Rare Human Missense Variants can affect the Function of Disease-Relevant Proteins by Loss and Gain of Peroxisomal Targeting Motifs

 , ,
, ,  and
and
Abstract
1. Introduction
2. Results
2.1. Mining of gnomAD for SNVs Causing Missense Mutations in C-terminal Tripeptides
2.2. Investigation of SNVs Resulting in PTS1 Ablation (Loss-of-Function)
2.2.1. Identification and Prioritization of SNVs Resulting in Loss-of-Function PTS1 for Experimental Testing
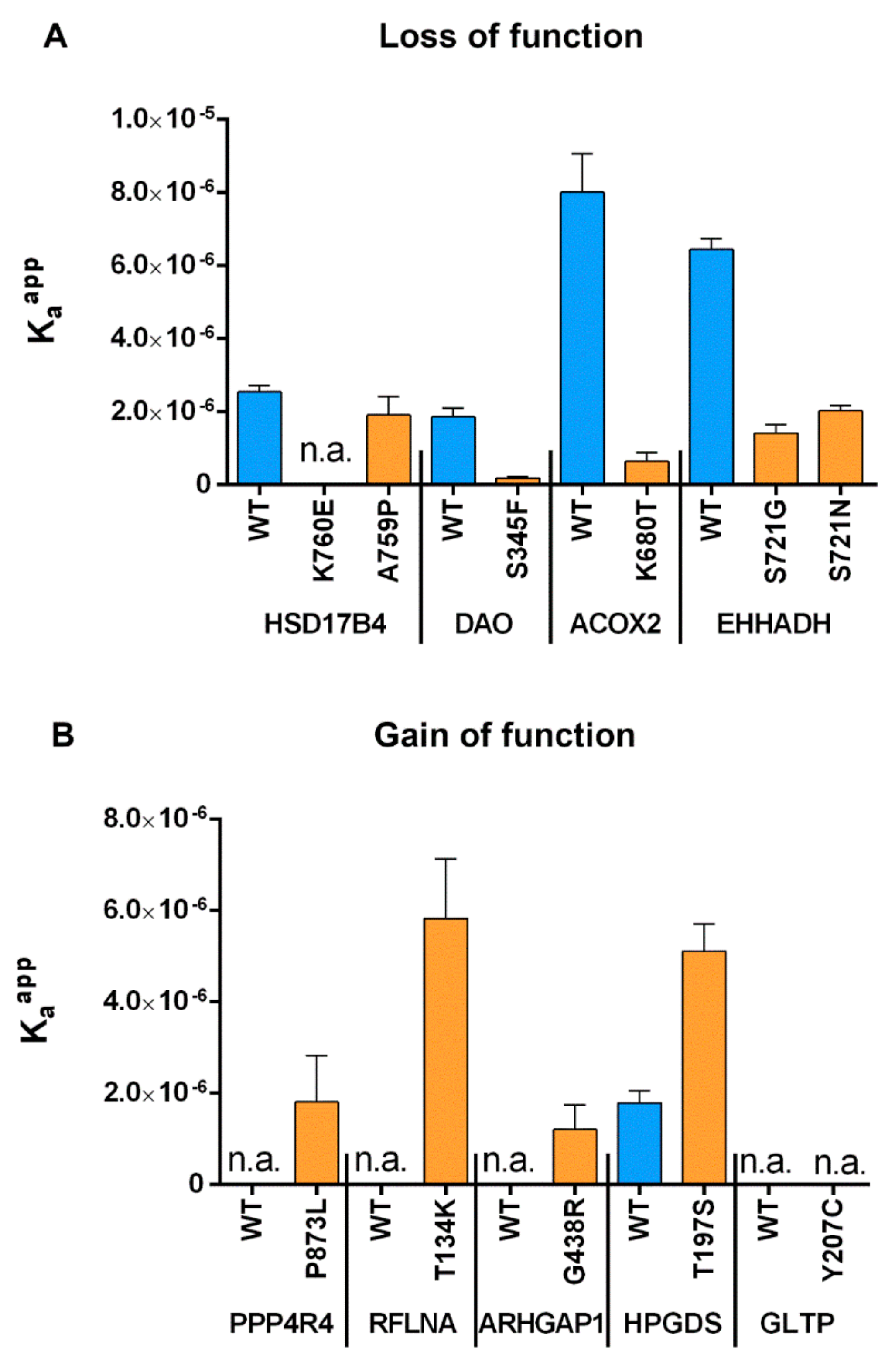
2.2.2. Experimental Investigation of Functional Consequences of SNVs in PTS1 Motifs
2.3. Investigation of SNVs Resulting de novo PTS1 Generation in Cytosolic Proteins (Gain-of-Function)
2.3.1. Identification and Prioritization of Gain-of-Function SNVs Generating de novo PTS1 for Experimental Testing
2.3.2. Experimental Validation of Gain-of-function SNVs Leading to de novo Generation of PTS1
2.4. Measurement of Relative Affinity Before and After Point Mutations for both GoF and LoF Mutants
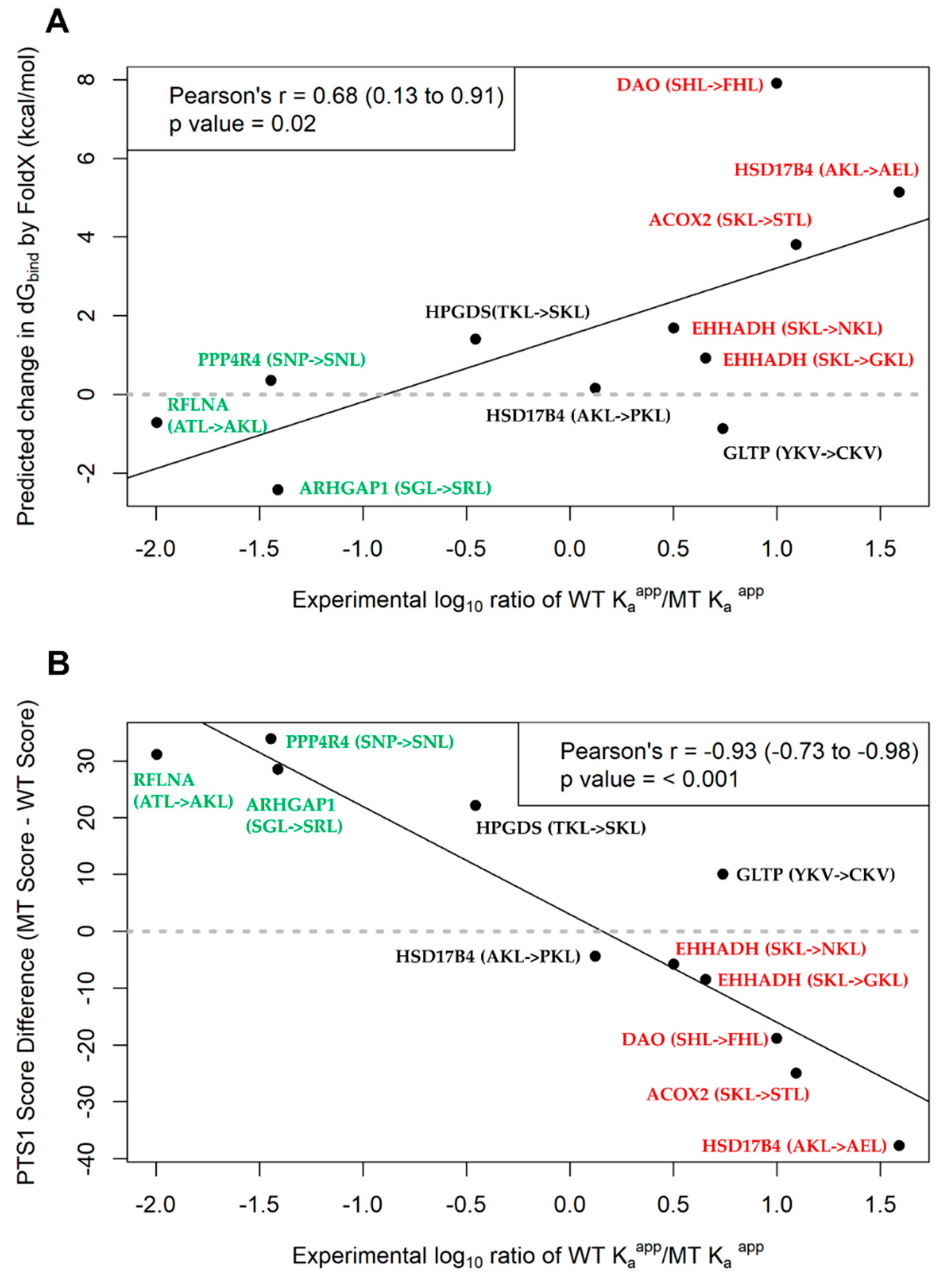
2.5. Assessment of PTS1 Predictor and FoldX Predictions of PTS1 Signal Quality
2.6. Disease-Relevance of Investigated SNVs as Consequence of Abolishment or Gain in Peroxisome Targeting
3. Discussion
4. Materials and Methods
4.1. Obtaining CCDS Protein Coding Genomic Coordinates and Sequence Information
4.2. Parsing gnomAD vcf Files to Obtain Relevant SNV Information
4.3. Obtaining Subcellular Localization Annotations from UniProt
4.4. Obtaining PTS1 Predictor Results for Native Transcripts and their Corresponding Point Mutants
4.5. Prioritization of SNV before Final Selection for Experimental Testing
4.6. FoldX Prediction of Change in Free Binding Energy due to Point Mutations made at PTS1 Ligand-Receptor Interface
4.7. Statistical Analyses
4.8. Cloning
4.9. Cell Culture
4.10. FRET Affinity Measurements
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| gnomAD | Genome Aggregation Database |
| CCDS | Consensus Coding Sequence Project |
| UniProt | Universal Protein resource |
| PTS | Peroxisome targeting signal |
| PEX | Peroxin |
| SNV | Single nucleotide variant |
| dbSNP | The Single Nucleotide Polymorphism Database |
| vcf | Variant call file |
| tbi | Tabix-indexed file |
| GoF | Gain-of-function |
| LoF | Loss-of-function |
| EGFP | Enhanced green fluorescent protein |
| FRET | Fluorescence resonance energy transfer |
References
- Pinto, N.; Dolan, M.E. Clinically relevant genetic variations in drug metabolizing enzymes. Curr. Drug Metab. 2011, 12, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- Merkulov, V.M.; Leberfarb, E.Y.; Merkulova, T.I. Regulatory SNPs and their widespread effects on the transcriptome. J. Biosci. 2018, 43, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.; Cooper, D.N. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 2014, 133, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Kucukkal, T.G.; Petukh, M.; Li, L.; Alexov, E. Structural and physico-chemical effects of disease and non-disease nsSNPs on proteins. Curr. Opin. Struct. Biol. 2015, 32, 18–24. [Google Scholar] [CrossRef]
- Yates, C.M.; Sternberg, M.J. The effects of non-synonymous single nucleotide polymorphisms (nsSNPs) on protein-protein interactions. J. Mol. Biol. 2013, 425, 3949–3963. [Google Scholar] [CrossRef]
- David, A.; Sternberg, M.J. The Contribution of Missense Mutations in Core and Rim Residues of Protein-Protein Interfaces to Human Disease. J. Mol. Biol. 2015, 427, 2886–2898. [Google Scholar] [CrossRef]
- Laurila, K.; Vihinen, M. Prediction of disease-related mutations affecting protein localization. BMC Genom. 2009, 10, 122. [Google Scholar] [CrossRef]
- Fodor, K.; Wolf, J.; Erdmann, R.; Schliebs, W.; Wilmanns, M. Molecular requirements for peroxisomal targeting of alanine-glyoxylate aminotransferase as an essential determinant in primary hyperoxaluria type 1. PLoS Biol. 2012, 10, e1001309. [Google Scholar] [CrossRef] [PubMed]
- Schrader, M.; Fahimi, H.D. The peroxisome: Still a mysterious organelle. Histochem. Cell Biol. 2008, 129, 421–440. [Google Scholar] [CrossRef] [PubMed]
- Waterham, H.R.; Ferdinandusse, S.; Wanders, R.J. Human disorders of peroxisome metabolism and biogenesis. Biochim. Biophys. Acta 2016, 1863, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.J.; Dodt, G.; Raymond, G.V.; Braverman, N.E.; Moser, A.B.; Moser, H.W. Peroxisome biogenesis disorders. Biochim. Biophys. Acta 2006, 1763, 1733–1748. [Google Scholar] [CrossRef] [PubMed]
- Braverman, N.E.; Raymond, G.V.; Rizzo, W.B.; Moser, A.B.; Wilkinson, M.E.; Stone, E.M.; Steinberg, S.J.; Wangler, M.F.; Rush, E.T.; Hacia, J.G.; et al. Peroxisome biogenesis disorders in the Zellweger spectrum: An overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol. Genet. Metab. 2016, 117, 313–321. [Google Scholar] [CrossRef]
- Wanders, R.J.; Waterham, H.R. Peroxisomal disorders: The single peroxisomal enzyme deficiencies. Biochim. Biophys. Acta 2006, 1763, 1707–1720. [Google Scholar] [CrossRef]
- Gould, S.G.; Keller, G.A.; Subramani, S. Identification of a peroxisomal targeting signal at the carboxy terminus of firefly luciferase. J. Cell Biol. 1987, 105, 2923–2931. [Google Scholar] [CrossRef]
- Gould, S.J.; Keller, G.A.; Hosken, N.; Wilkinson, J.; Subramani, S. A conserved tripeptide sorts proteins to peroxisomes. J. Cell Biol. 1989, 108, 1657–1664. [Google Scholar] [CrossRef]
- Lametschwandtner, G.; Brocard, C.; Fransen, M.; Van Veldhoven, P.; Berger, J.; Hartig, A. The difference in recognition of terminal tripeptides as peroxisomal targeting signal 1 between yeast and human is due to different affinities of their receptor Pex5p to the cognate signal and to residues adjacent to it. J. Biol. Chem. 1998, 273, 33635–33643. [Google Scholar] [CrossRef]
- Brocard, C.; Hartig, A. Peroxisome targeting signal 1: Is it really a simple tripeptide? Biochim. Biophys. Acta 2006, 1763, 1565–1573. [Google Scholar] [CrossRef]
- Klootwijk, E.D.; Reichold, M.; Helip-Wooley, A.; Tolaymat, A.; Broeker, C.; Robinette, S.L.; Reinders, J.; Peindl, D.; Renner, K.; Eberhart, K.; et al. Mistargeting of peroxisomal EHHADH and inherited renal Fanconi’s syndrome. N. Engl. J. Med. 2014, 370, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Assmann, N.; Dettmer, K.; Simbuerger, J.M.B.; Broeker, C.; Nuernberger, N.; Renner, K.; Courtneidge, H.; Klootwijk, E.D.; Duerkop, A.; Hall, A.; et al. Renal Fanconi Syndrome Is Caused by a Mistargeting-Based Mitochondriopathy. Cell Rep. 2016, 15, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Leiper, J.M. Inhibition of alanine:glyoxylate aminotransferase 1 dimerization is a prerequisite for its peroxisome-to-mitochondrion mistargeting in primary hyperoxaluria type 1. J. Cell Biol. 1996, 135, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Kunze, M. Predicting Peroxisomal Targeting Signals to Elucidate the Peroxisomal Proteome of Mammals. Subcell. Biochem. 2018, 89, 157–199. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, G.; Maurer-Stroh, S.; Eisenhaber, B.; Hartig, A.; Eisenhaber, F. Prediction of Peroxisomal Targeting Signal 1 Containing Proteins from Amino Acid Sequence. J. Mol. Biol. 2003, 328, 581–592. [Google Scholar] [CrossRef]
- Hochreiter, B.; Chong, C.S.; Hartig, A.; Maurer-Stroh, S.; Berger, J.; Schmid, J.A.; Kunze, M. A novel FRET approach quantifies the interaction strength of peroxisomal targeting signals and their receptor in living cells. Nat. Chem. Biol. 2019. [Google Scholar]
- Laurila, K.; Vihinen, M. PROlocalizer: Integrated web service for protein subcellular localization prediction. Amino Acids 2011, 40, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 2019, 531210. [Google Scholar] [CrossRef]
- Stanley, W.A.; Filipp, F.V.; Kursula, P.; Schuller, N.; Erdmann, R.; Schliebs, W.; Sattler, M.; Wilmanns, M. Recognition of a functional peroxisome type 1 target by the dynamic import receptor pex5p. Mol. Cell 2006, 24, 653–663. [Google Scholar] [CrossRef]
- Schluter, A.; Real-Chicharro, A.; Gabaldon, T.; Sanchez-Jimenez, F.; Pujol, A. PeroxisomeDB 2.0: An integrative view of the global peroxisomal metabolome. Nucleic Acids Res. 2010, 38, D800–D805. [Google Scholar] [CrossRef]
- Skoulding, N.S.; Chowdhary, G.; Deus, M.J.; Baker, A.; Reumann, S.; Warriner, S.L. Experimental validation of plant peroxisomal targeting prediction algorithms by systematic comparison of in vivo import efficiency and in vitro PTS1 binding affinity. J. Mol. Biol. 2015, 427, 1085–1101. [Google Scholar] [CrossRef] [PubMed]
- Hochreiter, B.; Kunze, M.; Moser, B.; Schmid, J.A. Advanced FRET normalization allows quantitative analysis of protein interactions including stoichiometries and relative affinities in living cells. Sci. Rep. 2019, 9, 8233. [Google Scholar] [CrossRef] [PubMed]
- Guerois, R.; Nielsen, J.E.; Serrano, L. Predicting Changes in the Stability of Proteins and Protein Complexes: A Study of More Than 1000 Mutations. J. Mol. Biol. 2002, 320, 369–387. [Google Scholar] [CrossRef]
- Corvin, A.; McGhee, K.A.; Murphy, K.; Donohoe, G.; Nangle, J.M.; Schwaiger, S.; Kenny, N.; Clarke, S.; Meagher, D.; Quinn, J.; et al. Evidence for association and epistasis at the DAOA/G30 and D-amino acid oxidase loci in an Irish schizophrenia sample. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2007, 144B, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Chumakov, I.; Blumenfeld, M.; Guerassimenko, O.; Cavarec, L.; Palicio, M.; Abderrahim, H.; Bougueleret, L.; Barry, C.; Tanaka, H.; La Rosa, P.; et al. Genetic and physiological data implicating the new human gene G72 and the gene for D-amino acid oxidase in schizophrenia. Proc. Natl. Acad. Sci. USA 2002, 99, 13675–13680. [Google Scholar] [CrossRef] [PubMed]
- Caldinelli, L.; Sacchi, S.; Molla, G.; Nardini, M.; Pollegioni, L. Characterization of human DAAO variants potentially related to an increased risk of schizophrenia. Biochim. Biophys. Acta 2013, 1832, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Watanabe, S.; Kinoshita, A.; Mishima, H.; Nishimura, G.; Moriuchi, H.; Yoshiura, K.I.; Dateki, S. Identification of a homozygous frameshift variant in RFLNA in a patient with a typical phenotype of spondylocarpotarsal synostosis syndrome. J. Hum. Genet. 2019, 64, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Huyghe, S.; Mannaerts, G.P.; Baes, M.; Van Veldhoven, P.P. Peroxisomal multifunctional protein-2: The enzyme, the patients and the knockout mouse model. Biochim. Biophys. Acta 2006, 1761, 973–994. [Google Scholar] [CrossRef] [PubMed]
- Vilarinho, S.; Sari, S.; Mazzacuva, F.; Bilguvar, K.; Esendagli-Yilmaz, G.; Jain, D.; Akyol, G.; Dalgic, B.; Gunel, M.; Clayton, P.T.; et al. ACOX2 deficiency: A disorder of bile acid synthesis with transaminase elevation, liver fibrosis, ataxia, and cognitive impairment. Proc. Natl. Acad. Sci. USA 2016, 113, 11289–11293. [Google Scholar] [CrossRef]
- Mitchell, J.; Paul, P.; Chen, H.J.; Morris, A.; Payling, M.; Falchi, M.; Habgood, J.; Panoutsou, S.; Winkler, S.; Tisato, V.; et al. Familial amyotrophic lateral sclerosis is associated with a mutation in D-amino acid oxidase. Proc. Natl. Acad. Sci. USA 2010, 107, 7556–7561. [Google Scholar] [CrossRef]
- Ferdinandusse, S.; Denis, S.; Overmars, H.; Van Eeckhoudt, L.; Van Veldhoven, P.P.; Duran, M.; Wanders, R.J.; Baes, M. Developmental changes of bile acid composition and conjugation in l- and d-bifunctional protein single and double knockout mice. J. Biol. Chem. 2005, 280, 18658–18666. [Google Scholar] [CrossRef] [PubMed]
- Houten, S.M.; Denis, S.; Argmann, C.A.; Jia, Y.; Ferdinandusse, S.; Reddy, J.K.; Wanders, R.J. Peroxisomal L-bifunctional enzyme (Ehhadh) is essential for the production of medium-chain dicarboxylic acids. J. Lipid Res. 2012, 53, 1296–1303. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.I.; Tisayakorn, S.; Jorgensen, C.; D’Ambrosio, L.M.; Goudreault, M.; Gingras, A.C. PP4R4/KIAA1622 forms a novel stable cytosolic complex with phosphoprotein phosphatase 4. J. Biol. Chem. 2008, 283, 29273–29284. [Google Scholar] [CrossRef] [PubMed]
- Ramos, F.; Villoria, M.T.; Alonso-Rodriguez, E.; Clemente-Blanco, A. Role of protein phosphatases PP1, PP2A, PP4 and Cdc14 in the DNA damage response. Cell Stress 2019, 3, 70–85. [Google Scholar] [CrossRef]
- Sneider, A.; Hah, J.; Wirtz, D.; Kim, D.H. Recapitulation of molecular regulators of nuclear motion during cell migration. Cell Adh. Migr. 2019, 13, 50–62. [Google Scholar] [CrossRef]
- Gay, O.; Gilquin, B.; Assard, N.; Stuelsatz, P.; Delphin, C.; Lachuer, J.; Gidrol, X.; Baudier, J. Refilins are short-lived Actin-bundling proteins that regulate lamellipodium protrusion dynamics. Biol. Open 2016, 5, 1351–1361. [Google Scholar] [CrossRef]
- Hashimoto, K.; Ochi, H.; Sunamura, S.; Kosaka, N.; Mabuchi, Y.; Fukuda, T.; Yao, K.; Kanda, H.; Ae, K.; Okawa, A.; et al. Cancer-secreted hsa-miR-940 induces an osteoblastic phenotype in the bone metastatic microenvironment via targeting ARHGAP1 and FAM134A. Proc. Natl. Acad. Sci. USA 2018, 115, 2204–2209. [Google Scholar] [CrossRef]
- Satterfield, L.; Shuck, R.; Kurenbekova, L.; Allen-Rhoades, W.; Edwards, D.; Huang, S.; Rajapakshe, K.; Coarfa, C.; Donehower, L.A.; Yustein, J.T. miR-130b directly targets ARHGAP1 to drive activation of a metastatic CDC42-PAK1-AP1 positive feedback loop in Ewing sarcoma. Int. J. Cancer 2017, 141, 2062–2075. [Google Scholar] [CrossRef]
- Li, J.P.; Liu, Y.; Yin, Y.H. ARHGAP1 overexpression inhibits proliferation, migration and invasion of C-33A and SiHa cell lines. Onco Targets Ther. 2017, 10, 691–701. [Google Scholar] [CrossRef]
- Urade, Y.; Hayaishi, O. Prostaglandin D synthase: Structure and function. Vitam. Horm. 2000, 58, 89–120. [Google Scholar]
- Matsuoka, T.; Hirata, M.; Tanaka, H.; Takahashi, Y.; Murata, T.; Kabashima, K.; Sugimoto, Y.; Kobayashi, T.; Ushikubi, F.; Aze, Y.; et al. Prostaglandin D2 as a mediator of allergic asthma. Science 2000, 287, 2013–2017. [Google Scholar] [CrossRef]
- Mendez-Enriquez, E.; Hallgren, J. Mast Cells and Their Progenitors in Allergic Asthma. Front. Immunol. 2019, 10, 821. [Google Scholar] [CrossRef]
- Rittchen, S.; Heinemann, A. Therapeutic Potential of Hematopoietic Prostaglandin D2 Synthase in Allergic Inflammation. Cells 2019, 8, 619. [Google Scholar] [CrossRef]
- Liu, M.; Eguchi, N.; Yamasaki, Y.; Urade, Y.; Hattori, N.; Urabe, T. Protective role of hematopoietic prostaglandin D synthase in transient focal cerebral ischemia in mice. Neuroscience 2009, 163, 296–307. [Google Scholar] [CrossRef]
- Mohri, I.; Eguchi, N.; Suzuki, K.; Urade, Y.; Taniike, M. Hematopoietic prostaglandin D synthase is expressed in microglia in the developing postnatal mouse brain. Glia 2003, 42, 263–274. [Google Scholar] [CrossRef]
- Chung, C.C.; Kanetsky, P.A.; Wang, Z.; Hildebrandt, M.A.; Koster, R.; Skotheim, R.I.; Kratz, C.P.; Turnbull, C.; Cortessis, V.K.; Bakken, A.C.; et al. Meta-analysis identifies four new loci associated with testicular germ cell tumor. Nat. Genet. 2013, 45, 680–685. [Google Scholar] [CrossRef]
- Inoue, T.; Irikura, D.; Okazaki, N.; Kinugasa, S.; Matsumura, H.; Uodome, N.; Yamamoto, M.; Kumasaka, T.; Miyano, M.; Kai, Y.; et al. Mechanism of metal activation of human hematopoietic prostaglandin D synthase. Nat. Struct. Biol. 2003, 10, 291–296. [Google Scholar] [CrossRef]
- Kumar, S.; Dudley, J.T.; Filipski, A.; Liu, L. Phylomedicine: An evolutionary telescope to explore and diagnose the universe of disease mutations. Trends Genet. 2011, 27, 377–386. [Google Scholar] [CrossRef]
- Leon, S.; Goodman, J.M.; Subramani, S. Uniqueness of the mechanism of protein import into the peroxisome matrix: Transport of folded, co-factor-bound and oligomeric proteins by shuttling receptors. Biochim. Biophys. Acta 2006, 1763, 1552–1564. [Google Scholar] [CrossRef]
- Islinger, M.; Li, K.W.; Seitz, J.; Volkl, A.; Luers, G.H. Hitchhiking of Cu/Zn superoxide dismutase to peroxisomes--evidence for a natural piggyback import mechanism in mammals. Traffic 2009, 10, 1711–1721. [Google Scholar] [CrossRef]
- Schueren, F.; Lingner, T.; George, R.; Hofhuis, J.; Dickel, C.; Gartner, J.; Thoms, S. Peroxisomal lactate dehydrogenase is generated by translational readthrough in mammals. eLife 2014, 3, e03640. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002, 12, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77 (Suppl. 9), 114–122. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| rsID. (Nucleotide Change) | Gene | CCDS ID | PTS1 Motif | UniProt Subcellular Localization * | |
|---|---|---|---|---|---|
| WT | MT | ||||
| rs1049954328 (A>G) | HSD17B4 ** | CCDS56379.1 | AKL | AEL | Peroxisome |
| rs751064948 (G>C) | HSD17B4 ** | CCDS56379.1 | AKL | PKL | Peroxisome |
| rs143732132 (C>T) | DAO | CCDS9122.1 | SHL | FHL | Peroxisome |
| rs1182979757 (T>G) | ACOX2 | CCDS33775.1 | SKL | STL | Peroxisome |
| rs138945273 (T>C) | EHHADH | CCDS33901.1 | SKL | GKL | Peroxisome |
| rs968361154 (C>T) | EHHADH | CCDS33901.1 | SKL | NKL | Peroxisome |
| rsID (Nucleotide Change) | Gene | CCDS ID | Motif | PTS1 Prediction (WT) | PTS1 Prediction (MT) | UniProt Subcellular Localization * | |
|---|---|---|---|---|---|---|---|
| WT | MT | ||||||
| rs576288488 (C>T) | PPP4R4 | CCDS9921.1 | SNP | SNL | Not targeted | Targeted | Cytoplasm |
| rs760206157 (C>A) | RFLNA | CCDS9258.1 | ATL | AKL | Not targeted | Targeted | Cytoplasm, cytoskeleton |
| rs747965330 (C>T) | ARHGAP1 | CCDS7922.1 | SGL | SRL | Not targeted | Targeted | Cytoplasm |
| rs765481546 (G>C) | HPGDS | CCDS3640.1 | TKL | SKL | Not targeted | Targeted | Cytoplasm |
| rs773190605 (T>C) | GLTP | CCDS9136.1 | YKV | CKV | Twilight zone | Targeted | Cytoplasm |
| Gene | Motif | WT Kaapp (10−6) | MT Kaapp (10−6) | log Kaapp Ratio | FoldX ddGbind (kcal/mol) | PTS1 Predictor Score Difference (MT-WT) | |
|---|---|---|---|---|---|---|---|
| WT | MT | ||||||
| HSD17B4 | AKL | AEL | 2.544 | 0.065 | 1.592 | 5.138 | −37.8 |
| HSD17B4 | AKL | PKL | 2.544 | 1.915 | 0.123 | 0.154 | −4.4 |
| DAO | SHL | FHL | 1.859 | 0.186 | 1.000 | 7.910 | −18.9 |
| ACOX2 | SKL | STL | 8.015 | 0.646 | 1.094 | 3.800 | −25.0 |
| EHHADH | SKL | GKL | 6.444 | 1.424 | 0.655 | 0.920 | −8.5 |
| EHHADH | SKL | NKL | 6.444 | 2.027 | 0.502 | 1.687 | −5.8 |
| PPP4R4 | SNP | SNL | 0.065 | 1.808 | −1.444 | 0.359 | 33.9 |
| RFLNA | ATL | AKL | 0.059 | 5.826 | −1.995 | −0.723 | 31.1 |
| ARHGAP1 | SGL | SRL | 0.047 | 1.209 | −1.410 | −2.432 | 28.5 |
| HPGDS | TKL | SKL | 1.792 | 5.110 | −0.455 | 1.397 | 22.2 |
| GLTP | YKV | CKV | 0.108 | 0.020 | 0.738 | −0.877 | 10.1 |
| Gene | Associated Disease (OMIM) | rsID | ClinVar | gnomAD Allele Frequency | PolyPhen-2 | SIFT |
|---|---|---|---|---|---|---|
| HSD17B4 | DBP deficiency (type I-III), Perrault Syndrome 1 | rs1049954328 | NA | Singleton | Possibly damaging | Deleterious |
| rs751064948 | NA | Singleton | Probably damaging | Deleterious | ||
| DAO | Schizophrenia * | rs143732132 | NA | 0.018%, No homozygotes | Possibly damaging | Deleterious |
| ACOX2 | Bile acid synthesis defect, congenital | rs1182979757 | NA | Singleton | Possibly damaging | Deleterious |
| EHHADH | Fanconi renotubular syndrome 3 | rs138945273 | Likely benign | 0.028%, No homozygotes | Benign | Deleterious |
| rs968361154 | NA | Singleton | Benign | Tolerated | ||
| PPP4R4 | None | rs576288488 | NA | 0.009%, No homozygotes | Benign | Deleterious ** |
| RFLNA | None | rs760206157 | NA | 0.003%, No homozygotes | Benign | Tolerated |
| ARHGAP1 | None | rs747965330 | NA | <0.001%, No homozygotes | Probably damaging | Deleterious ** |
| HPGDS | None | rs765481546 | NA | Singleton | Possibly damaging | Deleterious |
| GLTP | None | rs773190605 | NA | Singleton | Possibly damaging | Deleterious |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chong, C.-S.; Kunze, M.; Hochreiter, B.; Krenn, M.; Berger, J.; Maurer-Stroh, S. Rare Human Missense Variants can affect the Function of Disease-Relevant Proteins by Loss and Gain of Peroxisomal Targeting Motifs. Int. J. Mol. Sci. 2019, 20, 4609. https://doi.org/10.3390/ijms20184609
Chong C-S, Kunze M, Hochreiter B, Krenn M, Berger J, Maurer-Stroh S. Rare Human Missense Variants can affect the Function of Disease-Relevant Proteins by Loss and Gain of Peroxisomal Targeting Motifs. International Journal of Molecular Sciences. 2019; 20(18):4609. https://doi.org/10.3390/ijms20184609
Chicago/Turabian StyleChong, Cheng-Shoong, Markus Kunze, Bernhard Hochreiter, Martin Krenn, Johannes Berger, and Sebastian Maurer-Stroh. 2019. "Rare Human Missense Variants can affect the Function of Disease-Relevant Proteins by Loss and Gain of Peroxisomal Targeting Motifs" International Journal of Molecular Sciences 20, no. 18: 4609. https://doi.org/10.3390/ijms20184609
APA StyleChong, C.-S., Kunze, M., Hochreiter, B., Krenn, M., Berger, J., & Maurer-Stroh, S. (2019). Rare Human Missense Variants can affect the Function of Disease-Relevant Proteins by Loss and Gain of Peroxisomal Targeting Motifs. International Journal of Molecular Sciences, 20(18), 4609. https://doi.org/10.3390/ijms20184609