A Novel Prognostic DNA Methylation Panel for Colorectal Cancer

 , and
, and
Abstract
1. Introduction
2. Results
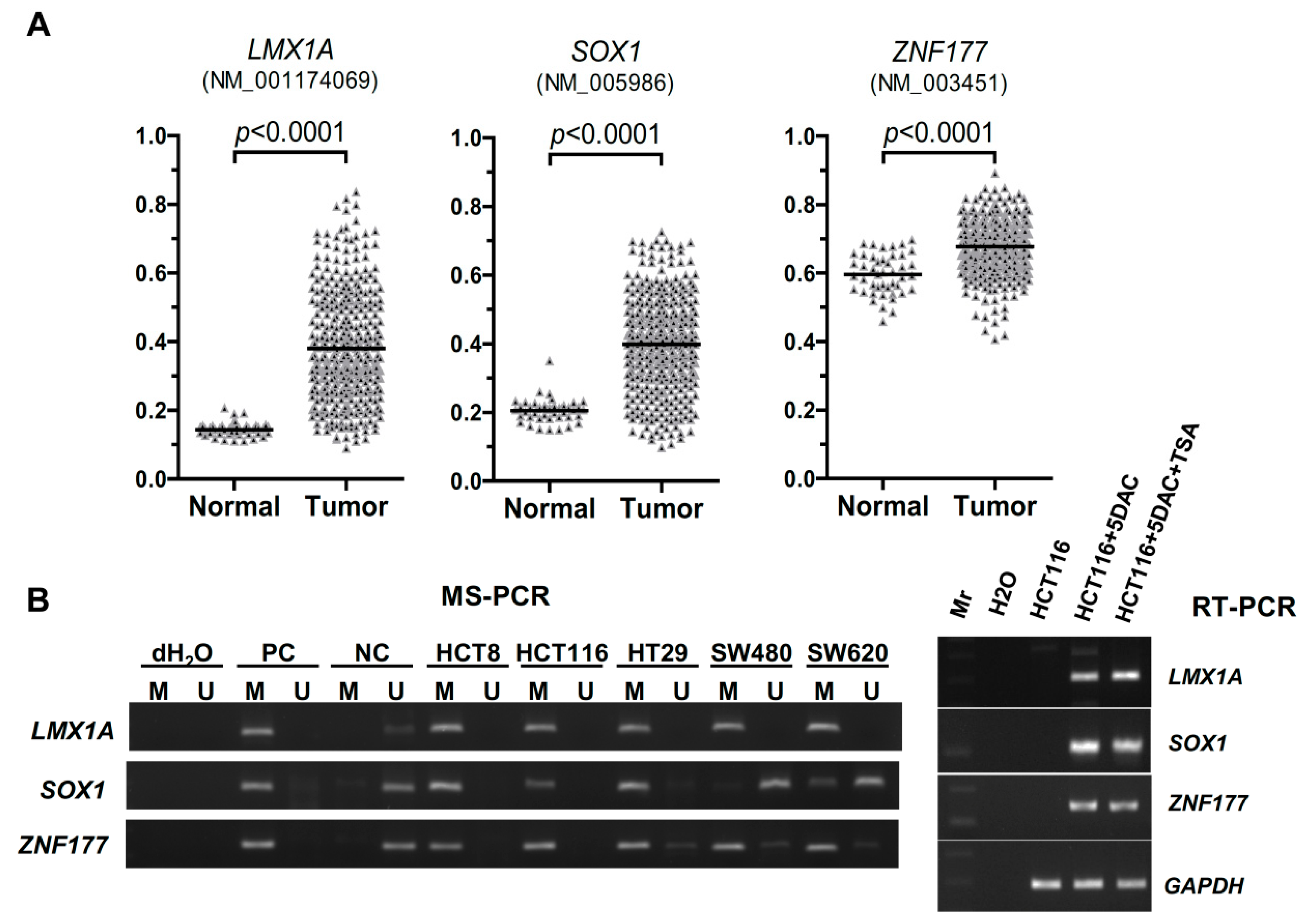
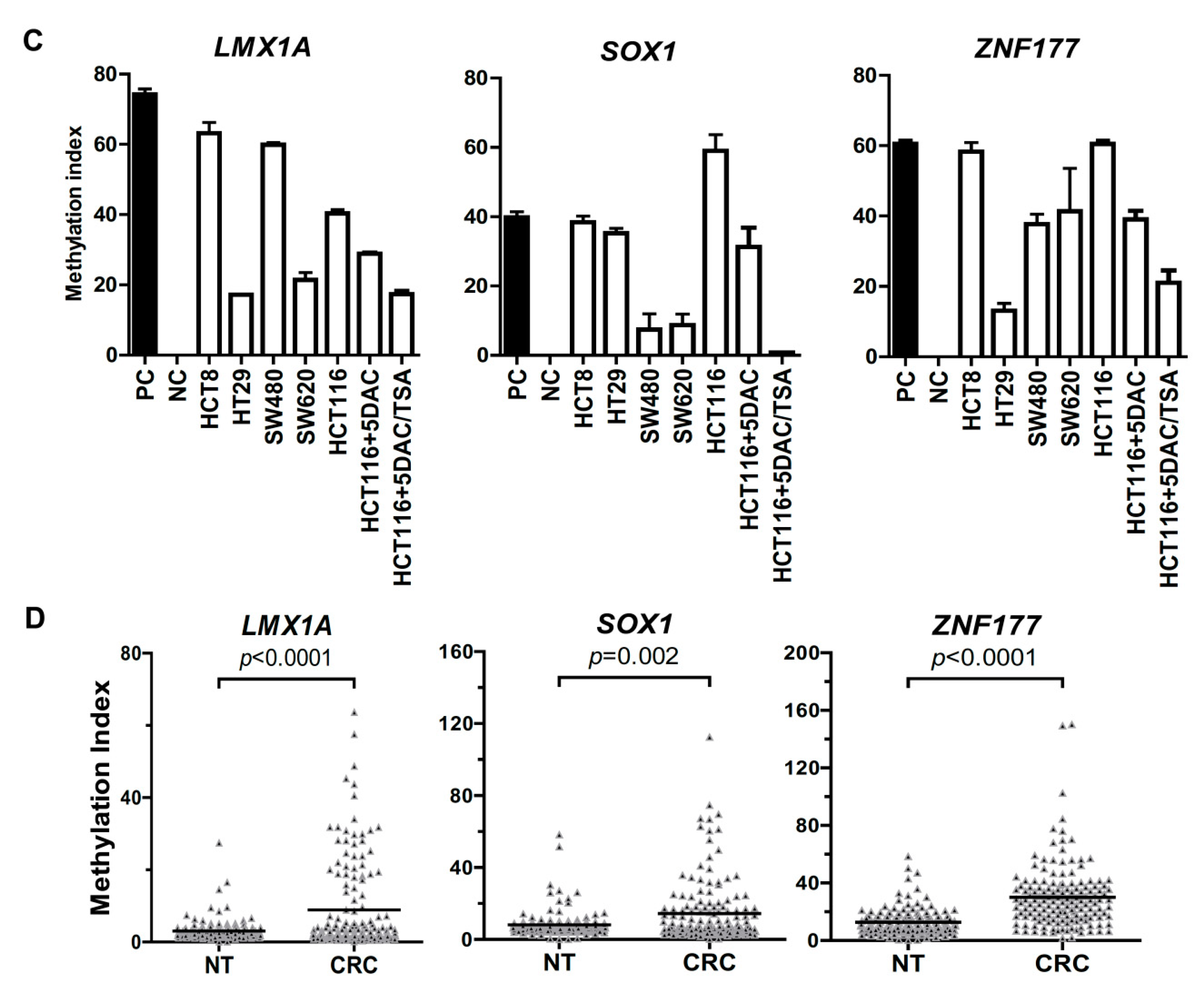
2.1. Abnormal Methylation of LMX1A, SOX1, and ZNF177 in CRC
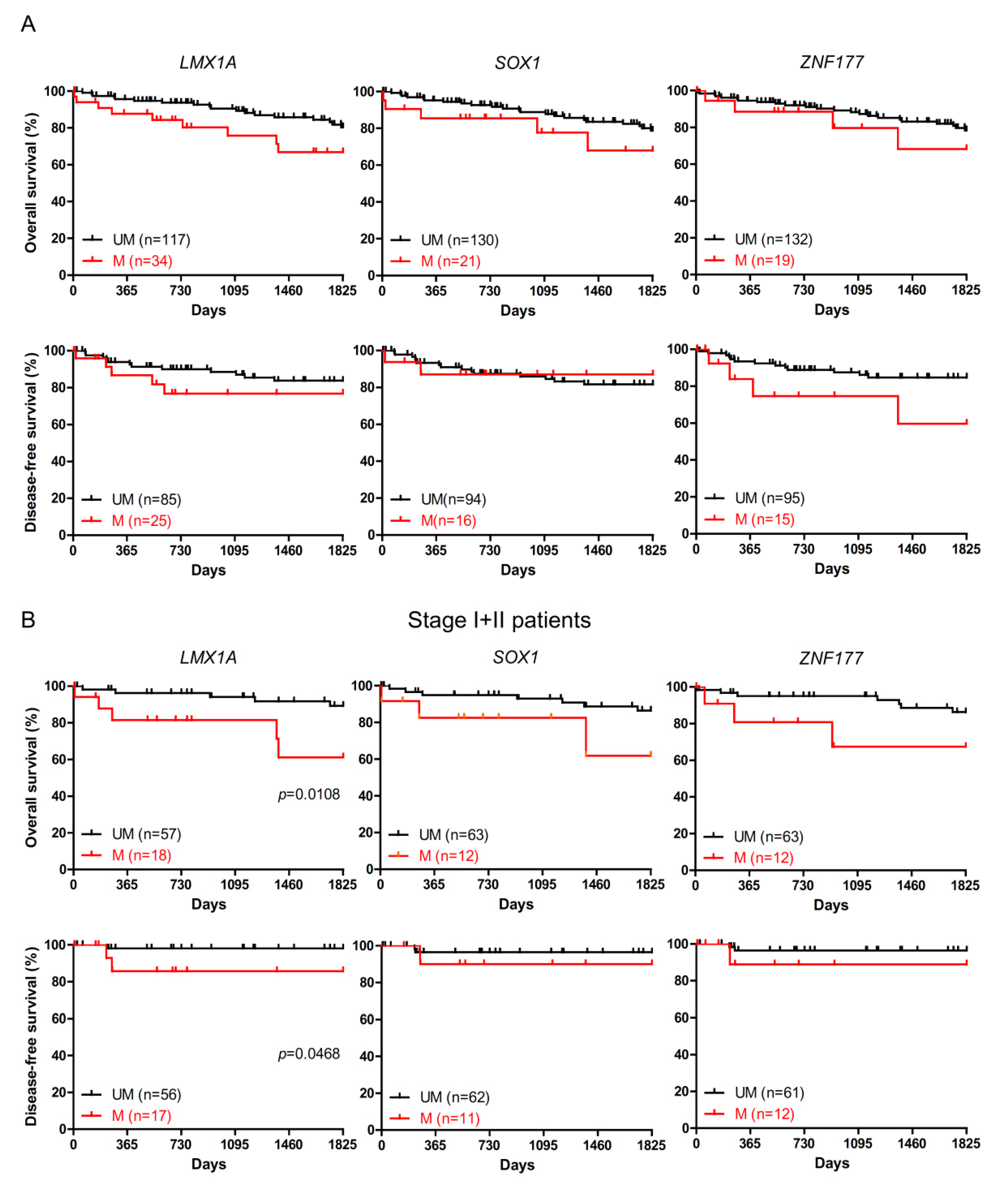
2.2. The Association of LMX1A Methylation and Patient Survival
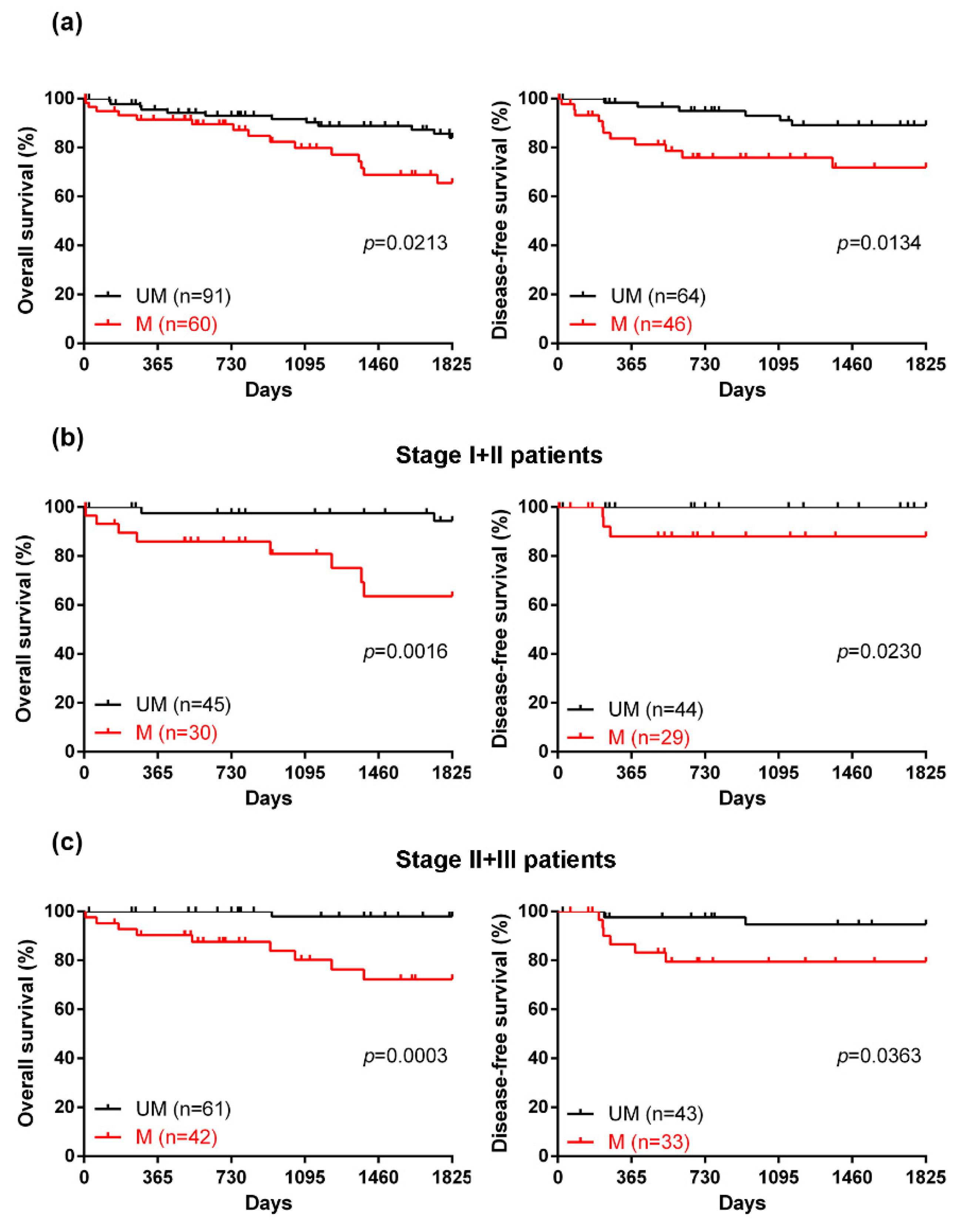
2.3. The Efficacy of a Novel DNA Methylation Panel for Predicting the Prognosis of CRC
3. Discussion
4. Materials and Methods
4.1. MethHC Database
4.2. Clinical Samples and Cell Lines
4.3. DNA Methylation and Gene Expression Analysis
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA-Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal cancer. Lancet 2014, 383, 1490–1502. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, J.; He, X.; Wang, C.; Lian, L.; Liu, H.; Wang, J.; Lan, P. Postoperative adjuvant chemotherapy for stage II colorectal cancer: A systematic review of 12 randomized controlled trials. J. Gastrointest. Surg. Off. J. Soc. Surg. Aliment. Tract 2012, 16, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Fakhr, M.G.; Hagh, M.F.; Shanehbandi, D.; Baradaran, B. DNA methylation pattern as important epigenetic criterion in cancer. Genet. Res. Int. 2013, 2013, 317569. [Google Scholar] [CrossRef]
- Silva, T.D.; Vidigal, V.M.; Felipe, A.V.; De Lima, J.M.; Neto, R.A.; Saad, S.S.; Forones, N.M. DNA methylation as an epigenetic biomarker in colorectal cancer. Oncol. Lett. 2013, 6, 1687–1692. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.; Pan, K.; Linnekamp, J.F.; Medema, J.P.; Kandimalla, R. DNA methylation based biomarkers in colorectal cancer: A systematic review. Biochim. Biophys. Acta 2016, 1866, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Imperiale, T.F.; Ransohoff, D.F.; Itzkowitz, S.H. Multitarget stool DNA testing for colorectal-cancer screening. N. Engl. J. Med. 2014, 371, 187–188. [Google Scholar] [CrossRef] [PubMed]
- Toth, K.; Wasserkort, R.; Sipos, F.; Kalmar, A.; Wichmann, B.; Leiszter, K.; Valcz, G.; Juhasz, M.; Miheller, P.; Patai, A.V.; et al. Detection of methylated septin 9 in tissue and plasma of colorectal patients with neoplasia and the relationship to the amount of circulating cell-free DNA. PLoS ONE 2014, 9, e115415. [Google Scholar] [CrossRef]
- Wasserkort, R.; Kalmar, A.; Valcz, G.; Spisak, S.; Krispin, M.; Toth, K.; Tulassay, Z.; Sledziewski, A.Z.; Molnar, B. Aberrant septin 9 DNA methylation in colorectal cancer is restricted to a single CpG island. BMC Cancer 2013, 13, 398. [Google Scholar] [CrossRef]
- Molnar, B.; Toth, K.; Bartak, B.K.; Tulassay, Z. Plasma methylated septin 9: A colorectal cancer screening marker. Expert Rev. Mol. Diagn. 2015, 15, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.Y.; Kuo, C.C.; Wu, C.C.; Hsiao, C.W.; Hu, J.M.; Hsu, C.H.; Chou, Y.C.; Shih, Y.L.; Lin, Y.W. NKX6.1 hypermethylation predicts the outcome of stage II colorectal cancer patients undergoing chemotherapy. Genes Chromosom. Cancer 2018, 57, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.C.; Lin, C.Y.; Shih, Y.L.; Hsieh, C.B.; Lin, P.Y.; Guan, S.B.; Hsieh, M.S.; Lai, H.C.; Chen, C.J.; Lin, Y.W. Frequent methylation of HOXA9 gene in tumor tissues and plasma samples from human hepatocellular carcinomas. Clin. Chem. Lab. Med. 2014, 52, 1235–1245. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Lagares, A.; Mendez-Gonzalez, J.; Hervas, D.; Saigi, M.; Pajares, M.J.; Garcia, D.; Crujerias, A.B.; Pio, R.; Montuenga, L.M.; Zulueta, J.; et al. A Novel Epigenetic Signature for Early Diagnosis in Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 3361–3371. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Tsao, C.M.; Kuo, C.C.; Yu, M.H.; Lin, Y.W.; Yang, C.Y.; Li, H.J.; Yan, M.D.; Wang, T.J.; Chou, Y.C.; et al. Quantitative DNA methylation analysis of selected genes in endometrial carcinogenesis. Taiwan. J. Obstet. Gynecol. 2015, 54, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.C.; Lin, Y.W.; Huang, T.H.; Yan, P.; Huang, R.L.; Wang, H.C.; Liu, J.; Chan, M.W.; Chu, T.Y.; Sun, C.A.; et al. Identification of novel DNA methylation markers in cervical cancer. Int. J. Cancer 2008, 123, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Feng, L.; Xie, Y.; Zhang, H.; Wu, Y. Hypermethylation-mediated reduction of LMX1A expression in gastric cancer. Cancer Sci. 2011, 102, 361–366. [Google Scholar] [CrossRef]
- Zhao, Y.; Guo, S.; Sun, J.; Huang, Z.; Zhu, T.; Zhang, H.; Gu, J.; He, Y.; Wang, W.; Ma, K.; et al. Methylcap-seq reveals novel DNA methylation markers for the diagnosis and recurrence prediction of bladder cancer in a Chinese population. PLoS ONE 2012, 7, e35175. [Google Scholar] [CrossRef]
- Su, H.Y.; Lai, H.C.; Lin, Y.W.; Chou, Y.C.; Liu, C.Y.; Yu, M.H. An epigenetic marker panel for screening and prognostic prediction of ovarian cancer. Int. J. Cancer 2009, 124, 387–393. [Google Scholar] [CrossRef]
- Shih, Y.L.; Hsieh, C.B.; Yan, M.D.; Tsao, C.M.; Hsieh, T.Y.; Liu, C.H.; Lin, Y.W. Frequent concomitant epigenetic silencing of SOX1 and secreted frizzled-related proteins (SFRPs) in human hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2013, 28, 551–559. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhou, H.; Ma, K.; Sun, J.; Feng, X.; Geng, J.; Gu, J.; Wang, W.; Zhang, H.; He, Y.; et al. Abnormal methylation of seven genes and their associations with clinical characteristics in early stage non-small cell lung cancer. Oncol. Lett. 2013, 5, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Kuo, I.Y.; Chang, J.M.; Jiang, S.S.; Chen, C.H.; Chang, I.S.; Sheu, B.S.; Lu, P.J.; Chang, W.L.; Lai, W.W.; Wang, Y.C. Prognostic CpG methylation biomarkers identified by methylation array in esophageal squamous cell carcinoma patients. Int. J. Med. Sci. 2014, 11, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Li, S. Epigenetic inactivation of SOX1 promotes cell migration in lung cancer. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 4603–4610. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.I.; Angulo, J.C.; Martin, A.; Sanchez-Chapado, M.; Gonzalez-Corpas, A.; Colas, B.; Ropero, S. A DNA hypermethylation profile reveals new potential biomarkers for the evaluation of prognosis in urothelial bladder cancer. Acta Pathol. Microbiol. Immunol. Scand. 2017, 125, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Herzig, D.O.; Tsikitis, V.L. Molecular markers for colon diagnosis, prognosis and targeted therapy. J. Surg. Oncol. 2015, 111, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Mahasneh, A.; Al-Shaheri, F.; Jamal, E. Molecular biomarkers for an early diagnosis, effective treatment and prognosis of colorectal cancer: Current updates. Exp. Mol. Pathol. 2017, 102, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Toyota, M.; Kondo, Y.; Lin, E.; Zhang, L.; Guo, Y.; Hernandez, N.S.; Chen, X.; Ahmed, S.; Konishi, K.; et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 18654–18659. [Google Scholar] [CrossRef]
- Ang, P.W.; Loh, M.; Liem, N.; Lim, P.L.; Grieu, F.; Vaithilingam, A.; Platell, C.; Yong, W.P.; Iacopetta, B.; Soong, R. Comprehensive profiling of DNA methylation in colorectal cancer reveals subgroups with distinct clinicopathological and molecular features. BMC Cancer 2010, 10, 227. [Google Scholar] [CrossRef]
- Lee, S.; Cho, N.Y.; Yoo, E.J.; Kim, J.H.; Kang, G.H. CpG island methylator phenotype in colorectal cancers: Comparison of the new and classic CpG island methylator phenotype marker panels. Arch. Pathol. Lab. Med. 2008, 132, 1657–1665. [Google Scholar] [CrossRef]
- Barault, L.; Charon-Barra, C.; Jooste, V.; de la Vega, M.F.; Martin, L.; Roignot, P.; Rat, P.; Bouvier, A.M.; Laurent-Puig, P.; Faivre, J.; et al. Hypermethylator phenotype in sporadic colon cancer: Study on a population-based series of 582 cases. Cancer Res. 2008, 68, 8541–8546. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Catalano, P.J.; Benson, A.B., 3rd; O’Dwyer, P.; Hamilton, S.R.; Issa, J.P. Association between DNA methylation and shortened survival in patients with advanced colorectal cancer treated with 5-fluorouracil based chemotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 6093–6098. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Cho, N.Y.; Choi, M.; Yoo, E.J.; Kim, J.H.; Kang, G.H. Clinicopathological features of CpG island methylator phenotype-positive colorectal cancer and its adverse prognosis in relation to KRAS/BRAF mutation. Pathol. Int. 2008, 58, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Shin, S.H.; Kwon, H.J.; Cho, N.Y.; Kang, G.H. Prognostic implications of CpG island hypermethylator phenotype in colorectal cancers. Virchows Arch. Int. J. Pathol. 2009, 455, 485–494. [Google Scholar] [CrossRef]
- Sanchez, J.A.; Krumroy, L.; Plummer, S.; Aung, P.; Merkulova, A.; Skacel, M.; DeJulius, K.L.; Manilich, E.; Church, J.M.; Casey, G.; et al. Genetic and epigenetic classifications define clinical phenotypes and determine patient outcomes in colorectal cancer. Br. J. Surg. 2009, 96, 1196–1204. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.L.; Cheong, K.; Ku, S.L.; Meagher, A.; O’Connor, T.; Hawkins, N.J. Adverse prognostic effect of methylation in colorectal cancer is reversed by microsatellite instability. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2003, 21, 3729–3736. [Google Scholar] [CrossRef] [PubMed]
- Hobert, O.; Westphal, H. Functions of LIM-homeobox genes. Trends Genet. 2000, 16, 75–83. [Google Scholar] [CrossRef]
- Buescher, M.; Hing, F.S.; Chia, W. Formation of neuroblasts in the embryonic central nervous system of Drosophila melanogaster is controlled by SoxNeuro. Development 2002, 129, 4193–4203. [Google Scholar] [PubMed]
- Kan, L.; Israsena, N.; Zhang, Z.; Hu, M.; Zhao, L.R.; Jalali, A.; Sahni, V.; Kessler, J.A. Sox1 acts through multiple independent pathways to promote neurogenesis. Dev. Biol. 2004, 269, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Laity, J.H.; Lee, B.M.; Wright, P.E. Zinc finger proteins: New insights into structural and functional diversity. Curr. Opin. Struct. Biol. 2001, 11, 39–46. [Google Scholar] [CrossRef]
- Cassandri, M.; Smirnov, A.; Novelli, F.; Pitolli, C.; Agostini, M.; Malewicz, M.; Melino, G.; Raschella, G. Zinc-finger proteins in health and disease. Cell Death Discov. 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Chao, T.K.; Su, P.H.; Lee, H.Y.; Shih, Y.L.; Su, H.Y.; Chu, T.Y.; Yu, M.H.; Lin, Y.W.; Lai, H.C. Characterization of LMX-1A as a metastasis suppressor in cervical cancer. J. Pathol. 2009, 219, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.M.; Yan, M.D.; Shih, Y.L.; Yu, P.N.; Kuo, C.C.; Lin, W.C.; Li, H.J.; Lin, Y.W. SOX1 functions as a tumor suppressor by antagonizing the WNT/beta-catenin signaling pathway in hepatocellular carcinoma. Hepatology 2012, 56, 2277–2287. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.W.; Tsao, C.M.; Yu, P.N.; Shih, Y.L.; Lin, C.H.; Yan, M.D. SOX1 suppresses cell growth and invasion in cervical cancer. Gynecol. Oncol. 2013, 131, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Tan, Z.R.; Yu, J.; Li, H.; Lv, Q.L.; Shao, Y.Y.; Zhou, H.H. DNA hypermethylated status and gene expression of PAX1/SOX1 in patients with colorectal carcinoma. OncoTargets Ther. 2017, 10, 4739–4751. [Google Scholar] [CrossRef] [PubMed]
- Molnar, B.; Galamb, O.; Peterfia, B.; Wichmann, B.; Csabai, I.; Bodor, A.; Kalmar, A.; Szigeti, K.A.; Bartak, B.K.; Nagy, Z.B.; et al. Gene promoter and exon DNA methylation changes in colon cancer development—mRNA expression and tumor mutation alterations. BMC Cancer 2018, 18, 695. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Set | Best Cut-Off Value | Sensitivity | Specificity |
|---|---|---|---|
| NKX6.1 | MI > 18.42 | 23.2% | 99.3% |
| LMX1A | MI > 16.84 | 22.5% | 99.3% |
| SOX1 | MI > 30.26 | 13.9% | 98.7% |
| ZNF177 | MI > 51.14 | 12.6% | 99.3% |
| NKX6.1 or ZNF177 | 33.1% | 98.7% | |
| NKX6.1 or LMX1A | 31.1% | 98.7% | |
| LMX1A or ZNF177 | 30.5% | 98.7% | |
| NKX6.1 or SOX1 | 29.1% | 98.0% | |
| LMX1A or SOX1 | 27.2% | 98.0% | |
| SOX1 or ZNF177 | 21.9% | 98.7% | |
| NKX6.1 or LMX1A or ZNF177 | 37.7% | 98.0% | |
| NKX6.1 or SOX1 or ZNF177 | 36.4% | 98.0% | |
| LMX1A or SOX1 or ZNF177 | 33.8% | 98.0% | |
| NKX6.1 or LMX1A or SOX1 | 33.8% | 97.4% | |
| NKX6.1 or LMX1A or SOX1 or ZNF177 | 39.7% | 97.4% |
| Symbol | LMX1A | SOX1 | ZNF177 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| (MI > 16.84) | (MI > 30.26) | (MI > 51.14) | ||||||||
| UM | M | p | UM | M | p | UM | M | p | ||
| Age (63.44 ± 14.46) | <64 years | 62 | 12 | 0.0508 | 67 | 10 | 0.8162 | 69 | 8 | 0.4673 |
| ≥64 years | 55 | 22 | 63 | 11 | 63 | 11 | ||||
| Sex | Female | 61 | 18 | 1.0000 | 66 | 13 | 0.3597 | 70 | 9 | 0.8067 |
| Male | 56 | 16 | 64 | 8 | 62 | 10 | ||||
| Stage | І | 20 | 2 | 0.2885 | 22 | 0 | 0.0510 | 18 | 4 | 0.5910 |
| ІІ | 38 | 15 | 41 | 12 | 45 | 8 | ||||
| ІІІ | 40 | 10 | 43 | 7 | 46 | 4 | ||||
| ІV | 19 | 7 | 24 | 2 | 23 | 3 | ||||
| Tumor grade | Well differentiated | 4 | 0 | 0.5387 | 4 | 0 | 0.6122 | 4 | 0 | 0.1327 |
| Moderately differentiated | 95 | 27 | 104 | 18 | 104 | 18 | ||||
| Poorly or undifferentiated | 15 | 5 | 18 | 2 | 20 | 0 | ||||
| Missing data | 4 | 1 | 4 | 1 | 4 | 1 | ||||
| Tumor size | ≤5 cm | 72 | 10 | 0.0048 | 72 | 10 | 0.6151 | 74 | 8 | 0.5894 |
| >5 cm | 37 | 18 | 46 | 9 | 48 | 7 | ||||
| Missing data | 9 | 5 | 12 | 2 | 10 | 4 | ||||
| No. of lymph node | ≥12 | 95 | 22 | 0.3755 | 101 | 16 | 1.0000 | 105 | 12 | 0.2676 |
| 0–11 | 15 | 6 | 18 | 3 | 17 | 4 | ||||
| Missing data | 8 | 5 | 11 | 2 | 10 | 3 | ||||
| Chemotherapy | No | 31 | 8 | 1.0000 | 32 | 7 | 0.4141 | 34 | 5 | 0.7729 |
| Yes | 79 | 20 | 87 | 12 | 88 | 11 | ||||
| Missing data | 8 | 5 | 11 | 2 | 10 | 3 | ||||
| Recurrence | No | 74 | 19 | 0.5483 | 79 | 14 | 0.8095 | 82 | 11 | 0.8025 |
| Yes | 43 | 15 | 51 | 7 | 50 | 8 | ||||
| Survival | Alive | 100 | 24 | 0.0722 | 108 | 16 | 0.5381 | 109 | 15 | 0.7494 |
| Dead | 17 | 10 | 22 | 5 | 23 | 4 | ||||
| Symbol | NKX6.1 or LMX1A or SOX1 or ZNF177 | |||
|---|---|---|---|---|
| UM | M | p | ||
| Age (63.44 ± 14.46) | <64 years | 52 | 21 | 0.0083 |
| ≥64 years | 39 | 39 | ||
| Sex | Female | 48 | 31 | 1.0000 |
| Male | 43 | 29 | ||
| Stage | І | 16 | 6 | 0.3678 |
| ІІ | 30 | 23 | ||
| ІІІ | 32 | 18 | ||
| ІV | 13 | 13 | ||
| Tumor grade | Well differentiated | 4 | 0 | 0.2355 |
| Moderately differentiated | 72 | 50 | ||
| Poorly or undifferentiated | 13 | 7 | ||
| Missing data | 4 | 1 | ||
| Tumor size | ≤5 cm | 57 | 25 | 0.0323 |
| >5 cm | 28 | 27 | ||
| Missing data | 7 | 7 | ||
| No. of lymph node | ≥12 | 74 | 43 | 0.4653 |
| 0–11 | 11 | 10 | ||
| Missing data | 7 | 6 | ||
| Chemotherapy | No | 24 | 15 | 1.0000 |
| Yes | 61 | 38 | ||
| Missing data | 7 | 6 | ||
| Recurrence | No | 59 | 34 | 0.3928 |
| Yes | 32 | 26 | ||
| Survival | Alive | 80 | 44 | 0.0297 |
| Dead | 11 | 16 | ||
| Variable | Univariate Analysis Hazard Ratio | Multivariate Analysis Hazard Ratio |
|---|---|---|
| (95% Confidence Interval) | (95% Confidence Interval) | |
| Age (years) | 0.99 (0.96–1.02) | 0.98 (0.95–1.01) |
| Sex (female versus male) | 1.05 (0.49–2.22) | 1.35 (0.57–3.21) |
| Gene methylation | ||
| Unmethylation | Reference | Reference |
| Methylation | 2.95 (1.36–6.39) ** | 3.43 (1.37–8.57) ** |
| Stage | ||
| I + II | Reference | Reference |
| III + IV | 1.77 (0.81–3.87) | 2.66 (0.98–7.26) |
| Tumor grade | ||
| Well + moderately | Reference | Reference |
| Poorly or undifferentiated | 0.67 (0.16–2.86) | 0.74 (0.16–3.36) |
| Tumor size | ||
| ≤5 cm | Reference | Reference |
| >5 cm | 0.97 (0.42–2.21) | 0.63 (0.25–1.61) |
| No. of lymph node | ||
| ≥12 | Reference | Reference |
| 0–11 | 0.42 (0.17–1.07) | 0.49 (0.17–1.40) |
| Chemotherapy | ||
| No | Reference | Reference |
| Yes | 0.51 (0.23–1.13) | 0.32 (0.12–0.85) * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, H.-H.; Kuo, C.-C.; Hsiao, C.-W.; Chen, C.-Y.; Hu, J.-M.; Hsu, C.-H.; Chou, Y.-C.; Lin, Y.-W.; Shih, Y.-L. A Novel Prognostic DNA Methylation Panel for Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 4672. https://doi.org/10.3390/ijms20194672
Chung H-H, Kuo C-C, Hsiao C-W, Chen C-Y, Hu J-M, Hsu C-H, Chou Y-C, Lin Y-W, Shih Y-L. A Novel Prognostic DNA Methylation Panel for Colorectal Cancer. International Journal of Molecular Sciences. 2019; 20(19):4672. https://doi.org/10.3390/ijms20194672
Chicago/Turabian StyleChung, Hsin-Hua, Chih-Chi Kuo, Cheng-Wen Hsiao, Chao-Yang Chen, Je-Ming Hu, Chih-Hsiung Hsu, Yu-Ching Chou, Ya-Wen Lin, and Yu-Lueng Shih. 2019. "A Novel Prognostic DNA Methylation Panel for Colorectal Cancer" International Journal of Molecular Sciences 20, no. 19: 4672. https://doi.org/10.3390/ijms20194672
APA StyleChung, H.-H., Kuo, C.-C., Hsiao, C.-W., Chen, C.-Y., Hu, J.-M., Hsu, C.-H., Chou, Y.-C., Lin, Y.-W., & Shih, Y.-L. (2019). A Novel Prognostic DNA Methylation Panel for Colorectal Cancer. International Journal of Molecular Sciences, 20(19), 4672. https://doi.org/10.3390/ijms20194672