Role of GOLPH3 and TPX2 in Neuroblastoma DNA Damage Response and Cell Resistance to Chemotherapy


Abstract
1. Introduction
2. Results
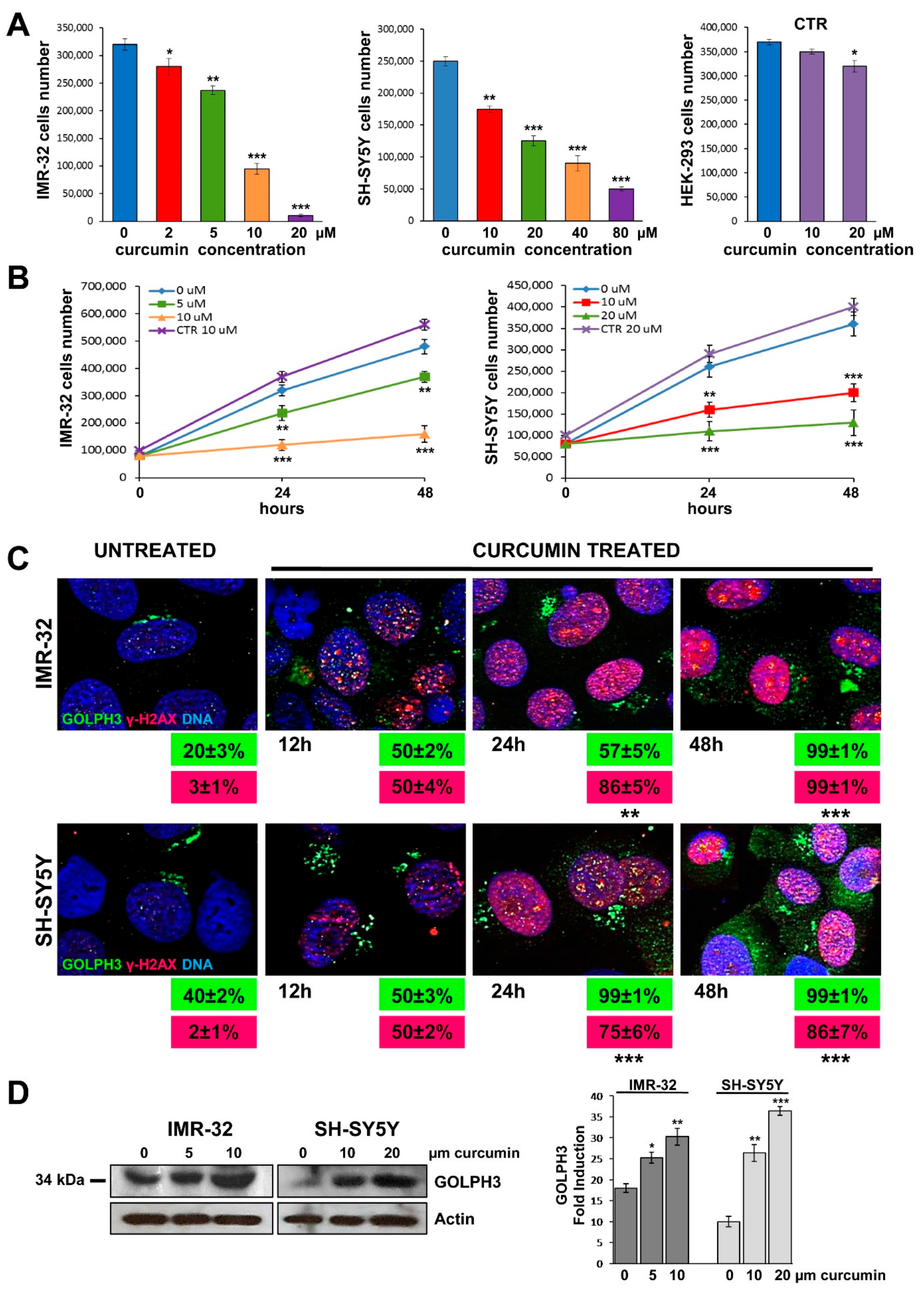
2.1. Cytotoxicity Induced by Curcumin
2.2. Curcumin Induces DNA Damage in Neuroblastoma Cells
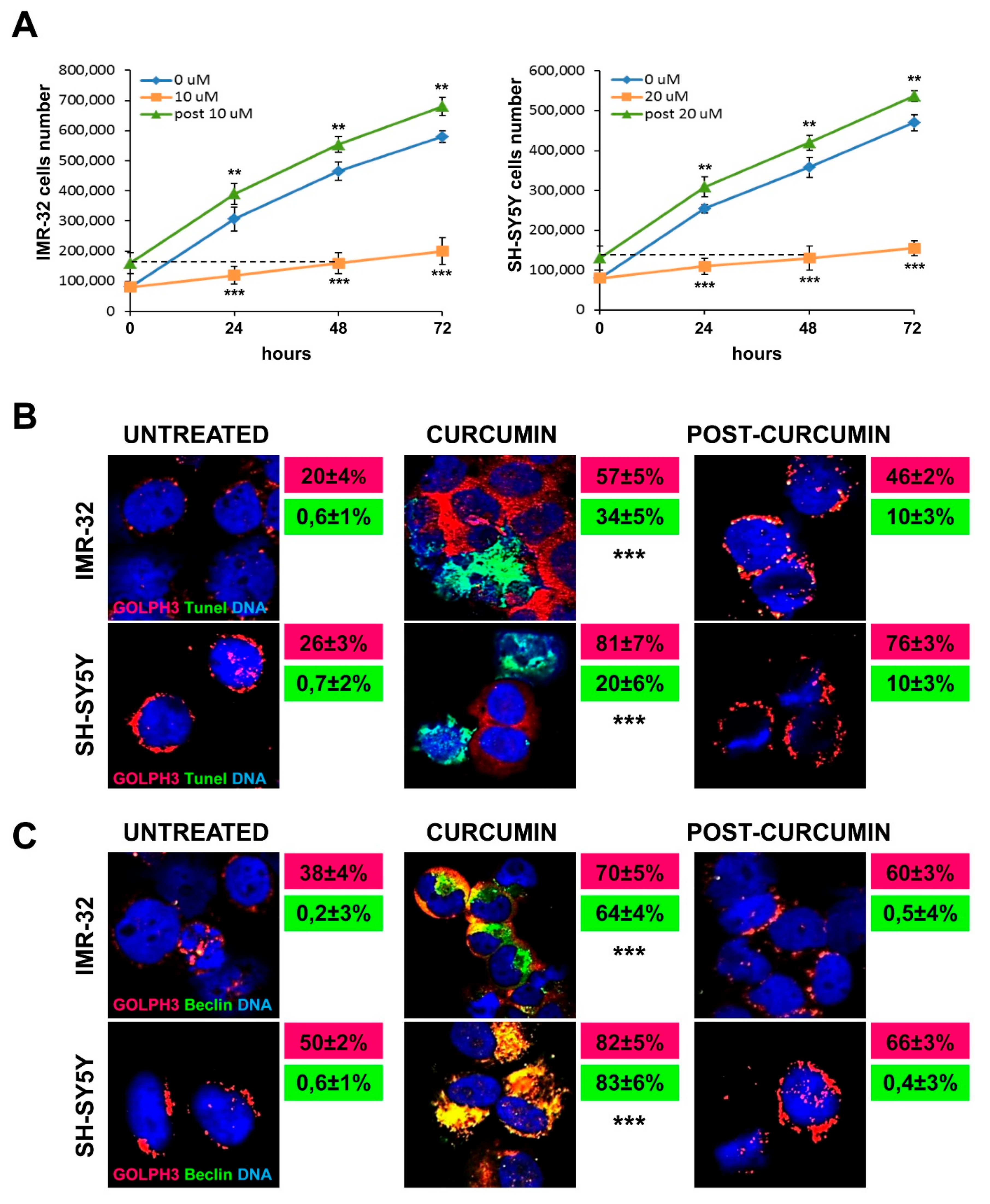
2.3. Curcumin Promotes GOLPH3 Up-Regulation and Dispersal of Golgi in Neuroblastoma Cells
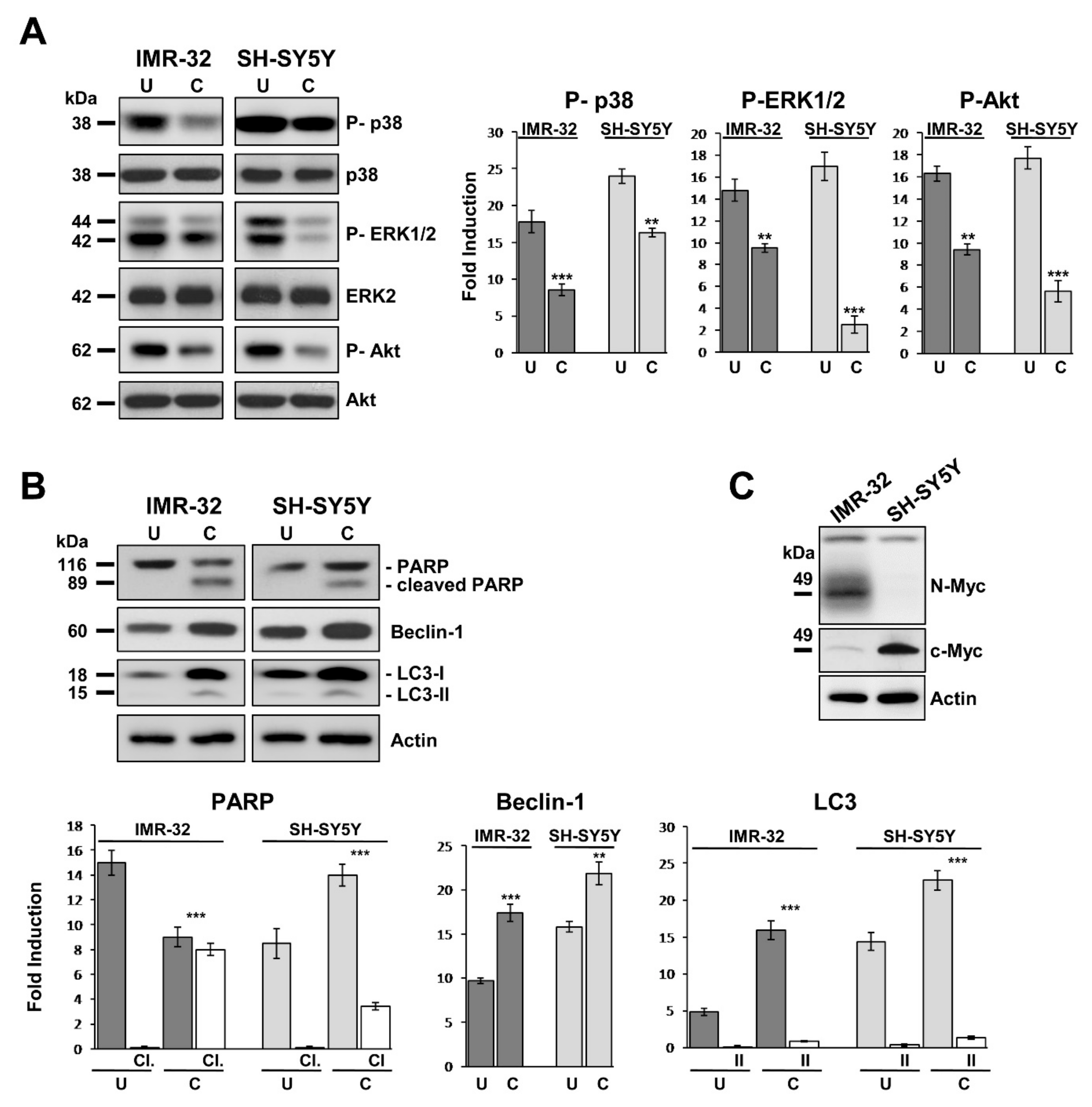
2.4. Curcumin Reduces Activation of MAP-Kinases and Induces Apoptosis and Autophagy in Neuroblastoma Cells
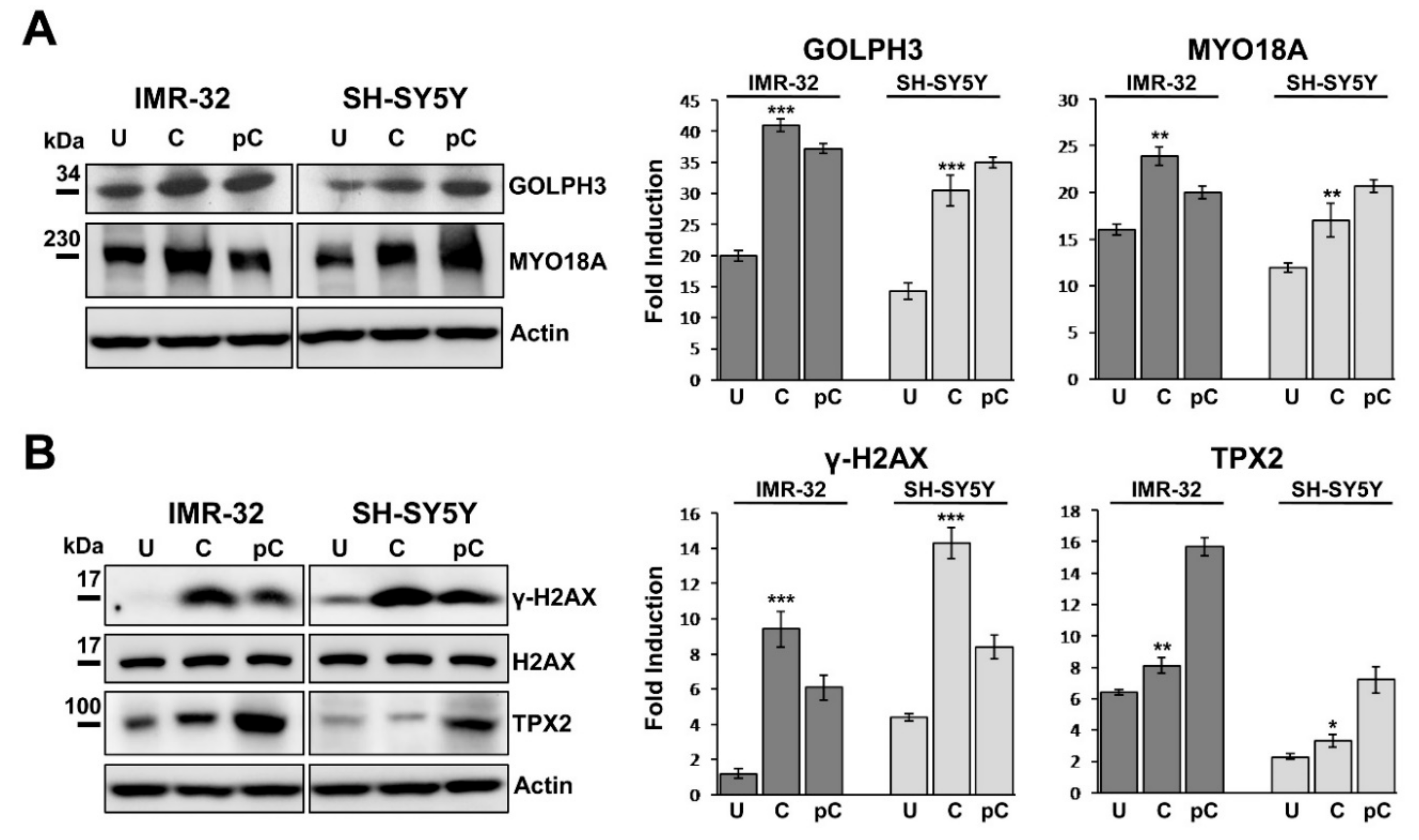
2.5. GOLPH3 Up-Regulation is Related With Higher Expression of MYO18A and TPX2 in Neuroblastoma Cells
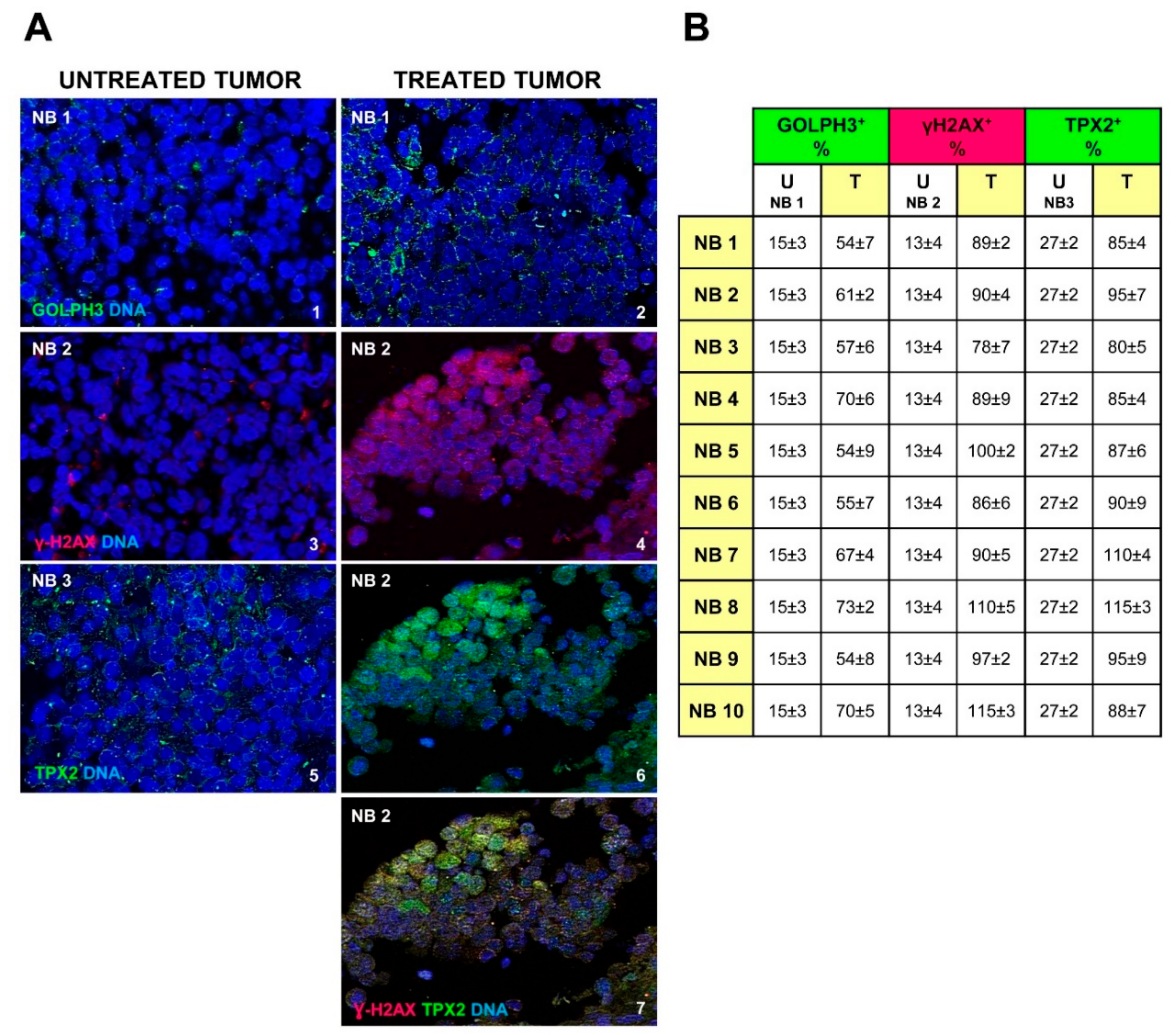
2.6. Chemotherapy Increases GOLPH3, γH2AX and TPX2 Expression in Neuroblastoma Cells
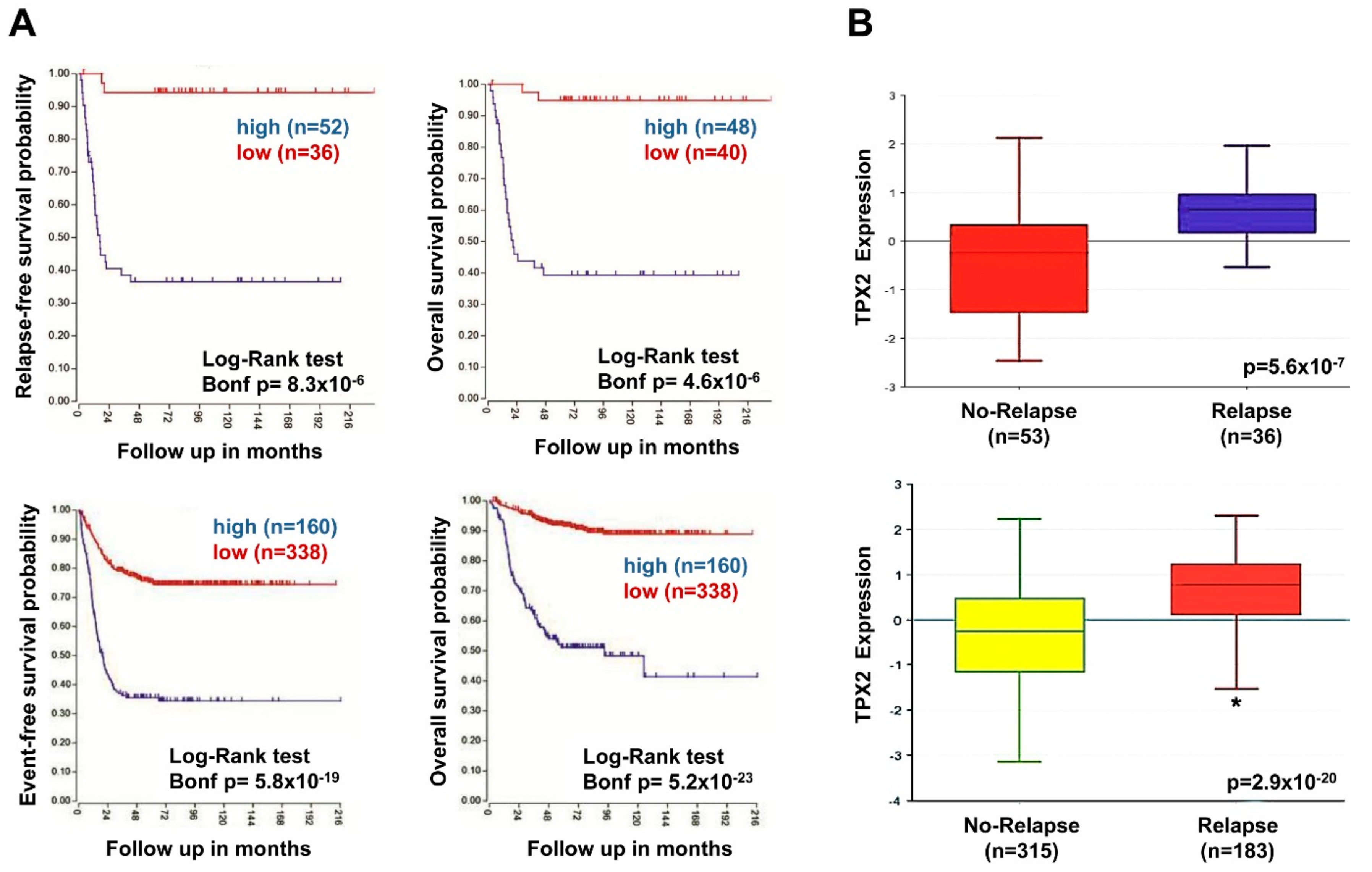
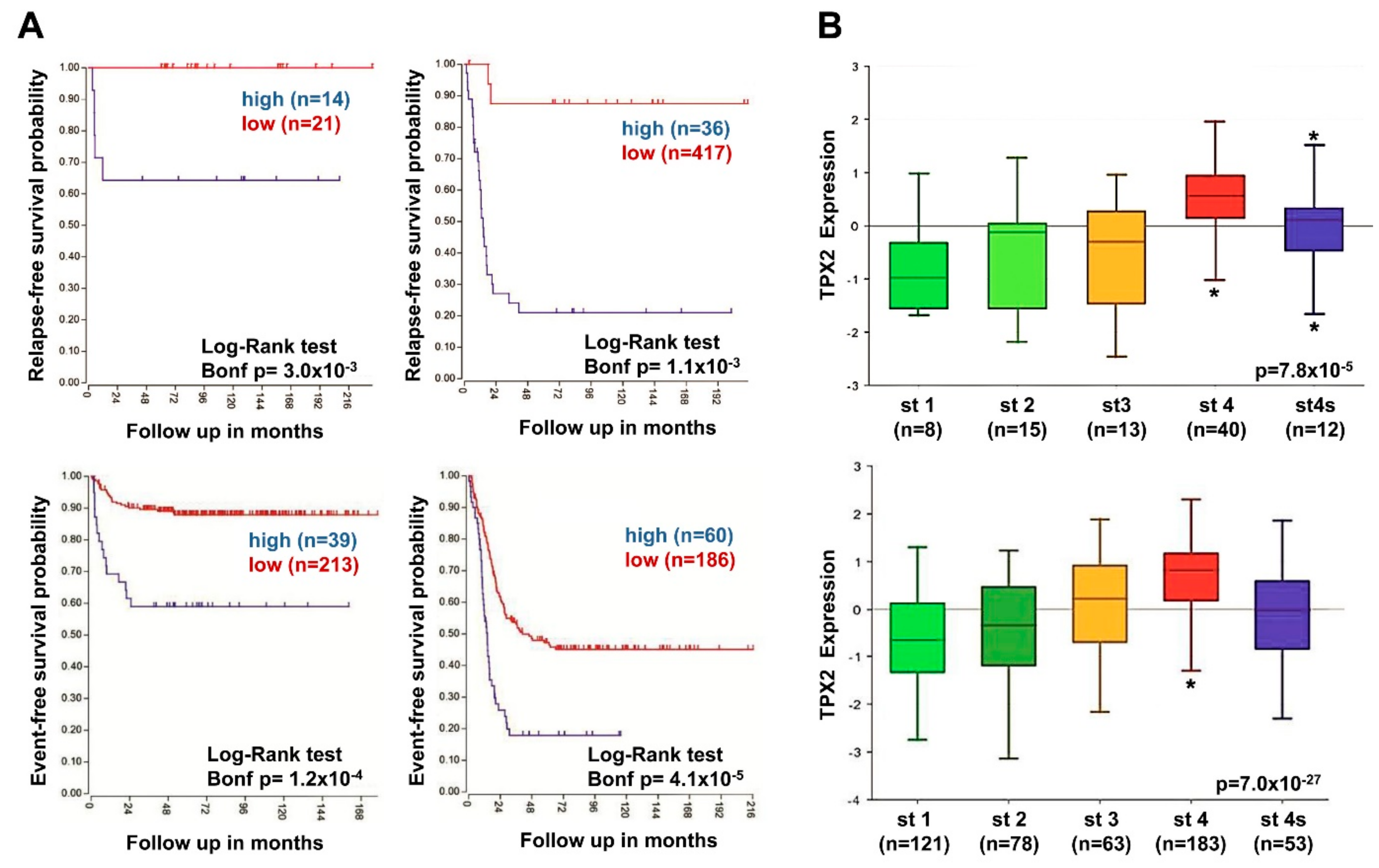
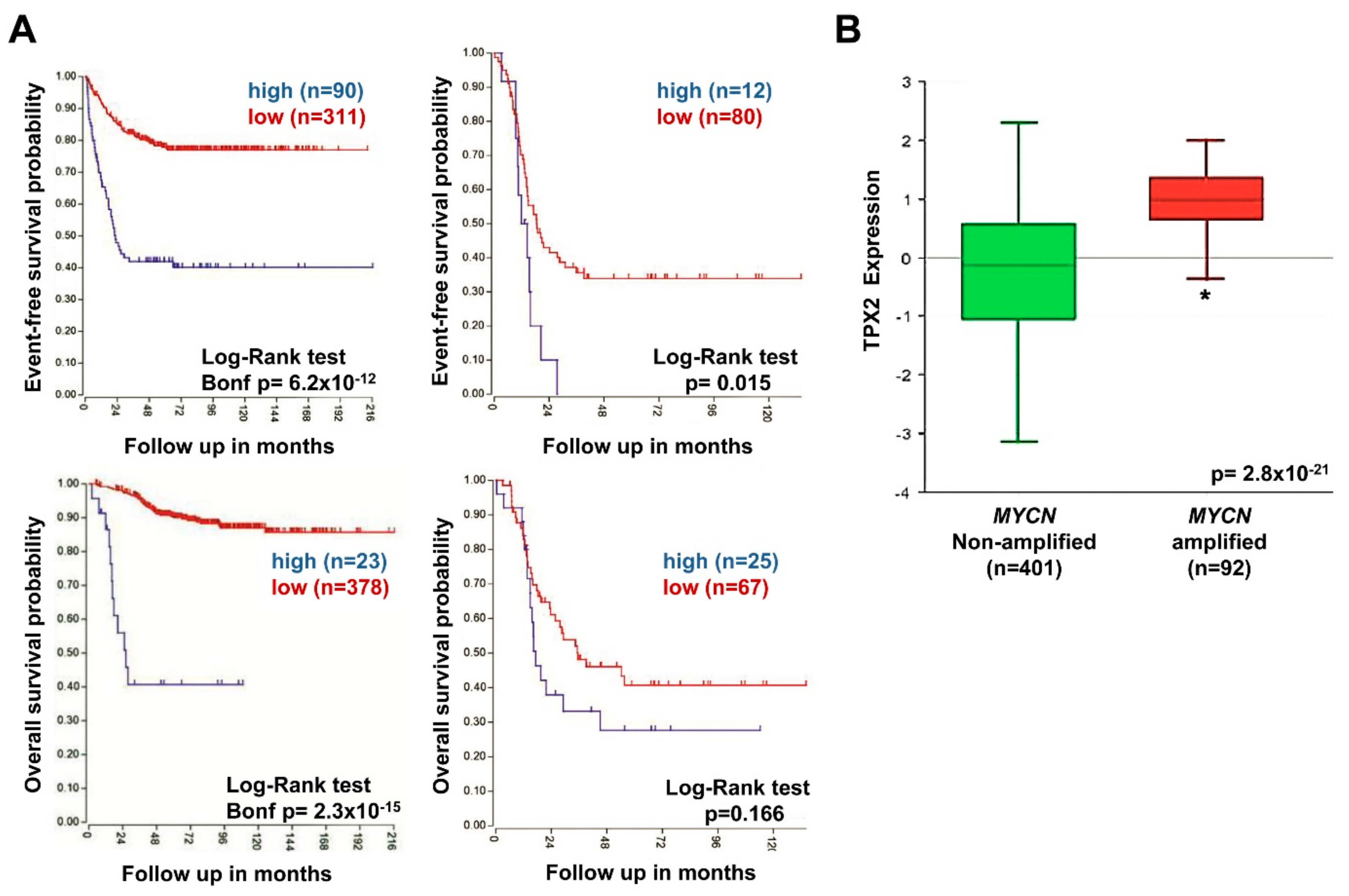
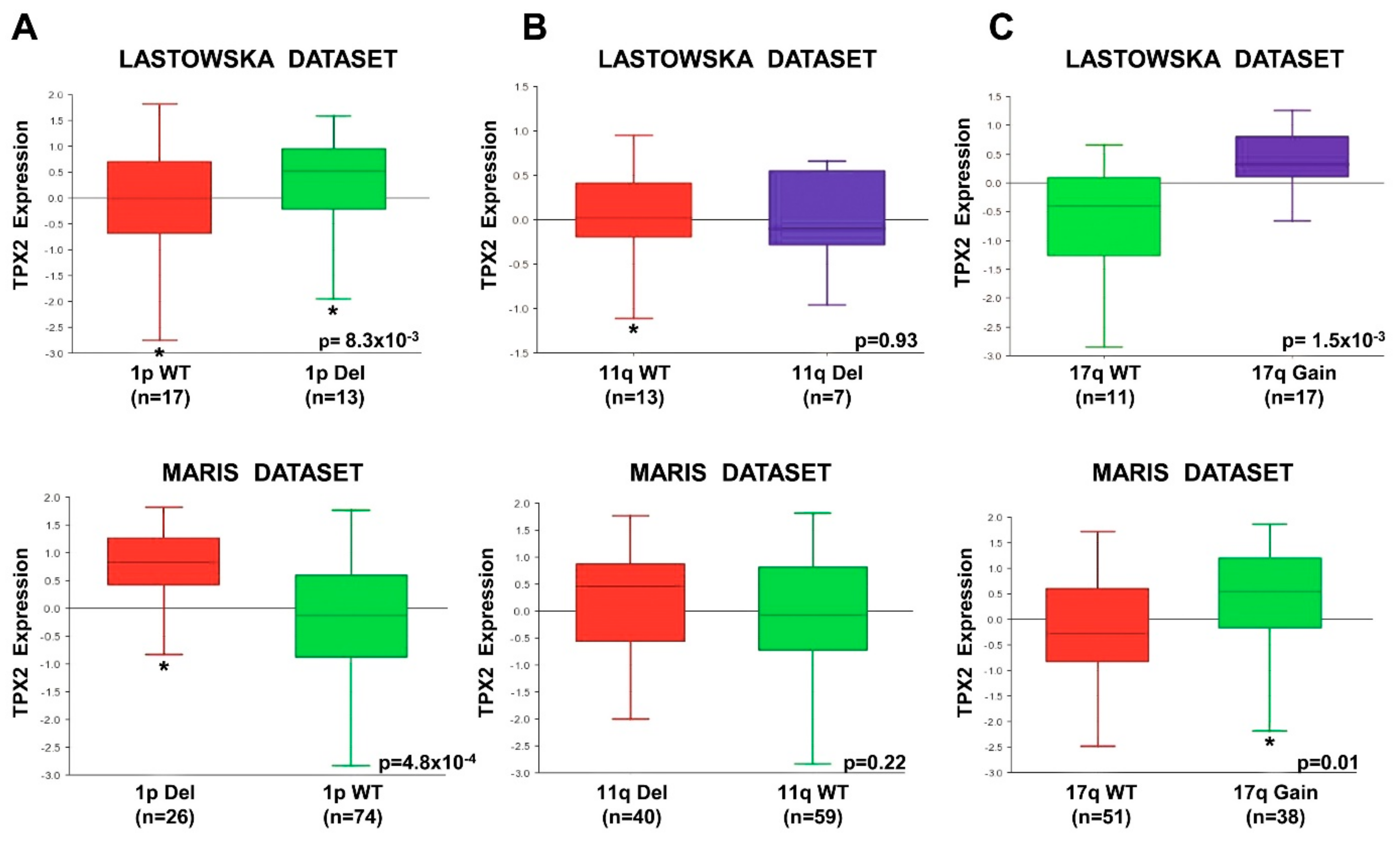
2.7. High TPX2 Gene Expression is Significantly Associated With Poor Prognosis in Neuroblastoma Patients
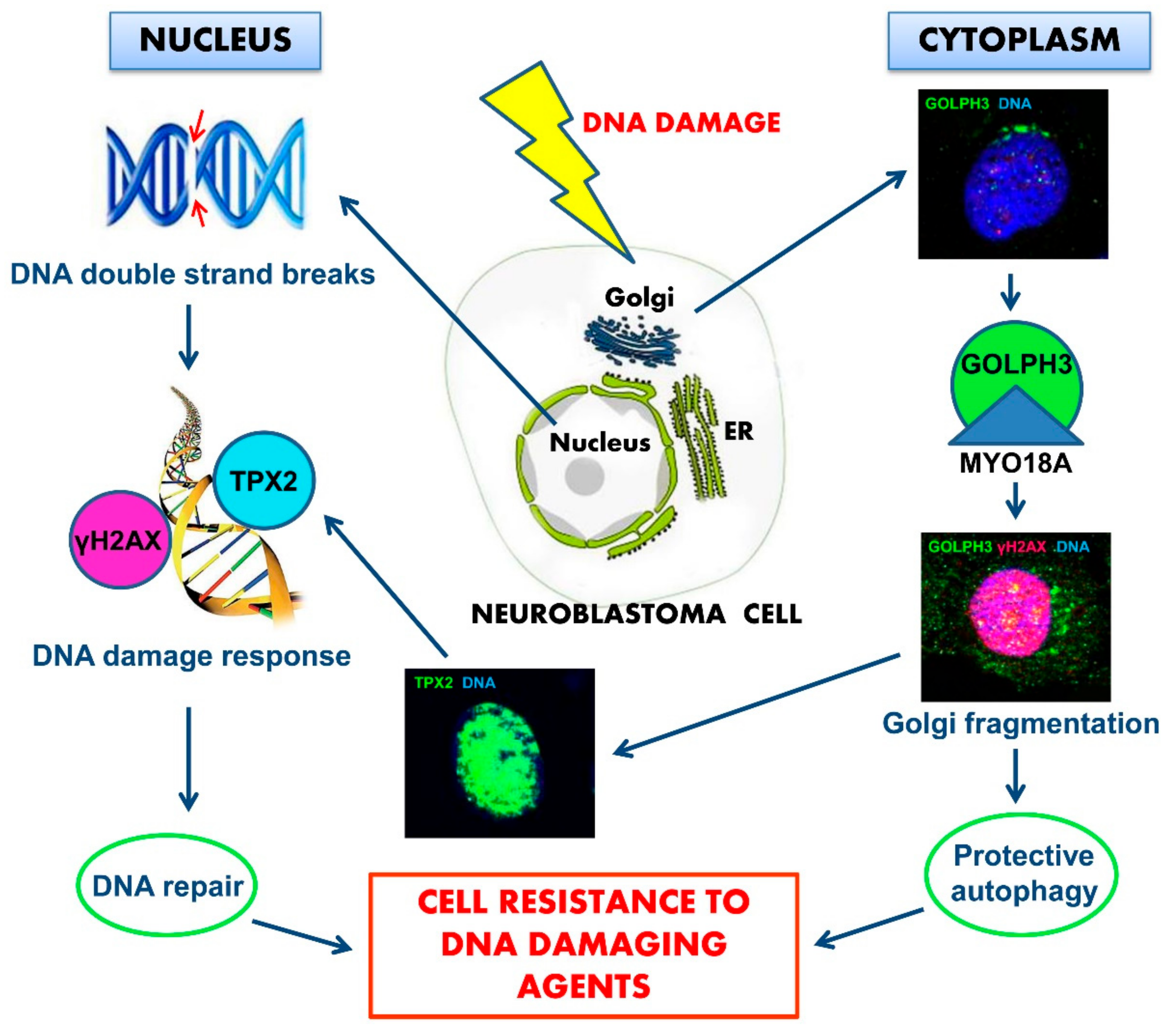
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Curcumin and Cells Growth
4.3. Western Blot Analysis
4.4. Kinase Activation Assay
4.5. Patients and Tumor Samples
4.6. Immunofluorescence Analysis
4.7. Detection of Apoptotic Cells
4.8. Genomic Profile Determination
4.9. TPX2 Gene Expression Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NB | Neuroblastoma |
| MYCN | v-myc myelocytomatosis viral related oncogene, neuroblastoma derived |
| GOLPH3 | Golgi phosphoprotein 3 |
| MYO18A | Myosin XVIIIA |
| H2AX | H2A histone family, member X |
| TPX2 | microtubule-associated, homolog (Xenopus laevis) |
| DNA-PK | DNA dependent protein kinase complex |
| MAPKs | mitogen-activated protein kinases |
| p38 | p38 MAPK |
| ERK1/2 | extracellular signal-regulated kinase 1 and 2 |
| Akt | RAC-alpha serine-threonine-protein kinase |
| Ras | Rat Sarcoma family of GTP-ases |
| GAP | GTPase-activating protein |
| PARP | Poly ADP-Ribose Polymerase |
| LC3A | Microtubule Associated Protein 1 Light Chain 3 Alpha |
| ER | endoplasmic reticulum |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| CGH | Comparative Genomic Hybridization |
| INSS | International Neuroblastoma Staging System |
| INRG | International Neuroblastoma Risk Group |
| SIOPEN | International Society of Pediatric Oncology European Neuroblastoma |
| AIEOP | Italian Association of Pediatric Hematology and Oncology |
Appendix A
Rapamycin, Bortezomib, and Colchicine Treatments
References
- Cheung, N.K.; Dyer, M.A. Neuroblastoma: Developmental biology, cancer genomics and immunotherapy. Nat. Rev. Cancer 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Speleman, F.; Park, J.R.; Henderson, T.O. Neuroblastoma: A Tough Nut to Crack. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, e548–e557. [Google Scholar] [CrossRef] [PubMed]
- Bosse, K.R.; Maris, J.M. Advances in the translational genomics of neuroblastoma: From improving risk stratification and revealing novel biology to identifying actionable genomic alterations. Cancer 2016, 122, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef]
- Cohn, S.L.; Pearson, A.D.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. INRG Task Force. The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Monclair, T.; Brodeur, G.M.; Ambros, P.F.; Brisse, H.J.; Cecchetto, G.; Holmes, K.; Kaneko, M.; London, W.B.; Matthay, K.K.; Nuchtern, J.G.; et al. INRG Task Force. The International Neuroblastoma Risk Group (INRG) staging system: An INRG Task Force report. J. Clin. Oncol. 2009, 27, 298–303. [Google Scholar] [CrossRef]
- Louis, C.U.; Shohet, J.M. Neuroblastoma: Molecular pathogenesis and therapy. Annu. Rev. Med. 2015, 66, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Ni Chonghaile, T.; Sarosiek, K.A.; Vo, T.T.; Ryan, J.A.; Tammareddi, A.; Moore, V.G.; Deng, J.; Anderson, K.C.; Richardson, P.; Tai, Y.T.; et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science 2011, 334, 1129–1133. [Google Scholar] [CrossRef]
- Keshelava, N.; Zuo, J.J.; Waidyaratne, N.S.; Triche, T.J.; Reynolds, C.P. p53 mutations and loss of p53 function confer multidrug resistance in neuroblastoma. Med. Pediatr. Oncol. 2000, 35, 563–568. [Google Scholar] [CrossRef]
- Khalil, M.A.; Hrabeta, J.; Cipro, S.; Stiborova, M.; Vicha, A.; Eckschlager, T. Neuroblastoma stem cells-mechanisms of chemoresistance and histone deacetylase inhibitors. Neoplasma 2012, 59, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.A.; Hraběta, J.; Groh, T.; Procházka, P.; Doktorová, H.; Eckschlager, T. Valproic Acid Increases CD133 Positive Cells that Show Low Sensitivity to Cytostatics in Neuroblastoma. PLoS ONE 2016, 11, e0162916. [Google Scholar] [CrossRef] [PubMed]
- Bresler, S.C.; Weiser, D.A.; Huwe, P.J.; Park, J.H.; Krytska, K.; Ryles, H.; Laudenslager, M.; Rappaport, E.F.; Wood, A.C.; McGrady, P.W.; et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 2014, 26, 682–694. [Google Scholar] [CrossRef] [PubMed]
- Murakami-Tonami, Y.; Ikeda, H.; Yamagishi, R.; Inayoshi, M.; Inagaki, S.; Kishida, S.; Komata, Y.; Koster, J.; Takeuchi, I.; Kondo, Y.; et al. SGO1 is involved in the DNA damage response in MYCN-amplified neuroblastoma cells. Sci. Rep. 2016, 6, 31615. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Dippold, H.C.; Ng, M.M.; Farber-Katz, S.E.; Lee, S.K.; Kerr, M.L.; Peterman, M.C.; Sim, R.; Wiharto, P.A.; Galbraith, K.A.; Madhavarapu, S.; et al. GOLPH3 bridges phosphatidylinositol-4- phosphate and actomyosin to stretch and shape the Golgi to promote budding. Cell 2009, 139, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Bishé, B.; Syed, G.H.; Field, S.J.; Siddiqui, A. Role of phosphatidylinositol 4-phosphate (PI4P) and its binding protein GOLPH3 in hepatitis C virus secretion. J. Biol. Chem. 2012, 287, 27637–27647. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.M.; Dippold, H.C.; Buschman, M.D.; Noakes, C.J.; Field, S.J. GOLPH3L antagonizes GOLPH3 to determine Golgi morphology. Mol. Biol. Cell 2013, 24, 796–808. [Google Scholar] [CrossRef]
- Makowski, S.L.; Tran, T.T.; Field, S.J. Emerging themes of regulation at the Golgi. Curr. Opin. Cell Biol. 2017, 45, 17–23. [Google Scholar] [CrossRef]
- Buschman, M.D.; Field, S.J. MYO18A: An unusual myosin. Adv. Biol. Regul. 2018, 67, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Rahajeng, J.; Kuna, R.S.; Makowski, S.L.; Tran, T.T.T.; Buschman, M.D.; Li, S.; Cheng, N.; Ng, M.M.; Field, S.J. Efficient Golgi Forward Trafficking Requires GOLPH3-Driven, PI4P-Dependent Membrane Curvature. Dev. Cell. 2019, 19, 30451–30454. [Google Scholar] [CrossRef] [PubMed]
- Farber-Katz, S.E.; Dippold, H.C.; Buschman, M.D.; Peterman, M.C.; Xing, M.; Noakes, C.J.; Tat, J.; Ng, M.M.; Rahajeng, J.; Cowan, D.M.; et al. DNA damage triggers Golgi dispersal via DNA-PK and GOLPH3. Cell 2014, 156, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.L.; Chin, L. Signaling from the Golgi: Mechanisms and models for Golgi phosphoprotein 3-mediated oncogenesis. Clin. Cancer Res. 2010, 16, 2229–2234. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.L.; Kabbarah, O.; Liang, M.C.; Ivanova, E.; Anagnostou, V.; Wu, J.; Dhakal, S.; Wu, M.; Chen, S.; Feinberg, T.; et al. GOLPH3 modulates mTOR signalling and rapamycin sensitivity in cancer. Nature 2009, 459, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Petrosyan, A. Onco-Golgi: Is Fragmentation a Gate to Cancer Progression? Biochem. Mol. Biol. J. 2015, 1, 1–16. [Google Scholar] [CrossRef]
- Zhou, B.; Wang, G.; Gao, S.; Chen, Y.; Jin, C.; Wang, Z.; Yang, Y.; Ma, Z.; Zhang, W.; Feng, X. Expression of GOLPH3 protein in colon cancer tissues and its association with the prognosis of patients. Oncol. Lett. 2016, 12, 3936–3940. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell. 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Zeng, Z.; Lin, H.; Zhao, X.; Liu, G.; Wang, X.; Xu, R.; Chen, K.; Li, J.; Song, L. Overexpression of GOLPH3 promotes proliferation and tumorigenicity in breast cancer via suppression of the FOXO1 transcription factor. Clin. Cancer Res. 2012, 18, 4059–4069. [Google Scholar] [CrossRef]
- Kunigou, O.; Nagao, H.; Kawabata, N.; Ishidou, Y.; Nagano, S.; Maeda, S.; Komiya, S.; Setoguchi, T. Role of GOLPH3 and GOLPH3L in the proliferation of human rhabdomyosarcoma. Oncol. Rep. 2011, 26, 1337–1342. [Google Scholar] [CrossRef]
- Zhang, L.J.; Wang, K.B.; Liu, L.S.; Chen, L.Z.; Peng, B.G.; Liang, L.J.; Li, Z.; Xue, L.; Li, W.; Xia, J.T. Overexpression of GOLPH3 is associated with poor prognosis and clinical progression in pancreatic ductal adenocarcinoma. BMC Cancer 2014, 14, 571–577. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, Q.; Zhuang, J.; Deng, Y.; Zhao, X.; Tang, B.; Yao, D.; Zhao, W.; Chang, C.; Lu, Q.; Chen, W.; et al. GOLPH3 is a potential therapeutic target and a prognostic indicator of poor survival in bladder cancer treated by cystectomy. Oncotarget 2015, 6, 32177–32192. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Wu, G.; Liao, Y.; Xiao, G.; Ma, X.; Zou, X.; Zhang, G.; Xiao, R.; Wang, X.; Liu, Q.; et al. GOLPH3 is a novel marker of poor prognosis and a potential therapeutic target in human renal cell carcinoma. Br. J. Cancer 2014, 110, 2250–2260. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, X.; Wu, Y.; Sun, B.; Lv, H.; Rong, F.; Zheng, X. Overexpression of GOLPH3 protein is associated with worse prognosis in patients with epithelial ovarian cancer. Tumour Biol. 2014, 35, 11845–11849. [Google Scholar] [CrossRef] [PubMed]
- Buschman, M.D.; Rahajeng, J.; Field, S.J. GOLPH3 links the Golgi, DNA damage, and cancer. Cancer Res. 2015, 75, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Buschman, M.D.; Xing, M.; Field, S.J. The GOLPH3 pathway regulates Golgi shape and function and is activated by DNA damage. Front. Neurosci. 2015, 9, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Neumayer, G.; Belzil, C.; Gruss, O.J.; Nguyen, M.D. TPX2: Of spindle assembly, DNA damage response, and cancer. Cell Mol. Life. Sci. 2014, 71, 3027–3047. [Google Scholar] [CrossRef] [PubMed]
- Neumayer, G.; Nguyen, M.D. TPX2 impacts acetylation of histone H4 at lysine 16: Implications for DNA damage response. PLoS ONE 2014, 9, e110994. [Google Scholar] [CrossRef]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Portoles, C.; Bird, A.W.; Hyman, A.; Cañamero, M.; Pérez de Castro, I.; Malumbres, M. Tpx2 controls spindle integrity, genome stability, and tumor development. Cancer Res. 2012, 72, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.C.; Zhang, Y.; Wang, X.L.; Qin, W.S.; Liu, Y.H.; Zhang, L.; Zhu, C.L. Upregulation of the TPX2 gene is associated with enhanced tumor malignance of esophageal squamous cell carcinoma. Biomed. Pharmacother. 2013, 67, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Wang, J.; Tian, Y.; Xu, J.; Gou, X.; Cheng, J. The TPX2 gene is a promising diagnostic and therapeutic target for cervical cancer. Oncol. Rep. 2012, 27, 1353–1359. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Zheng, B.; Li, J.; Lv, X.; Zhang, H.; Yu, F.; Kong, L.; Li, Y.; Yu, M.; Fang, L.; et al. TPX2 level correlates with cholangiocarcinoma cell proliferation, apoptosis, and EMT. Biomed. Pharmacother. 2018, 107, 1286–1293. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zuo, J.; Wei, Y. Targeting of TPX2 by miR-330-3p in melanoma inhibits proliferation. Biomed. Pharmacother. 2018, 107, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Huang, R.Y.; Jiang, F.N.; Chen, D.X.; Wang, C.; Han, Z.D.; Liang, Y.X.; Zhong, W.D. Overexpression of TPX2 is associated with progression and prognosis of prostate cancer. Oncol. Lett. 2018, 16, 2823–2832. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Zhang, N.; Xu, Y.; Li, X.; Shi, D.; Wang, Y.; Li, D.; Cai, S. TPX2 is a novel prognostic marker for the growth and metastasis of colon cancer. J. Transl. Med. 2013, 11, 313–315. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Capetillo, O.; Lee, A.; Nussenzweig, M.; Nussenzweig, A. H2AX: The histone guardian of the genome. DNA Repair. 2004, 3, 959–967. [Google Scholar] [CrossRef]
- Ivashkevich, A.N.; Martin, O.A.; Smith, A.J.; Redon, C.E.; Bonner, W.M.; Martin, R.F.; Lobachevsky, P.N. γH2AX foci as a measure of DNA damage: A computational approach to automatic analysis. Mutat. Res. 2011, 711, 49–60. [Google Scholar] [CrossRef]
- Marimpietri, D.; Brignole, C.; Nico, B.; Pastorino, F.; Pezzolo, A.; Piccardi, F.; Cilli, M.; Di Paolo, D.; Pagnan, G.; Longo, L.; et al. Combined therapeutic effects of vinblastine and rapamycin on human neuroblastoma growth, apoptosis, and angiogenesis. Clin. Cancer Res. 2007, 13, 3977–3988. [Google Scholar] [CrossRef]
- Brignole, C.; Marimpietri, D.; Pastorino, F.; Nico, B.; Di Paolo, D.; Cioni, M.; Piccardi, F.; Cilli, M.; Pezzolo, A.; Corrias, M.V.; et al. Effect of bortezomib on human neuroblastoma cell growth, apoptosis, and angiogenesis. J. Natl. Cancer Inst. 2006, 98, 1142–1157. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa-Yagi, Y. Induction of apoptotic cell death in differentiating neuroblastoma SH-SY5Y cells by colchicine. Biochem Biophys Res Commun. 1994, 199, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Neumayer, G.; Helfricht, A.; Shim, S.Y.; Le, H.T.; Lundin, C.; Belzil, C.; Chansard, M.; Yu, Y.; Lees-Miller, S.P.; Gruss, O.J.; et al. Targeting protein for xenopus kinesin-like protein 2 (TPX2) regulates γ-histone 2AX (γ-H2AX) levels upon ionizing radiation. J. Biol. Chem. 2012, 287, 42206–42222. [Google Scholar] [CrossRef] [PubMed]
- Janoueix-Lerosey, I.; Schleiermacher, G.; Michels, E.; Mosseri, V.; Ribeiro, A.; Lequin, D.; Vermeulen, J.; Couturier, J.; Peuchmaur, M.; Valent, A.; et al. Overall genomic pattern is a predictor of outcome in neuroblastoma. J. Clin. Oncol. 2009, 27, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.W.; Liu, Y.; He, S.; Durbin, A.D.; Abraham, B.J.; Easton, J.; Shao, Y.; Xu, B.; Zhu, S.; Zhang, X.; et al. MYC Drives a Subset of High-Risk Pediatric Neuroblastomas and Is Activated through Mechanisms Including Enhancer Hijacking and Focal Enhancer Amplification. Cancer Discov. 2018, 8, 320–335. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Cheung, N.K.; Vider, J.; Cheung, I.Y.; Gerald, W.L.; Tickoo, S.K.; Holland, E.C.; Blasberg, R.G. MYCN and MYC regulate tumor proliferation and tumorigenesis directly through BMI1 in human neuroblastomas. FASEB J. 2011, 25, 4138–4149. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Morandell, S.; Yaffe, M.B. Exploiting synthetic lethal interactions between DNA damage signaling, checkpoint control, and p53 for targeted cancer therapy. In Progress in Molecular Biology and Translational Science; Doetsch, P.V., Ed.; Academic Press: New York, NY, USA, 2012; Volume 110, pp. 289–314. [Google Scholar] [CrossRef]
- Shehzad, A.; Lee, Y.S. Molecular mechanisms of curcumin action: Signal transduction. BioFactors 2013, 39, 27–36. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Kumar, A.; Bharti, A.C. Anticancer potential of curcumin: Preclinical and clinical studies. Anticancer. Res. 2003, 23, 363–398. [Google Scholar]
- Epstein, J.; Sanderson, I.R.; Macdonald, T.T. Curcumin as a therapeutic agent: The evidence from in vitro, animal and human studies. Br. J. Nutr. 2010, 103, 1545–1557. [Google Scholar] [CrossRef] [PubMed]
- Freudlsperger, C.; Greten, J.; Schumacher, U. Curcumin induces apoptosis in human neuroblastoma cells via inhibition of NFkappaB. Anticancer Res. 2008, 28, 209–214. [Google Scholar] [PubMed]
- Ketron, A.C.; Gordon, O.N.; Schneider, C.; Osheroff, N. Oxidative metabolites of curcumin poison human type II topoisomerases. Biochemistry 2013, 52, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Song, X.; Shang, M.; Zou, W.; Zhang, M.; Wei, H.; Shao, H. Curcumin exerts cytotoxicity dependent on reactive oxygen species accumulation in non-small-cell lung cancer cells. Future Oncol. 2019, 15, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Zhou, Y.; Wen, C.; Zhou, C.; Zhang, W.; Hu, X.; Wang, L.; You, C.; Shao, J. Curcumin sensitizes glioblastoma to temozolomide by simultaneously generating ROS and disrupting AKT/mTOR signaling. Oncol. Rep. 2014, 32, 1610–1616. [Google Scholar] [CrossRef] [PubMed]
- Liontas, A.; Yeger, H. Curcumin and resveratrol induce apoptosis and nuclear translocation and activation of p53 in human neuroblastoma. Anticancer Res. 2004, 24, 987–998. [Google Scholar]
- Ting, C.Y.; Wang, H.E.; Yu, C.C.; Liu, H.C.; Liu, Y.C.; Chiang, I.T. Curcumin Triggers DNA Damage and Inhibits Expression of DNA Repair Proteins in Human Lung Cancer Cells. Anticancer Res. 2015, 35, 3867–3873. [Google Scholar]
- Ji, Z. Targeting DNA damage and repair by curcumin. Breast Cancer 2010, 4, 1–3. [Google Scholar]
- Blasiak, J.; Trzeciak, A.; Kowalik, J. Curcumin damages DNA in human gastric mucosa cells and lymphocytes. J. Environ. Pathol. Toxicol. Oncol. 1999, 18, 271–276. [Google Scholar]
- Cao, J.; Jia, L.; Zhou, H.M.; Liu, Y.; Zhong, L.F. Mitochondrial and nuclear DNA damage induced by curcumin in human hepatoma G2 cells. Toxicol. Sci. 2006, 91, 476–483. [Google Scholar] [CrossRef]
- Lu, J.J.; Cai, Y.J.; Ding, J. Curcumin induces DNA damage and caffeine-insensitive cell cycle arrest in colorectal carcinoma HCT116 cells. Mol. Cell. Biochem. 2011, 354, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Rowe, D.L.; Ozbay, T.; O’Regan, R.M.; Nahta, R. Modulation of the BRCA1 Protein and Induction of Apoptosis in Triple Negative Breast Cancer Cell Lines by the Polyphenolic Compound Curcumin. Breast Cancer 2009, 3, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, J.; Prasad, S.; Aggarwal, B.B. Curcumin and cancer cells: How many ways can curry kill tumor cells selectively? AAPS J. 2009, 11, 495–510. [Google Scholar] [CrossRef] [PubMed]
- Jaroonwitchawan, T.; Chaicharoenaudomrung, N.; Namkaew, J.; Noisa, P. Curcumin attenuates paraquat-induced cell death in human neuroblastoma cells through modulating oxidative stress and autophagy. Neurosci. Lett. 2017, 636, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Han, L.; Weng, J.; Wang, K.; Chen, T. Rapamycin inhibits proliferation and induces autophagy in human neuroblastoma cells. Biosci. Rep. 2018, 38, 6. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhang, J.; Jin, Y.; Yang, Y.; Shi, J.; Chen, F.; Han, S.; Chu, P.; Lu, J.; Wang, H.; et al. MiR-20a-5p suppresses tumor proliferation by targeting autophagy-related gene 7 in neuroblastoma. Cancer Cell Int. 2018, 18, 5. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Xu, Q.; Zhang, Y.; Shen, M.; Zhang, S.; Mao, F.; Li, B.; Yan, X.; Shi, Z.; Wang, L.; et al. miR-34a inhibits progression of neuroblastoma by targeting autophagy-related gene 5. Eur. J. Pharmacol. 2019, 850, 53–63. [Google Scholar] [CrossRef]
- Shukla, N.; Ameur, N.; Yilmaz, I.; Nafa, K.; Lau, C.Y.; Marchetti, A.; Borsu, L.; Barr, F.G.; Ladanyi, M. Oncogene Mutation Profiling of Pediatric Solid Tumors Reveals Significant Subsets of Embryonal Rhabdomyosarcoma and Neuroblastoma With Mutated Genes in Growth Signaling Pathways. Clin. Cancer Res. 2011, 18, 748–757. [Google Scholar] [CrossRef]
- Eleveld, T.F.; Oldridge, D.A.; Bernard, V.; Koster, J.; Colmet Daage, L.; Diskin, S.J.; Schild, L.; Bentahar, N.B.; Bellini, A.; Chicard, M.; et al. Relapsed Neuroblastomas Show Frequent RAS-MAPK Pathway Mutations. Nat. Genet. 2015, 47, 864–873. [Google Scholar] [CrossRef]
- Holzel, M.; Huang, S.; Koster, J.; Ora, I.; Lakeman, A.; Caron, H.; Nijkamp, W.; Xie, J.; Callens, T.; Asgharzadeh, S.; et al. NF1 Is a Tumor Suppressor in Neuroblastoma that Determines Retinoic Acid Response and Disease Outcome. Cell 2010, 142, 218–229. [Google Scholar] [CrossRef]
- Woodfield, S.E.; Zhang, L.; Scorsone, K.; Liu, Y.; Zage, P.E. Binimetinib Inhibits MEK and is Effective Against Neuroblastoma Tumor Cells with Low NF1 Expression. BMC Cancer 2016, 16, 172. [Google Scholar] [CrossRef] [PubMed]
- Eleveld, T.F.; Schild, L.; Koster, J.; Zwijnenburg, D.A.; Alles, L.K.; Ebus, M.E.; Volckmann, R.; Tijtgat, G.A.; van Sluis, P.; Versteeg, R.; et al. RAS-MAPK Pathway-Driven Tumor Progression Is Associated with Loss of CIC and Other Genomic Aberrations in Neuroblastoma. Cancer Res. 2018, 78, 6297–6307. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.H.; Zhang, Z.C.; Wynn, R.M.; Seemann, J. GM130 Regulates Golgi-Derived Spindle Assembly by Activating TPX2 and Capturing Microtubules. Cell 2015, 162, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Sheridan, P.; Niida, A.; Sawada, G.; Uchi, R.; Mizuno, H.; Kurashige, J.; Sugimachi, K.; Sasaki, S.; Shimada, Y.; et al. The AURKA/TPX2 axis drives colon tumorigenesis cooperatively with MYC. Ann. Oncol. 2015, 26, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Shaw, G.; Morse, S.; Ararat, M.; Graham, F.L. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 2002, 16, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Gu, M.; Song, C.; Miao, L.; Hu, L.; Liang, D.; Zheng, C. The tumorigenicity diversification in human embryonic kidney 293 cell line cultured in vitro. Biologicals 2008, 36, 263–268. [Google Scholar] [CrossRef]
- Shehzad, A.; Lee, Y.S. Curcumin: Multiple molecular targets mediate multiple pharmacological actions: A review. Drugs Fut. 2010, 35, 113–119. [Google Scholar] [CrossRef]
- Park, J.R.; Bagatell, R.; Cohn, S.L.; Pearson, A.D.; Villablanca, J.G.; Berthold, F.; Burchill, S.; Boubaker, A.; McHugh, K.; Nuchtern, J.G.; et al. Revisions to the International neuroblastoma response criteria: A consensus statement from the National Cancer Institute clinical trials planning meeting. J. Clin. Oncol. 2017, 35, 2580–2587. [Google Scholar] [CrossRef]
- Ladenstein, R.; Valteau-Couanet, D.; Brock, P.; Yaniv, I.; Castel, V.; Laureys, G.; Malis, J.; Papadakis, V.; Lacerda, A.; Ruud, E.; et al. Randomized Trial of prophylactic granulocyte colony-stimulating factor during rapid COJEC induction in pediatric patients with high-risk neuroblastoma: The European HR-NBL1/SIOPEN study. J. Clin. Oncol. 2010, 28, 3516–3524. [Google Scholar] [CrossRef]
- Pezzolo, A.; Parodi, F.; Marimpietri, D.; Raffaghello, L.; Cocco, C.; Pistorio, A.; Mosconi, M.; Gambini, C.; Cilli, M.; Deaglio, S.; et al. Oct-4+/Tenascin C+ neuroblastoma cells serve as progenitors of tumor-derived endothelial cells. Cell Res. 2011, 21, 1470–1486. [Google Scholar] [CrossRef]
- Ferreira-Facio, C.S.; Milito, C.; Botafogo, V.; Fontana, M.; Thiago, L.S.; Oliveira, E.; da Rocha-Filho, A.S.; Werneck, F.; Forny, D.N.; Dekermacher, S.; et al. Contribution of multiparameter flow cytometry immunophenotyping to the diagnostic screening and classification of pediatric cancer. PLoS ONE 2013, 8, e55534. [Google Scholar] [CrossRef] [PubMed]
- Ognibene, M.; Pagnan, G.; Marimpietri, D.; Cangelosi, D.; Cilli, M.; Benedetti, M.C.; Boldrini, R.; Garaventa, A.; Frassoni, F.; Eva, A.; et al. CHL1 gene acts as a tumor suppressor in human neuroblastoma. Oncotarget 2018, 9, 25903–25921. [Google Scholar] [CrossRef] [PubMed]
- Parodi, S.; Pistorio, A.; Erminio, G.; Ognibene, M.; Morini, M.; Garaventa, A.; Gigliotti, A.R.; Haupt, R.; Frassoni, F.; Pezzolo, A. Loss of whole chromosome X predicts prognosis of neuroblastoma patients with numerical genomic profile. Pediatr. Blood. Cancer. 2019, 66, e27635. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yu, Y.; Hertwig, F.; Thierry-Mieg, J.; Thierry-Mieg, D.; Wang, J.; Furlanello, C.; Devanarayan, V.; Cheng, J.; Deng, Y.; et al. Comparison of RNA-seq and microarray-based models for clinical endpoint prediction. Genome Biol. 2015, 16, 133. [Google Scholar] [CrossRef] [PubMed]
- Valentijn, L.J.; Koster, J.; Haneveld, F.; Aissa, R.A.; van Sluis, P.; Broekmans, M.E.; Molenaar, J.J.; van Nes, J.; Versteeg, R. Functional MYCN signature predicts outcome of neuroblastoma irrespective of MYCN amplification. Proc. Natl. Acad. Sci. USA 2012, 109, 19190–19195. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, J.I.; Segerström, L.; Orrego, A.; Elfman, L.; Henriksson, M.; Kågedal, B.; Eksborg, S.; Sveinbjörnsson, B.; Kogner, P. Inhibitors of mammalian target of rapamycin downregulate MYCN protein expression and inhibit neuroblastoma growth in vitro and in vivo. Oncogene 2008, 27, 2910–2922. [Google Scholar] [CrossRef] [PubMed]
- Mody, R.; Zhao, L.; Yanik, G.A.; Opipari, V. Phase I study of bortezomib in combination with irinotecan in patients with relapsed/refractory high-risk neuroblastoma. Pediatr. Blood. Cancer 2019, 64, e26563. [Google Scholar] [CrossRef] [PubMed]
- Massarotti, A.; Theeramunkong, S.; Mesenzani, O.; Caldarelli, A.; Genazzani, A.A.; Tron, G.C. Identification of novel antitubulin agents by using a virtual screening approach based on a 7-point pharmacophore model of the tubulin colchi-site. Chem. Biol. Drug Des. 2011, 78, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Park, G.B.; Jin, D.H.; Kim, D. Sequential treatment with celecoxib and bortezomib enhances the ER stress-mediated autophagy-associated cell death of colon cancer cells. Oncol. Lett. 2018, 16, 4526–4536. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Das, A.; Datta, S.; Ganguli, A.; Chakrabarti, G. Colchicine induces autophagy and senescence in lung cancer cells at clinically admissible concentration: Potential use of colchicine in combination with autophagy inhibitor in cancer therapy. Tumor Biol. 2016, 37, 10653–10664. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Age Months | Stage INSS | Therapy Protocol | MYCN Status | Del 1p | Del 11q | Gain 17q | Chemotherapy (Rapid COJEC) | Immunotherapy (Anti-GD2) | Response to Chemotherapy | Relapse | Overall Survival |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NB 1 | 66 | 3 | HR-NBL-1 | Amplified | Y | Y | Y | Y | N | Partial | Y | Dead of disease |
| NB 2 | 70 | 3 | HR-NBL-1 | Amplified | N | N | Y | Y | N | Partial | Y | Dead of disease |
| NB 3 | 178 | 3 | HR-NBL-1 | Amplified | N | Y | Y | Y | N | Partial | Y | Dead of disease |
| NB 4 | 80 | 3 | HR-NBL-1 | Amplified | Y | Y | Y | Y | N | Partial | Y | Dead of disease |
| NB 5 | 53 | 4 | HR-NBL-1 | Amplified | Y | N | Y | Y | N | Partial | Y | Dead of disease |
| NB 6 | 20 | 4 | HR-NBL-1 | Amplified | Y | N | Y | Y | Y | Partial | Y | Dead of disease |
| NB 7 | 37 | 3 | HR-NBL-1 | Amplified | Y | Y | Y | Y | N | Partial | Y | Dead of disease |
| NB 8 | 65 | 4 | HR-NBL-1 | Non-Amplified | N | N | Y | Y | Y | Partial | Y | Dead of disease |
| NB 9 | 63 | 4 | HR-NBL-1 | Non-Amplified | N | Y | Y | Y | N | Partial | N | Dead of disease |
| NB10 | 7 | 4 | HR-NBL-1 | Amplified | Y | N | Y | Y | N | Partial | Y | Dead of disease |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ognibene, M.; Podestà, M.; Garaventa, A.; Pezzolo, A. Role of GOLPH3 and TPX2 in Neuroblastoma DNA Damage Response and Cell Resistance to Chemotherapy. Int. J. Mol. Sci. 2019, 20, 4764. https://doi.org/10.3390/ijms20194764
Ognibene M, Podestà M, Garaventa A, Pezzolo A. Role of GOLPH3 and TPX2 in Neuroblastoma DNA Damage Response and Cell Resistance to Chemotherapy. International Journal of Molecular Sciences. 2019; 20(19):4764. https://doi.org/10.3390/ijms20194764
Chicago/Turabian StyleOgnibene, Marzia, Marina Podestà, Alberto Garaventa, and Annalisa Pezzolo. 2019. "Role of GOLPH3 and TPX2 in Neuroblastoma DNA Damage Response and Cell Resistance to Chemotherapy" International Journal of Molecular Sciences 20, no. 19: 4764. https://doi.org/10.3390/ijms20194764
APA StyleOgnibene, M., Podestà, M., Garaventa, A., & Pezzolo, A. (2019). Role of GOLPH3 and TPX2 in Neuroblastoma DNA Damage Response and Cell Resistance to Chemotherapy. International Journal of Molecular Sciences, 20(19), 4764. https://doi.org/10.3390/ijms20194764