:1 | 3 | 0.986 | 0.006 | 0.954 | 0.026 | 0.547 | 0.074 | 0.018 | 0.073 |
| 2:2:1 | 5 | 0.977 | 0.012 | 0.966 | 0.022 | 0.647 | 0.016 | 0.018 | 0.047 | |
| 3:3:1 | 6 | 0.966 | 0.028 | 0.942 | 0.037 | 0.705 | 0.074 | 0.025 | 0.065 | |
| 4:4:1 | 9 | 0.976 | 0.015 | 0.966 | 0.023 | 0.723 | 0.049 | 0.078 | 0.022 | |
| 5:5:1 | 11 | 0.967 | 0.017 | 0.962 | 0.018 | 0.749 | 0.055 | 0.057 | 0.055 | |
| 6:6:1 | 13 | 0.976 | 0.012 | 0.970 | 0.028 | 0.768 | 0.060 | 0.062 | 0.049 | |
| 7:7:1 | 15 | 0.970 | 0.012 | 0.966 | 0.031 | 0.755 | 0.072 | 0.069 | 0.079 | |
| 8:8:1 | 17 | 0.978 | 0.016 | 0.961 | 0.041 | 0.749 | 0.103 | 0.060 | 0.092 | |
| Angles on x-, y-, z-axes | AUC | Acc | MCC | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. of Picture | Pic1 | Pic2 | Pic3 | Pic4 | Average | SD | Average | SD | Average | SD |
| 4 | 0,0,0, | 280,0,0, | 0,280,0 | 0,0,280 | 0.999 | 0.000 | 0.994 | 0.002 | 0.967 | 0.012 |
| 4 | 280,280,280, | 0,280,280, | 280,0,280 | 280,280,0 | 0.998 | 0.002 | 0.988 | 0.004 | 0.941 | 0.021 |
| 4 | 0,0,0, | 0,280,280, | 280.0.280 | 280,280,0 | 0.998 | 0.001 | 0.990 | 0.003 | 0.952 | 0.014 |
| 4 | 0,0,0, | 280,0,0, | 280.0.280 | 280,280,0 | 0.997 | 0.003 | 0.988 | 0.006 | 0.943 | 0.027 |
| 4 | 0,0,0, | 280,0,0, | 0.280.0 | 280,280,0 | 0.996 | 0.002 | 0.991 | 0.004 | 0.953 | 0.018 |
| 3 | - | 280,0,0, | 0,280,0 | 0,0,280 | 0.995 | 0.004 | 0.984 | 0.006 | 0.921 | 0.027 |
| 3 | 0,0,0, | - | 0,280,0 | 0,0,280 | 0.998 | 0.001 | 0.987 | 0.005 | 0.935 | 0.024 |
| 3 | 0,0,0, | 280,0,0, | - | 0,0,280 | 0.998 | 0.001 | 0.988 | 0.008 | 0.943 | 0.037 |
| 3 | 0,0,0, | 280,0,0, | 0,280,0 | - | 0.995 | 0.002 | 0.984 | 0.007 | 0.921 | 0.032 |
| 2 | 0,0,0, | 280,0,0, | - | - | 0.995 | 0.002 | 0.976 | 0.012 | 0.890 | 0.048 |
| 2 | 0,0,0, | - | 0,280,0 | - | 0.993 | 0.002 | 0.970 | 0.015 | 0.864 | 0.055 |
| 2 | 0,0,0, | - | - | 0,0,280 | 0.996 | 0.000 | 0.978 | 0.009 | 0.896 | 0.034 |
| 2 | - | 280,0,0, | 0,280,0 | - | 0.982 | 0.008 | 0.960 | 0.006 | 0.817 | 0.010 |
| 2 | - | - | 0,280,0 | 0,0,280 | 0.998 | 0.001 | 0.986 | 0.002 | 0.931 | 0.010 |
| Auc | Parameters | ||||
|---|---|---|---|---|---|
| Model # | Average | SD | Max_Depth | Nestimators | Max_Features |
| XGB_1 | 0.8855 | 0.0071 | 3 | 100 | 29 |
| XGB_2 | 0.8862 | 0.0095 | 3 | 500 | 29 |
| XGB_3 | 0.8854 | 0.0073 | 3 | 1000 | 29 |
| XGB_4 | 0.8885 | 0.0033 | 3 | 5000 | 29 |
| XGB_5 | 0.8883 | 0.0040 | 30 | 1000 | 29 |
| XGB_6 | 0.8872 | 0.0089 | 3 | 5000 | 40 |
| XGB_7 | 0.8851 | 0.0026 | 3 | 5000 | 50 |
| XGB_8 | 0.8890 | 0.0072 | 3 | 5000 | 60 |
| XGB_9 | 0.8873 | 0.0069 | 3 | 5000 | 100 |
| XGB_10 | 0.8835 | 0.0075 | 3 | 5000 | 120 |
| RF_1 | 0.8069 | 0.0193 | 2 | 10 | 29 |
| RF_2 | 0.8314 | 0.0287 | 2 | 100 | 29 |
| RF_3 | 0.8416 | 0.0252 | 2 | 1000 | 29 |
| RF_4 | 0.8803 | 0.0053 | 20 | 1000 | 29 |
| RF_5 | 0.8781 | 0.0104 | 200 | 1000 | 29 |
| RF_6 | 0.8780 | 0.0083 | 20 | 5000 | 29 |
| RF_7 | 0.8702 | 0.0067 | 20 | 1000 | 5 |
| RF_8 | 0.8813 | 0.0032 | 20 | 1000 | 80 |
| RF_9 | 0.8842 | 0.0052 | 20 | 1000 | 120 |
| RF_10 | 0.8807 | 0.0055 | 20 | 1000 | 250 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsuzaka, Y.; Uesawa, Y. Prediction Model with High-Performance Constitutive Androstane Receptor (CAR) Using DeepSnap-Deep Learning Approach from the Tox21 10K Compound Library. Int. J. Mol. Sci. 2019, 20, 4855. https://doi.org/10.3390/ijms20194855
Matsuzaka Y, Uesawa Y. Prediction Model with High-Performance Constitutive Androstane Receptor (CAR) Using DeepSnap-Deep Learning Approach from the Tox21 10K Compound Library. International Journal of Molecular Sciences. 2019; 20(19):4855. https://doi.org/10.3390/ijms20194855
Chicago/Turabian StyleMatsuzaka, Yasunari, and Yoshihiro Uesawa. 2019. "Prediction Model with High-Performance Constitutive Androstane Receptor (CAR) Using DeepSnap-Deep Learning Approach from the Tox21 10K Compound Library" International Journal of Molecular Sciences 20, no. 19: 4855. https://doi.org/10.3390/ijms20194855
APA StyleMatsuzaka, Y., & Uesawa, Y. (2019). Prediction Model with High-Performance Constitutive Androstane Receptor (CAR) Using DeepSnap-Deep Learning Approach from the Tox21 10K Compound Library. International Journal of Molecular Sciences, 20(19), 4855. https://doi.org/10.3390/ijms20194855