Crystal Structure of Kluyveromyces lactis Glucokinase (KlGlk1)

 , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
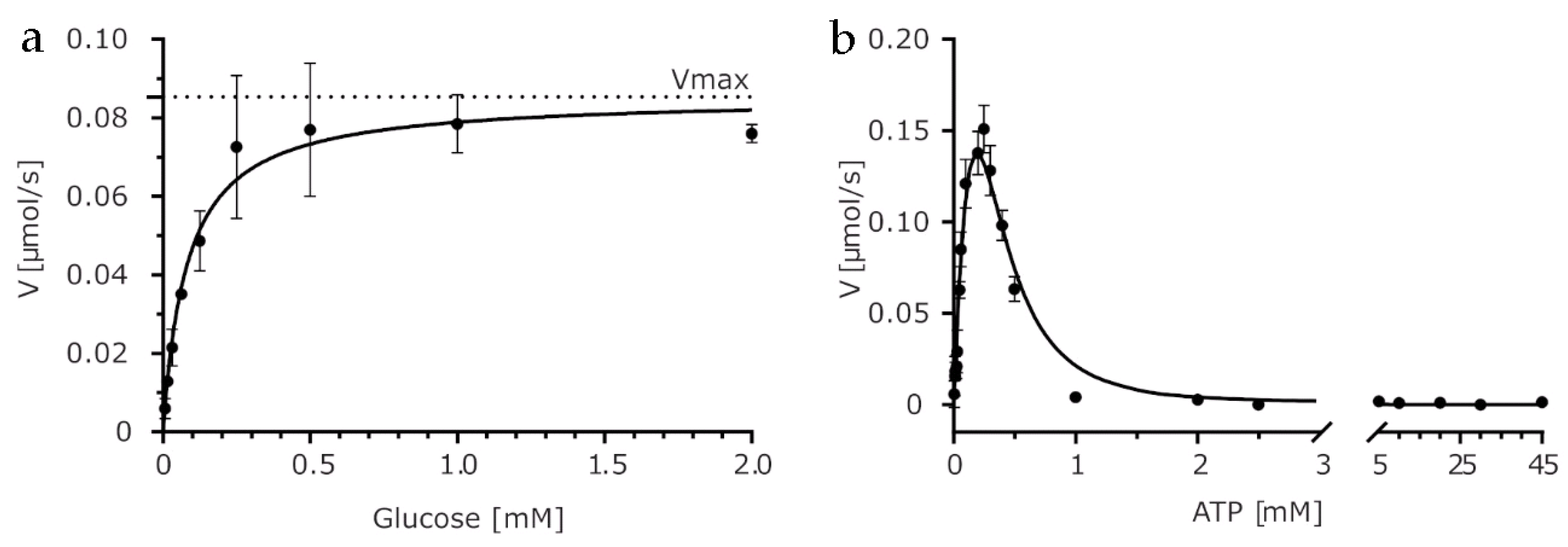
2.1. Biochemical Characterization
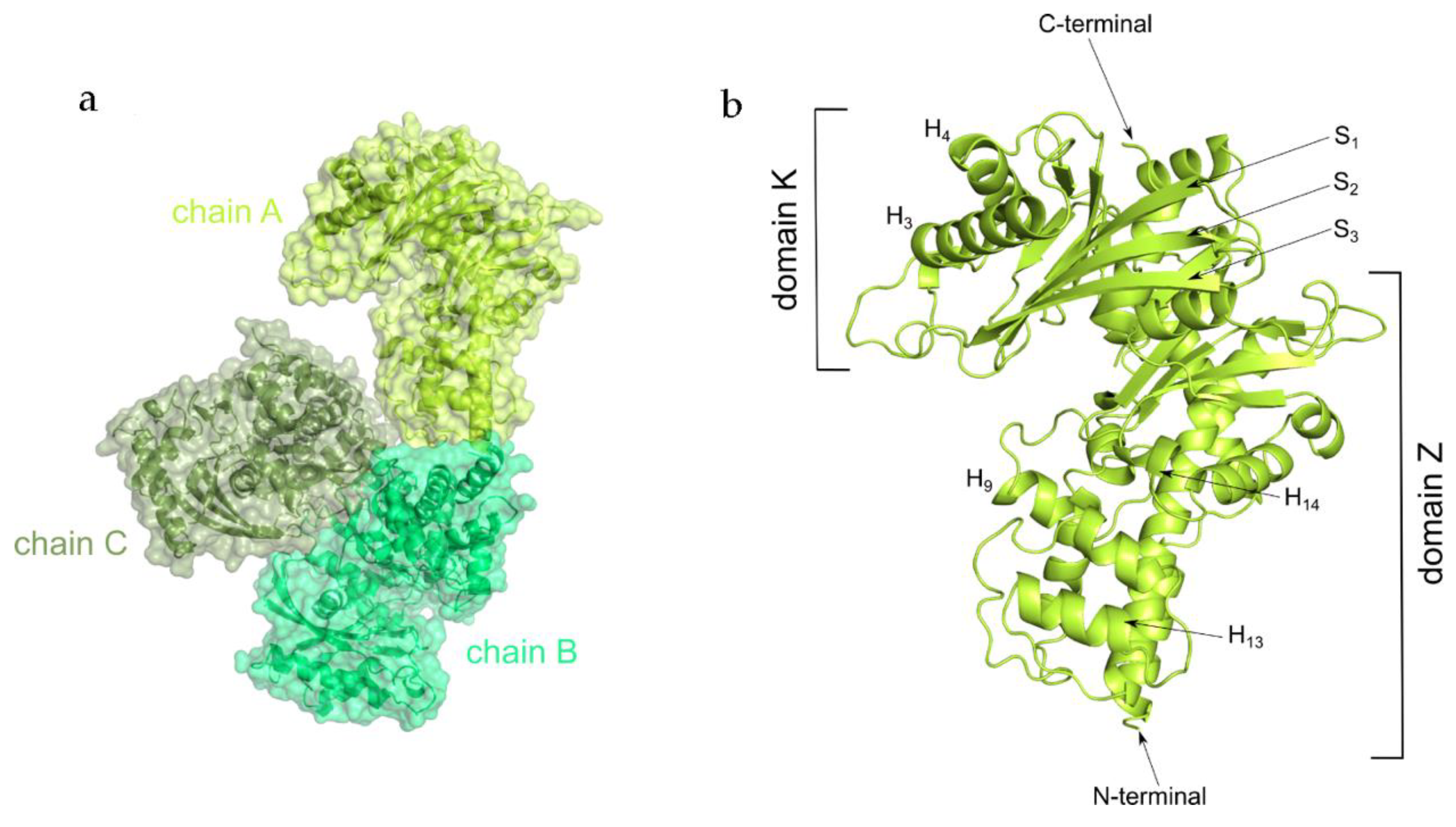
2.2. Overall Structure
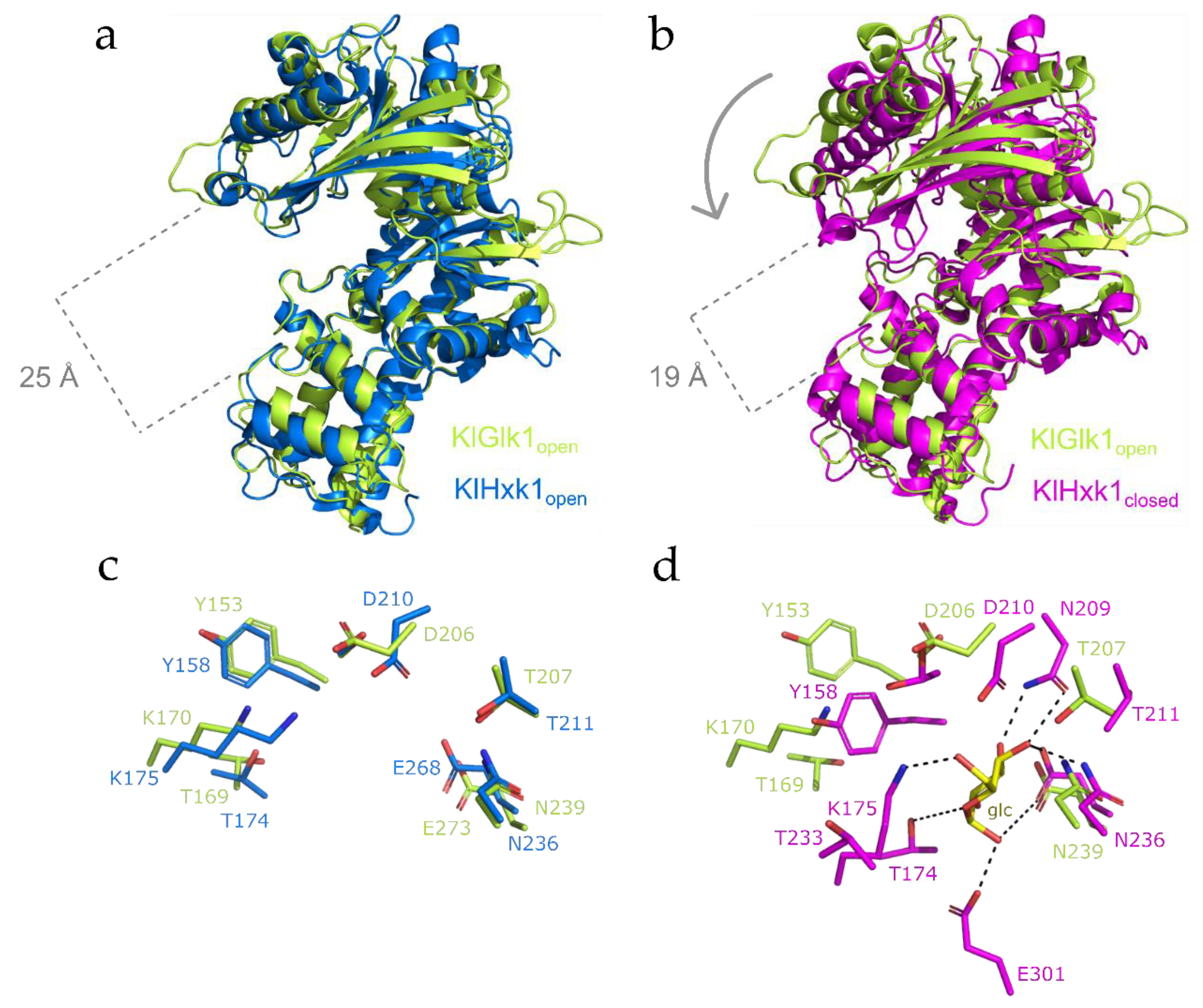
2.3. Structural Comparison between KlGlk1 and KlHxk1 Glucokinases
3. Materials and Methods
3.1. Cloning, Expression and Purification
3.2. Crystallization and Structure Determination
3.3. Activity Assay
3.4. RALS/LALS Analysis
3.5. Protein Stability Analysis
3.6. Protein Data Bank Accession Code
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | Adenosine triphosphate |
| ADP | Adenosine diphosphate |
| Å | Angstrom |
| TSA | Thermal Shift Assay |
| RMSD | Root Mean Square Deviation |
| IPTG | Isopropyl-β-D-thiogalactoside |
| HEPES | (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) |
| PEG | Poly(ethylene glycol) |
| RALS/LALS | Right-angle light scattering/low-angle light scattering |
| TRIS | Trisaminomethane |
| NADP | Nicotinamide adenine dinucleotide phosphate |
References
- Merritt, C.; Silva, L.E.; Tanner, A.L.; Stuart, K.; Pollastri, M.P. Kinases as druggable targets in trypanosomatid protozoan parasites. Chem. Rev. 2014, 114, 11280–11304. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, D.P.; Harris, M.T.; Schroeder, C.E.; Khan, H.; Kahney, E.W.; Hackler, A.L.; Patrick, S.L.; Weiner, W.S.; Aube, J.; Sharlow, E.R.; et al. Optimization and Evaluation of Antiparasitic Benzamidobenzoic Acids as Inhibitors of Kinetoplastid Hexokinase 1. ChemMedChem 2017, 12, 1994–2005. [Google Scholar] [CrossRef] [PubMed]
- Kalel, V.C.; Mäser, P.; Sattler, M.; Erdmann, R.; Popowicz, G.M. Come, sweet death: Targeting glycosomal protein import for antitrypanosomal drug development. Curr. Opin. Microbiol. 2018, 46, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Fushinobu, S.; Jeong, J.J.; Yoshioka, I.; Koga, S.; Shoun, H.; Wakagi, T. Crystal structure of an ADP-dependent glucokinase from Pyrococcus furiosus: implications for a sugar-induced conformational change in ADP-dependent kinase. J. Mol. Biol. 2003, 331, 871–883. [Google Scholar] [CrossRef]
- Grudnik, P.; Kaminski, M.M.; Rembacz, K.P.; Kuska, K.; Madej, M.; Potempa, J.; Dawidowski, M.; Dubin, G. Structural basis for ADP-dependent glucokinase inhibition by 8-bromo-substituted adenosine nucleotide. J. Biol. Chem. 2018, 293, 11088–11099. [Google Scholar] [CrossRef] [PubMed]
- Tokarz, P.; Wisniewska, M.; Kaminski, M.M.; Dubin, G.; Grudnik, P. Crystal structure of ADP-dependent glucokinase from Methanocaldococcus jannaschii in complex with 5-iodotubercidin reveals phosphoryl transfer mechanism. Protein Sci. 2018, 27, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Entian, K.D. Genetic and biochemical evidence for hexokinase PII as a key enzyme involved in catabolite repression in yeast. Mol. Gen. Genet. 1980, 178, 633–637. [Google Scholar] [CrossRef]
- Rolland, F.; Winderickx, J.; Thevelein, J.M. Glucose-sensing mechanisms in eukaryotic cells. Trends Biochem. Sci. 2001, 26, 310–317. [Google Scholar] [CrossRef]
- Rolland, F.; Baena-Gonzalez, E.; Sheen, J. Sugar sensing and signaling in plants: conserved and novel mechanisms. Annu. Rev. Plant. Biol. 2006, 57, 675–709. [Google Scholar] [CrossRef]
- Wilson, J.E. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J. Exp. Biol. 2003, 206, 2049–2057. [Google Scholar] [CrossRef]
- Moreno, F.; Ahuatzi, D.; Riera, A.; Palomino, C.A.; Herrero, P. Glucose sensing through the Hxk2-dependent signalling pathway. Biochem. Soc. Trans. 2005, 33, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.H.; Yoo, S.D.; Sheen, J. Regulatory functions of nuclear hexokinase 1 complex in glucose signaling. Cell 2006, 127, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Postic, C.; Shiota, M.; Magnuson, M.A. Cell-specific roles of glucokinase in glucose homeostasis. Recent Prog. Horm. Res. 2006, 56, 195–217. [Google Scholar] [CrossRef]
- van Schaftingen, E.; Veiga-da-Cunha, M.; Niculescu, L. The regulatory protein of glucokinase. Biochem. Soc. Trans. 1997, 25, 136–140. [Google Scholar] [CrossRef] [PubMed]
- de la Cera, T.; Herrero, P.; Moreno-Herrero, F.; Chaves, R.S.; Moreno, F. Mediator factor Med8p interacts with the hexokinase 2: implication in the glucose signalling pathway of Saccharomyces cerevisiae. J. Mol. Biol. 2002, 319, 703–714. [Google Scholar] [CrossRef]
- Anderson, C.M.; McDonald, R.C.; Steitz, T.A. Sequencing a protein by x-ray crystallography. I. Interpretation of yeast hexokinase B at 2.5 Å resolution by model building. J. Mol. Biol. 1978, 123, 1–13. [Google Scholar] [CrossRef]
- Bennett, W.S., Jr.; Steitz, T.A. Structure of a complex between yeast hexokinase A and glucose. II. Detailed comparisons of conformation and active site configuration with the native hexokinase B monomer and dimer. J. Mol. Biol. 1980, 140, 211–230. [Google Scholar] [CrossRef]
- Kuser, P.R.; Krauchenco, S.; Antunes, O.; Polikarpov, I. The high resolution crystal structure of yeast hexokinase PII with the correct primary sequence provides new insights into its mechanism of action. J. Biol. Chem. 2000, 275, 20814–20821. [Google Scholar] [CrossRef] [PubMed]
- Spohner, S.C.; Schaum, V.; Quitmann, H.; Czermak, P. Kluyveromyces lactis: An emerging tool in biotechnology. J. Biotechnol. 2016, 222, 104–116. [Google Scholar] [CrossRef]
- van Ooyen, A.J.; Dekker, P.; Huang, M.; Olsthoorn, M.M.; Jacobs, D.I.; Colussi, P.A.; Taron, C.H. Heterologous protein production in the yeast Kluyveromyces lactis. FEMS Yeast Res. 2006, 6, 381–392. [Google Scholar] [CrossRef]
- Kettner, K.; Müller, E.C.; Otto, A.; Rödel, G.; Breunig, K.D.; Kriegel, T.M. Identification and characterization of a novel glucose-phosphorylating enzyme in Kluyveromyces lactis. FEMS Yeast Res. 2007, 7, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Kuettner, E.B.; Kriegel, T.M.; Keim, A.; Naumann, M.; Sträter, N. Crystallization and preliminary X-ray diffraction studies of hexokinase KlHxk1 from Kluyveromyces lactis. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2007, 63, 430–433. [Google Scholar] [CrossRef] [PubMed]
- Kuettner, E.B.; Kettner, K.; Keim, A.; Svergun, D.I.; Volke, D.; Singer, D.; Hoffmann, R.; Müller, E.C.; Otto, A.; Kriegel, T.M.; et al. Crystal structure of hexokinase KlHxk1 of Kluyveromyces lactis: a molecular basis for understanding the control of yeast hexokinase functions via covalent modification and oligomerization. J. Biol. Chem. 2010, 285, 41019–41033. [Google Scholar] [CrossRef] [PubMed]
- Kettner, K.; Kuettner, E.B.; Otto, A.; Lilie, H.; Golbik, R.P.; Sträter, N.; Kriegel, T.M. In vivo phosphorylation and in vitro autophosphorylation-inactivation of Kluyveromyces lactis hexokinase KlHxk1. Biochem. Biophys. Res. Commun. 2013, 435, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Kopetzki, E.; Entian, K.D. Glucose repression and hexokinase isoenzymes in yeast. Isolation and characterization of a modified hexokinase PII isoenzyme. Eur. J. Biochem. 1985, 146, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, M.; Murakami, K. Analysis of the substrate inhibition of complete and partial types. Springerplus 2015, 4, 292. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ishikawa, H.; Ogino, H.; Oshida, H. Rates of reactions catalysed by a dimeric enzyme. Effects of the reaction scheme and the kinetic parameters on co-operativity. Biochem. J. 1991, 280, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Vagin, A.; Lebedev, A. MoRDa, an automatic molecular replacement pipeline. Acta Cryst. A 2015, 71, 19. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on TM-score. Nucl. Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Rivas-Pardo, J.A.; Herrera-Morande, A.; Castro-Fernandez, V.; Fernandez, F.J.; Vega, M.C.; Guixé, V. Crystal Structure, SAXS and Kinetic Mechanism of Hyperthermophilic ADP-Dependent Glucokinase from Thermococcus litoralis reveal a Conserved Mechanism for Catalysis. PLoS ONE 2013, 8, e66687. [Google Scholar] [CrossRef] [PubMed]
- Mueller, U.; Förster, R.; Hellmig, M.; Huschmann, F.U.; Kastner, A.; Malecki, P.; Pühringer, S.; Röwer, M.; Sparta, K.; Steffien, M.; et al. The macromolecular crystallography beamlines at BESSY II of the Helmholtz-Zentrum Berlin: Current status and perspectives. Eur. Phys. J. Plus 2015, 130, 141–150. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.R. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Reinhard, L.; Mayerhofer, H.; Geerlof, A.; Mueller- Dieckmann, J.; Weiss, M.S. Optimization of protein buffer cocktails using Thermofluor. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2013, 69, 209–214. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| KlGlk1 (PDB 6R2N) | |
| Data Collection | |
| Space group | C 2 2 21 |
| Total reflections/Unique reflections | 139134 (13371) |
| Cell dimensions | 102.0 Å, 122.7 Å, 360.3 Å 900, 900, 900 |
| Wavelength (Å) | 0.9184 |
| Resolution (Å) | 49.08-2.596 (2.689-2.596) |
| Mean I/sigma (I) | 13.6 (1.7) |
| Completeness (%) | 99.58 (97.10) |
| Multiplicity | 2.0 (2.0) |
| CC1/2 | 98.6 (52.8) |
| Refinement | |
| Reflections used in refinement | 69952 (6762) |
| Reflections used for R-free | 1099 (107) |
| R-work | 0.2045 (0.3300) |
| R-free | 0.2396 (0.3345) |
| Number of Atoms/Molecules | |
| non-hydrogen atoms | 11273 |
| macromolecules | 11032 |
| ligands♦ | 45 |
| solvent | 196 |
| B-factors | |
| average B-factor (Å) | 76.69 |
| macromolecules (Å) | 77.06 |
| ligands (Å) | 86.48 |
| solvent (Å) | 53.66 |
| wilson B-factor (Å) | 53.71 |
| Ramachandran plot | |
| Ramachandran favored (%) | 96.88 |
| Ramachandran allowed (%) | 3.05 |
| Ramachandran outliers (%) | 0.07 |
| R. m. s. deviations | |
| Bonds (Å) | 0.004 |
| Angles (°) | 0.96 |
| Rotamer outliers (%) | 0.92 |
| Clashscore | 3.08 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zak, K.M.; Kalińska, M.; Wątor, E.; Kuśka, K.; Krutyhołowa, R.; Dubin, G.; Popowicz, G.M.; Grudnik, P. Crystal Structure of Kluyveromyces lactis Glucokinase (KlGlk1). Int. J. Mol. Sci. 2019, 20, 4821. https://doi.org/10.3390/ijms20194821
Zak KM, Kalińska M, Wątor E, Kuśka K, Krutyhołowa R, Dubin G, Popowicz GM, Grudnik P. Crystal Structure of Kluyveromyces lactis Glucokinase (KlGlk1). International Journal of Molecular Sciences. 2019; 20(19):4821. https://doi.org/10.3390/ijms20194821
Chicago/Turabian StyleZak, Krzysztof M., Magdalena Kalińska, Elżbieta Wątor, Katarzyna Kuśka, Rościsław Krutyhołowa, Grzegorz Dubin, Grzegorz M. Popowicz, and Przemysław Grudnik. 2019. "Crystal Structure of Kluyveromyces lactis Glucokinase (KlGlk1)" International Journal of Molecular Sciences 20, no. 19: 4821. https://doi.org/10.3390/ijms20194821
APA StyleZak, K. M., Kalińska, M., Wątor, E., Kuśka, K., Krutyhołowa, R., Dubin, G., Popowicz, G. M., & Grudnik, P. (2019). Crystal Structure of Kluyveromyces lactis Glucokinase (KlGlk1). International Journal of Molecular Sciences, 20(19), 4821. https://doi.org/10.3390/ijms20194821