Key Topics in Molecular Docking for Drug Design


Abstract
1. Introduction
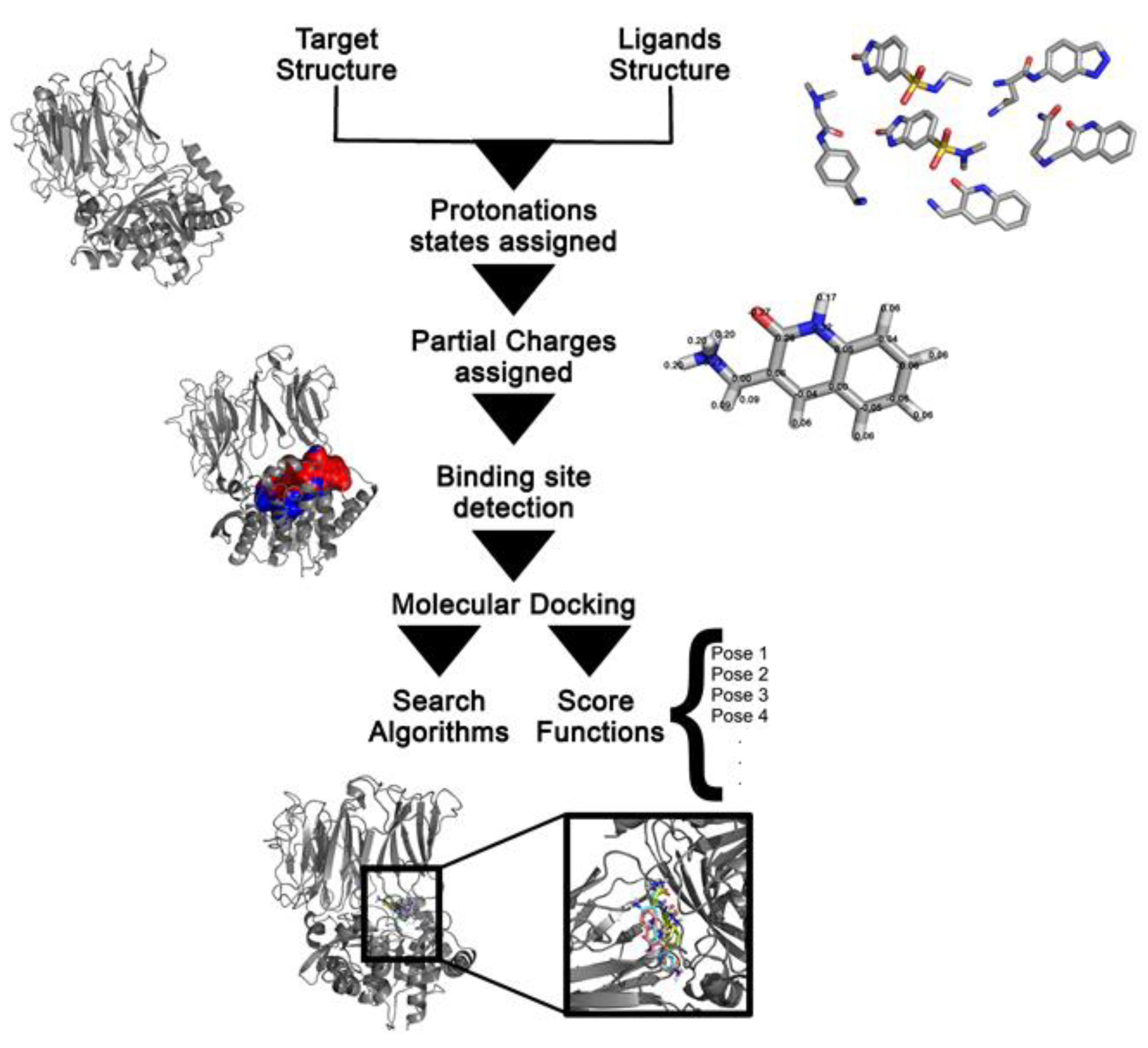
Molecular Docking in Drug Design
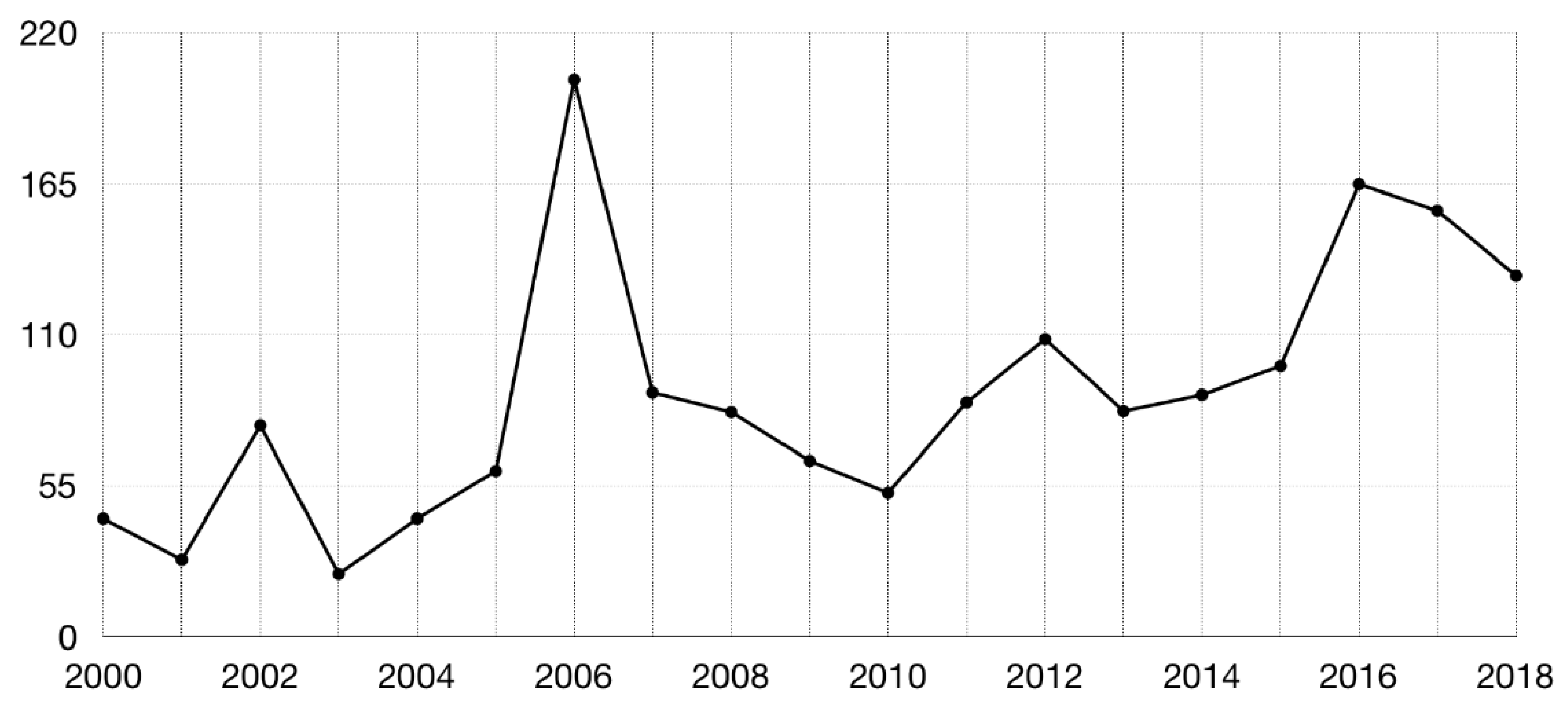
2. Benchmarking Sets
2.1. Benchmarking Sets for Pose Prediction and Binding Affinity Calculations
2.2. Benchmarking Sets for Virtual Screening
2.3. Evaluation Metrics
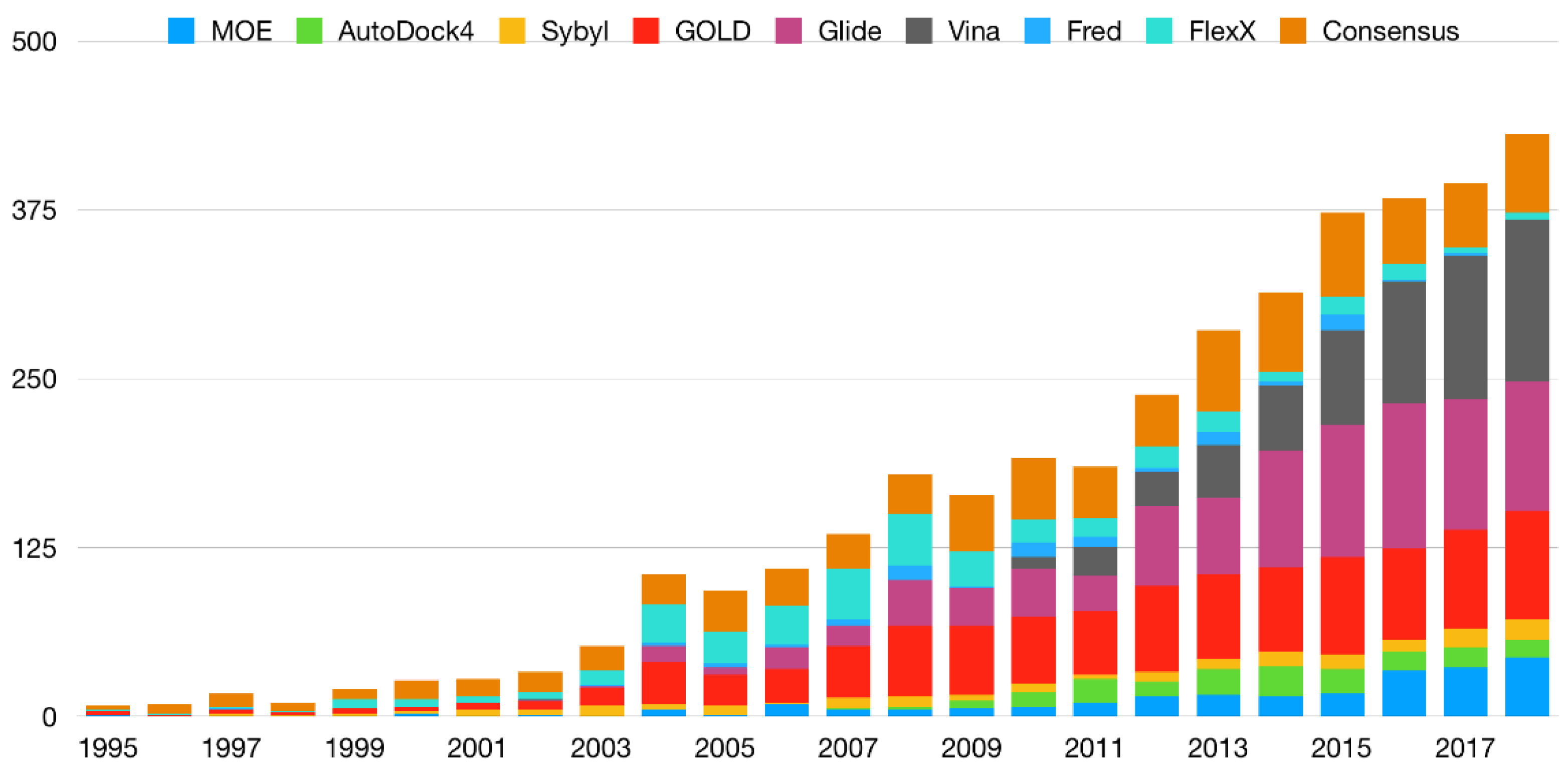
3. Consensus Methods
4. Efficient Exploration of Chemical Space: Fragment-Based Approaches
4.1. The Chemical Space
4.2. Fragment Libraries
4.3. Molecular Docking in FBDD
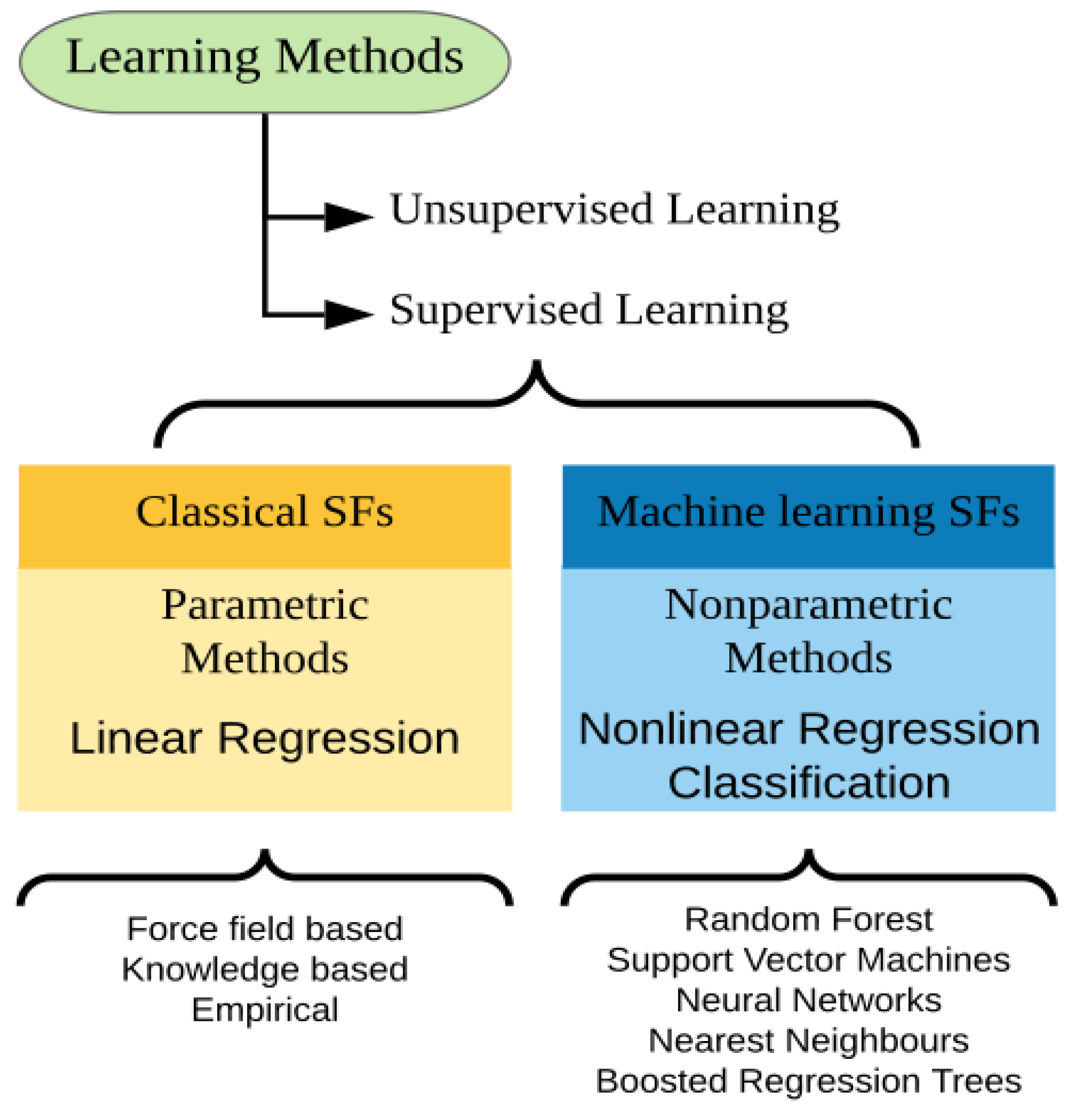
5. Machine Learning-Based Approaches
5.1. Protein Target Types: Generic and Family-Specific
5.2. Experiment Types: Binding Affinity Prediction and Virtual Screening
5.3. Algorithms and Feature Selection
5.4. Deep Learning
5.5. Recent Applications and Perspectives
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ML | Machine Learning |
| RF | Random Forest |
| MD | Molecular Dynamics |
| SF | Scoring Function |
| FBDD | Fragment-Based Drug Design |
| VS | Virtual Screening |
| MW | Molecular Weight |
| SAR | Structure-Activity Relationship |
| QSAR | Quantitative Structure-Activity Relationship |
| EF | Enrichment Factor |
| ROC | Receiver Operating Characteristic |
| PDB | Protein Data Bank |
| RMSD | Root-Mean-Square Deviation |
| MUV | Maximum Unbiased Validation |
| DUD | Directory of Useful Decoys |
| GPCR | G-Protein-Coupled Receptor |
| LADS | Latent Actives in the Decoy Set |
| BEDROC | Boltzmann-Enhanced Discrimination of Receiver Operating Characteristic |
| AUC | Area Under the Curve |
| RIE | Robust Initial Enhancement |
| DUD-E | Directory of Useful Decoys, Enhanced |
| DEKOIS | Demanding Evaluation Kits for Objective in Silico Screening |
| HTX | High Throughput X-ray Crystallography |
| RO3 | Rule of Three |
| DSF | Differential Scanning Fluorimetry |
| Rp | Pearson correlation coefficient |
| Rs | Spearman rank-correlation |
| BFGS | Broyden–Fletcher–Goldfarb–Shanno |
References
- Liu, Y.; Zhang, Y.; Zhong, H.; Jiang, Y.; Li, Z.; Zeng, G.; Chen, M.; Shao, B.; Liu, Z.; Liu, Y. Application of molecular docking for the degradation of organic pollutants in the environmental remediation: A review. Chemosphere 2018, 203, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 28, 455–461. [Google Scholar] [CrossRef] [PubMed]
- De Magalhães, C.S.; Almeida, D.M.; Barbosa, H.J.C.; Dardenne, L.E. A dynamic niching genetic algorithm strategy for docking highly flexible ligands. Inf. Sci. 2014, 289, 206–224. [Google Scholar]
- De Magalhães, C.S.; Barbosa, H.J.C.; Dardenne, L.E. Selection-Insertion Schemes in Genetic Algorithms for the Flexible Ligand Docking Problem. Lect. Notes Comput. Sci. 2004, 3102, 368–379. [Google Scholar]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R.; Uk, K.B.R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins Struct. Funct. Genet. 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef]
- Thomsen, R.; Christensen, M.H. MolDock: A new technique for high-accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef]
- Gioia, D.; Bertazzo, M.; Recanatini, M.; Masetti, M.; Cavalli, A. Dynamic docking: A paradigm shift in computational drug discovery. Molecules 2017, 22, 2029. [Google Scholar] [CrossRef]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Hetényi, C.; Van Der Spoel, D. Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS Lett. 2006, 580, 1447–1450. [Google Scholar] [CrossRef] [PubMed]
- Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, F.; Rarey, M. Combining global and local measures for structure-based druggability predictions. J. Chem. Inf. Model. 2012, 52, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Radoux, C.J.; Olsson, T.S.G.; Pitt, W.R.; Groom, C.R.; Blundell, T.L. Identifying Interactions that Determine Fragment Binding at Protein Hotspots. J. Med. Chem. 2016, 59, 4314–4325. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.Y.; Meiler, J. Predictive Power of Different Types of Experimental Restraints in Small Molecule Docking: A Review. J. Chem. Inf. Model. 2018, 58, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Brooijmans, N.; Kuntz, I.D. Molecular Recognition and Docking Algorithms. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 335–373. [Google Scholar] [CrossRef]
- Meng, E.C.; Shoichet, B.K.; Kuntz, I.D. Automated docking with grid-based energy evaluation. J. Comput. Chem. 1992, 13, 505–524. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2006, 45, 177–182. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeerschd, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Pearlman, R.S. Rapid Generation of High Quality Approximate 3-dimension Molecular Structures. Chem. Des. Auto. News 1987, 2, 1–7. [Google Scholar]
- McCammon, J.A.; Nielsen, J.E.; Baker, N.A.; Dolinsky, T.J. PDB2PQR: An automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef] [PubMed]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, L.E.; Barbosa, H.J.C.; De Magalhães, C.S.; Almeida, D.M.; da Silva, E.K.; Custódio, F.L.; Guedes, I.A. DockThor Portal. Available online: https://dockthor.lncc.br/v2/ (accessed on 22 March 2019).
- Guedes, I.A.; de Magalhães, C.S.; Dardenne, L.E. Receptor-ligand molecular docking. Biophys. Rev. 2014, 6, 75–87. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. DOCKING and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935. [Google Scholar] [CrossRef] [PubMed]
- Zsoldos, Z.; Reid, D.; Simon, A.; Sadjad, B.S.; Johnson, A.P. eHiTS: An Innovative Approach to the Docking and Scoring Function Problems. Curr. Protein Pept. Sci. 2006, 7, 421–435. [Google Scholar]
- Moitessier, N.; Englebienne, P.; Lee, D.; Lawandi, J.; Corbeil, C.R. Towards the development of universal, fast and highly accurate docking/scoring methods: A long way to go. Br. J. Pharmacol. 2008, 153, 7–26. [Google Scholar] [CrossRef]
- Hindle, S.A.; Rarey, M.; Buning, C.; Lengauer, T. Flexible docking under pharmacophore type constraints. J. Comput. Aided Mol. Des. 2002, 16, 129–149. [Google Scholar] [CrossRef]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical scoring functions for advanced Protein-Ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stützle, T.; Exner, T.E. An ant colony optimization approach to flexible protein–ligand docking. Swarm Intell. 2007, 1, 115–134. [Google Scholar] [CrossRef]
- Abagyan, R.; Kuznetsov, D.; Totrov, M. ICM-New Method for Protein Modeling and Design: Applications to Docking and Structure Prediction from. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Abagyan, R.; Totrov, M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 1994, 235, 983–1002. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.N. Surflex: Fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef]
- Jain, A.N. Surflex-Dock 2.1: Robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. J. Comput. Aided Mol. Des. 2007, 21, 281–306. [Google Scholar] [CrossRef]
- Yang, J.M.; Chen, C.C. GEMDOCK: A Generic Evolutionary Method for Molecular Docking. Proteins Struct. Funct. Bioinform. 2004, 55, 288–304. [Google Scholar] [CrossRef]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, C.; Gui, C.; Luo, X.; Chen, K.; Shen, J.; Wang, X.; Jiang, H. GAsDock: A new approach for rapid flexible docking based on an improved multi-population genetic algorithm. Bioorg. Med. Chem. Lett. 2004, 14, 4671–4676. [Google Scholar] [CrossRef] [PubMed]
- Rarey, M.; Wefing, S.; Lengauer, T. Placement of medium-sized molecular fragments into active sites of proteins. J. Comput. Aided Mol. Des. 1996, 10, 41–54. [Google Scholar] [CrossRef] [PubMed]
- McGann, M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef] [PubMed]
- Plewczynski, D.; Łaźniewski, M.; Augustyniak, R.; Ginalski, K. Can we trust docking results? Evaluation of seven commonly used programs on PDBbind database. J. Comput. Chem. 2011, 32, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.W.; Ayeni, C.; Breuer, S.; Torbett, B.E. Virtual screening for HIV protease inhibitors: A comparison of AutoDock 4 and Vina. PLoS ONE 2010, 5, e11955. [Google Scholar] [CrossRef] [PubMed]
- Capoferri, L.; Leth, R.; ter Haar, E.; Mohanty, A.K.; Grootenhuis, P.D.J.; Vottero, E.; Commandeur, J.N.M.; Vermeulen, N.P.E.; Jørgensen, F.S.; Olsen, L.; et al. Insights into regioselective metabolism of mefenamic acid by cytochrome P450 BM3 mutants through crystallography, docking, molecular dynamics, and free energy calculations. Proteins Struct. Funct. Bioinform. 2016, 84, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Pearce, L.V.; Xu, X.; Yang, X.; Yang, P.; Blumberg, P.M.; Xie, X.-Q. Structural Insight into Tetrameric hTRPV1 from Homology Modeling, Molecular Docking, Molecular Dynamics Simulation, Virtual Screening, and Bioassay Validations. J. Chem. Inf. Model. 2015, 55, 572–588. [Google Scholar] [CrossRef] [PubMed]
- Vadloori, B.; Sharath, A.K.; Prabhu, N.P.; Maurya, R. Homology modelling, molecular docking, and molecular dynamics simulations reveal the inhibition of Leishmania donovani dihydrofolate reductase-thymidylate synthase enzyme by Withaferin-A. BMC Res. Notes 2018, 11, 246. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.K.; Kumar, S.; Misra, S.; Yadav, L.; Teli, M.; Sharma, P.; Chaudhary, S.; Kumar, N.; Choi, E.H.; Kim, H.S.; et al. Molecular Insights into the Interaction of RONS and Thieno [3, 2-c]pyran Analogs with SIRT6/COX-2: A Molecular Dynamics Study. Sci. Rep. 2018, 8, 4777. [Google Scholar] [CrossRef]
- Makhouri, F.R.; Ghasemi, J.B. Combating Diseases with Computational Strategies Used for Drug Design and Discovery. Curr. Top. Med. Chem. 2019, 18, 2743–2773. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Hingerty, B.E.; Ritchie, R.H.; Ferrell, T.L.; Turner, J.E. Dielectric effects in biopolymers: The theory of ionic saturation revisited. Biopolymers 1985, 24, 427–439. [Google Scholar] [CrossRef]
- Halgren, T.A. The representation of van der Waals (vdW) interactions in molecular mechanics force fields: Potential form, combination rules, and vdW parameters. J. Am. Chem. Soc. 1992, 114, 7827–7843. [Google Scholar] [CrossRef]
- Hansch, C.; Fujita, T. ρ-σ-π Analysis. A Method for the Correlation of Biological Activity and Chemical Structure. J. Am. Chem. Soc. 1964, 86, 1616–1626. [Google Scholar] [CrossRef]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Velec, H.F.G.; Gohlke, H.; Klebe, G. DrugScore(CSD)-knowledge-based scoring function derived from small molecule crystal data with superior recognition rate of near-native ligand poses and better affinity prediction. J. Med. Chem. 2005, 48, 6296–6303. [Google Scholar] [CrossRef]
- Muegge, I. PMF scoring revisited. J. Med. Chem. 2006, 49, 5895–5902. [Google Scholar] [CrossRef]
- Bohacek, R.S.; McMartin, C.; Guida, W.C. The art and practice of structure-based drug design: A molecular modeling perspective. Med. Res. Rev. 1996, 16, 3–50. [Google Scholar] [CrossRef]
- Amaro, R.E.; Baudry, J.; Chodera, J.; Demir, Ö.; McCammon, J.A.; Miao, Y.; Smith, J.C. Ensemble Docking in Drug Discovery. Biophys. J. 2018, 114, 2271–2278. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Olsson, T.S.G.; Bowden, S.J.; Hall, R.J.; Verdonk, M.L.; Liebeschuetz, J.W.; Cole, J.C. Potential and limitations of ensemble docking. J. Chem. Inf. Model. 2012, 52, 1262–1274. [Google Scholar] [CrossRef]
- Totrov, M.; Abagyan, R. Flexible ligand docking to multiple receptor conformations: A practical alternative. Curr. Opin. Struct. Biol. 2008, 18, 178–184. [Google Scholar] [CrossRef] [PubMed]
- De Paris, R.; Vahl Quevedo, C.; Ruiz, D.D.; Gargano, F.; de Souza, O.N. A selective method for optimizing ensemble docking-based experiments on an InhA Fully-Flexible receptor model. BMC Bioinform. 2018, 19, 235. [Google Scholar] [CrossRef] [PubMed]
- De Paris, R.; Frantz, F.A.; Norberto de Souza, O.; Ruiz, D.D.A. wFReDoW: A Cloud-Based Web Environment to Handle Molecular Docking Simulations of a Fully Flexible Receptor Model. BioMed Res. Int. 2013, 2013, 469363. [Google Scholar] [CrossRef]
- Cavasotto, C.N.; Kovacs, J.A.; Abagyan, R.A. Representing receptor flexibility in ligand docking through relevant normal modes. J. Am. Chem. Soc. 2005, 127, 9632–9640. [Google Scholar] [CrossRef]
- Damm, K.L.; Carlson, H.A. Exploring experimental sources of multiple protein conformations in structure-based drug design. J. Am. Chem. Soc. 2007, 129, 8225–8235. [Google Scholar] [CrossRef]
- Leach, A.R.; Shoichet, B.K.; Peishoff, C.E. Prediction of protein-ligand interactions. Docking and scoring: successes and gaps. J. Med. Chem. 2006, 49, 5851–5855. [Google Scholar] [CrossRef]
- Wang, R.; Fang, X.; Lu, Y. The PDBbind Database: Collection of Binding Affinities for Protein–Ligand Complexes with Known Three-Dimensional Structures—Journal of Medicinal Chemistry (ACS Publications). J. Med. Chem. 2004, 47, 2977–2980. [Google Scholar] [CrossRef]
- Ahmed, A.; Smith, R.D.; Clark, J.J.; Dunbar, J.B., Jr.; Carlson, H.A. Recent improvements to Binding MOAD: A resource for protein-ligand Binding affinities and structures. Nucleic Acids Res. 2015, 43, D465–D469. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.D.; Ung, P.M.-U.; Esposito, E.X.; Wang, S.; Carlson, H.A.; Dunbar, J.B.; Yang, C.-Y. CSAR Benchmark Exercise of 2010: Combined Evaluation Across All Submitted Scoring Functions. J. Chem. Inf. Model. 2011, 51, 2115–2131. [Google Scholar] [CrossRef] [PubMed]
- Block, P. AffinDB: A freely accessible database of affinities for protein-ligand complexes from the PDB. Nucleic Acids Res. 2006, 34, D522–D526. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, R.; Fang, X.; Lu, Y.; Yang, C.Y.; Wang, S. The PDBbind database: Methodologies and updates. J. Med. Chem. 2005, 48, 4111–4119. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Liu, J.; Wang, R.; Liu, Z.; Liu, Y.; Han, L.; Li, Y.; Nie, W.; Li, J. PDB-wide collection of binding data: Current status of the PDBbind database. Bioinformatics 2014, 31, 405–412. [Google Scholar]
- Ballester, P.J.; Mitchell, J.B.O. A machine learning approach to predicting protein–ligand binding affinity with applications to molecular docking. Bioinformatics 2010, 26, 1169–1175. [Google Scholar] [CrossRef]
- Jiménez, J.; Škalič, M.; Martínez-Rosell, G.; De Fabritiis, G. KDEEP: Protein-Ligand Absolute Binding Affinity Prediction via 3D-Convolutional Neural Networks. J. Chem. Inf. Model. 2018, 58, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Durrant, J.D.; McCammon, J.A. NNScore: A neural-network-based scoring function for the characterization of protein-ligand complexes. J. Chem. Inf. Model. 2010, 50, 1865–1871. [Google Scholar] [CrossRef]
- Vreven, T.; Moal, I.H.; Vangone, A.; Pierce, B.G.; Kastritis, P.L.; Torchala, M.; Chaleil, R.; Jiménez-García, B.; Bates, P.A.; Fernandez-Recio, J.; et al. Updates to the Integrated Protein–Protein Interaction Benchmarks: Docking Benchmark Version 5 and Affinity Benchmark Version 2. J. Mol. Biol. 2015, 427, 3031–3041. [Google Scholar] [CrossRef]
- Koukos, P.I.; Faro, I.; van Noort, C.W.; Bonvin, A.M.J.J. A Membrane Protein Complex Docking Benchmark. J. Mol. Biol. 2018, 430, 5246–5256. [Google Scholar] [CrossRef]
- Li, H.; Leung, K.S.; Wong, M.H.; Ballester, P.J. Improving autodock vina using random forest: The growing accuracy of binding affinity prediction by the effective exploitation of larger data sets. Mol. Inform. 2015, 34, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Ain, Q.U.; Aleksandrova, A.; Roessler, F.D.; Ballester, P.J. Machine-learning scoring functions to improve structure-based binding affinity prediction and virtual screening. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2015, 5, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J. Community benchmarks for virtual screening. J. Comput. Aided Mol. Des. 2008, 22, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Kirchmair, J.; Markt, Æ.P.; Distinto, S.; Wolber, Æ.G. Evaluation of the performance of 3D virtual screening protocols: RMSD comparisons, enrichment assessments, and decoy selection—What can we learn from earlier mistakes? J. Comput. Aided Mol. Des. 2008, 22, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Berdini, V.; Hartshorn, M.J.; Mooij, W.T.M.; Murray, C.W.; Taylor, R.D.; Watson, P. Virtual screening using protein-ligand docking: Avoiding artificial enrichment. J. Chem. Inf. Comput. Sci. 2004, 44, 793–806. [Google Scholar] [CrossRef]
- Good, A.C.; Oprea, T.I. Optimization of CAMD techniques 3. Virtual screening enrichment studies: A help or hindrance in tool selection? J. Comput. Aided Mol. Des. 2008, 22, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Lovell, T.; Chen, H.; Lyne, P.D.; Giordanetto, F.; Li, J. On Evaluating Molecular-Docking Methods for Pose Prediction and Enrichment Factors. [J. Chem. Inf. Model. 46, 401–415 (2006)] by. J. Chem. Inf. Model. 2008, 48, 246. [Google Scholar] [CrossRef][Green Version]
- Vogel, S.M.; Bauer, M.R.; Boeckler, F.M. DEKOIS: Demanding evaluation kits for objective in silico screening—A versatile tool for benchmarking docking programs and scoring functions. J. Chem. Inf. Model. 2011, 51, 2650–2665. [Google Scholar] [CrossRef]
- Huang, N.; Shoichet, B.K.; Irwin, J.J. Benchmarking Sets for Molecular Docking Benchmarking Sets for Molecular Docking. Society 2006, 49, 6789–6801. [Google Scholar]
- Wallach, I.; Lilien, R. Virtual decoy sets for molecular docking benchmarks. J. Chem. Inf. Model. 2011, 51, 196–202. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.R.; Ibrahim, T.M.; Vogel, S.M.; Boeckler, F.M. Evaluation and optimization of virtual screening workflows with DEKOIS 2.0—A public library of challenging docking benchmark sets. J. Chem. Inf. Model. 2013, 53, 1447–1462. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, S.G.; Baumann, K. Maximum unbiased validation (MUV) data sets for virtual screening based on PubChem bioactivity data. J. Chem. Inf. Model. 2009, 49, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Gatica, E.A.; Cavasotto, C.N. Ligand and decoy sets for docking to G protein-coupled receptors. J. Chem. Inf. Model. 2012, 52, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lagarde, N.; Ben Nasr, N.; Jérémie, A.; Guillemain, H.; Laville, V.; Labib, T.; Zagury, J.F.; Montes, M. NRLiSt BDB, the manually curated nuclear receptors ligands and structures benchmarking database. J. Med. Chem. 2014, 57, 3117–3125. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Tilahun, E.L.; Kebede, E.H.; Reid, T.E.; Zhang, L.; Wang, X.S. Comparative modeling and benchmarking data sets for human histone deacetylases and sirtuin families. J. Chem. Inf. Model. 2015, 55, 374–388. [Google Scholar] [CrossRef] [PubMed]
- Cereto-Massagué, A.; Guasch, L.; Valls, C.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. DecoyFinder: An easy-to-use python GUI application for building target-specific decoy sets. Bioinformatics 2012, 28, 1661–1662. [Google Scholar] [CrossRef]
- Wang, L.; Pang, X.; Li, Y.; Zhang, Z.; Tan, W. RADER: A RApid DEcoy Retriever to facilitate decoy based assessment of virtual screening. Bioinformatics 2017, 33, 1235–1237. [Google Scholar] [CrossRef]
- Fawcett, T. An introduction to ROC analysis. Pattern Recognit. Lett. 2006, 27, 861–874. [Google Scholar] [CrossRef]
- Triballeau, N.; Acher, F.; Brabet, I.; Pin, J.-P.; Bertrand, H.-O. Virtual Screening Workflow Development Guided by the “Receiver Operating Characteristic” Curve Approach. Application to High-Throughput Docking on Metabotropic Glutamate Receptor Subtype 4. J. Med. Chem. 2005, 48, 2534–2547. [Google Scholar] [CrossRef]
- Truchon, J.F.; Bayly, C.I. Evaluating virtual screening methods: Good and bad metrics for the “early recognition” problem. J. Chem. Inf. Model. 2007, 47, 488–508. [Google Scholar] [CrossRef] [PubMed]
- Empereur-Mot, C.; Guillemain, H.; Latouche, A.; Zagury, J.F.; Viallon, V.; Montes, M. Predictiveness curves in virtual screening. J. Cheminform. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Alghamedy, F.; Bopaiah, J.; Jones, D.; Zhang, X.; Weiss, H.L.; Ellingson, S.R. Incorporating Protein Dynamics Through Ensemble Docking in Machine Learning Models to Predict Drug Binding. AMIA Summits Transl. Sci. Proc. 2018, 2017, 26–34. [Google Scholar] [PubMed]
- Sheridan, R.P.; Singh, S.B.; Fluder, E.M.; Kearsley, S.K. Protocols for Bridging the Peptide to Nonpeptide Gap in Topological Similarity Searches. J. Chem. Inf. Comput. Sci. 2002, 41, 1395–1406. [Google Scholar] [CrossRef]
- Charifson, P.S.; Corkery, J.J.; Murcko, M.A.; Walters, W.P. Consensus scoring: A method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J. Med. Chem. 1999, 42, 5100–5109. [Google Scholar] [CrossRef]
- Wang, R.; Wang, S. How does consensus scoring work for virtual library screening? An idealized computer experiment. J. Chem. Inf. Comput. Sci. 2001, 41, 1422–1426. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Li, H.; Jiang, H.; Wang, X.; Zheng, M.; Luo, J.; Zhang, H.; Liu, X. An effective docking strategy for virtual screening based on multi-objective optimization algorithm. BMC Bioinform. 2009, 10, 58. [Google Scholar]
- Nguyen, D.D.; Cang, Z.; Wu, K.; Wang, M.; Cao, Y.; Wei, G.W. Mathematical deep learning for pose and binding affinity prediction and ranking in D3R Grand Challenges. J. Comput. Aided Mol. Des. 2018, 33, 71–82. [Google Scholar] [CrossRef]
- Wang, R.; Lu, Y.; Wang, S. Comparative evaluation of 11 scoring functions for molecular docking. J. Med. Chem. 2003, 46, 2287–2303. [Google Scholar] [CrossRef]
- Ren, X.; Shi, Y.-S.; Zhang, Y.; Liu, B.; Zhang, L.-H.; Peng, Y.-B.; Zeng, R. Novel Consensus Docking Strategy to Improve Ligand Pose Prediction. J. Chem. Inf. Model. 2018, 58, 1662–1668. [Google Scholar] [CrossRef]
- Poli, G.; Martinelli, A.; Tuccinardi, T. Reliability analysis and optimization of the consensus docking approach for the development of virtual screening studies. J. Enzyme Inhib. Med. Chem. 2016, 31, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Tuccinardi, T.; Poli, G.; Romboli, V.; Giordano, A.; Martinelli, A. Extensive consensus docking evaluation for ligand pose prediction and virtual screening studies. J. Chem. Inf. Model. 2014, 54, 2980–2986. [Google Scholar] [CrossRef] [PubMed]
- Houston, D.R.; Walkinshaw, M.D. Consensus docking: Improving the reliability of docking in a virtual screening context. J. Chem. Inf. Model. 2013, 53, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Plewczynski, D.; Łażniewski, M.; Von Grotthuss, M.; Rychlewski, L.; Ginalski, K. VoteDock: Consensus docking method for prediction of protein-ligand interactions. J. Comput. Chem. 2011, 32, 568–581. [Google Scholar] [CrossRef] [PubMed]
- Perez-castillo, Y.; Sotomayor-burneo, S.; Jimenes-vargas, K.; Gonzalez-, M. CompScore: Boosting structure-based virtual screening performance by incorporating docking scoring functions components into consensus scoring. BioRxiv 2019. [Google Scholar] [CrossRef] [PubMed]
- Onawole, A.T.; Kolapo, T.U.; Sulaiman, K.O.; Adegoke, R.O. Structure based virtual screening of the Ebola virus trimeric glycoprotein using consensus scoring. Comput. Biol. Chem. 2018, 72, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Aliebrahimi, S.; Karami, L.; Arab, S.S.; Montasser Kouhsari, S.; Ostad, S.N. Identification of Phytochemicals Targeting c-Met Kinase Domain using Consensus Docking and Molecular Dynamics Simulation Studies. Cell Biochem. Biophys. 2017, 76, 135–145. [Google Scholar] [CrossRef]
- Li, D.D.; Meng, X.F.; Wang, Q.; Yu, P.; Zhao, L.G.; Zhang, Z.P.; Wang, Z.Z.; Xiao, W. Consensus scoring model for the molecular docking study of mTOR kinase inhibitor. J. Mol. Graph. Model. 2018, 79, 81–87. [Google Scholar] [CrossRef]
- Oda, A.; Tsuchida, K.; Takakura, T.; Yamaotsu, N.; Hirono, S. Comparison of consensus scoring strategies for evaluating computational models of protein-ligand complexes. J. Chem. Inf. Model. 2006, 46, 380–391. [Google Scholar] [CrossRef]
- Chaput, L.; Martinez-Sanz, J.; Quiniou, E.; Rigolet, P.; Saettel, N.; Mouawad, L. VSDC: A method to improve early recognition in virtual screening when limited experimental resources are available. J. Cheminform. 2016, 8. [Google Scholar] [CrossRef]
- Mavrogeni, M.E.; Pronios, F.; Zareifi, D.; Vasilakaki, S.; Lozach, O.; Alexopoulos, L.; Meijer, L.; Myrianthopoulos, V.; Mikros, E. A facile consensus ranking approach enhances virtual screening robustness and identifies a cell-active DYRK1α inhibitor. Future Med. Chem. 2018, 10, 2411–2430. [Google Scholar] [CrossRef] [PubMed]
- Zhan, W.; Li, D.; Che, J.; Zhang, L.; Yang, B.; Hu, Y.; Liu, T.; Dong, X. Integrating docking scores, interaction profiles and molecular descriptors to improve the accuracy of molecular docking: Toward the discovery of novel Akt1 inhibitors. Eur. J. Med. Chem. 2014, 75, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Ericksen, S.S.; Wu, H.; Zhang, H.; Michael, L.A.; Newton, M.A.; Hoffmann, F.M.; Wildman, S.A. Machine Learning Consensus Scoring Improves Performance Across Targets in Structure-Based Virtual Screening. J. Chem. Inf. Model. 2017, 57, 1579–1590. [Google Scholar] [CrossRef] [PubMed]
- Teramoto, R.; Fukunishi, H. Supervised consensus scoring for docking and virtual screening. J. Chem. Inf. Model. 2007, 47, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; He, W.; Zhou, X.; Chen, X. Optimization of molecular docking scores with support vector rank regression. Proteins Struct. Funct. Bioinform. 2013, 81, 1386–1398. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.M.; Hsu, D.F. Consensus scoring criteria in structure-based virtual screening. Emerg. Inf. Technol. Conf. 2005 2005, 2005, 165–167. [Google Scholar]
- Liu, S.; Fu, R.; Zhou, L.-H.; Chen, S.-P. Application of Consensus Scoring and Principal Component Analysis for Virtual Screening against β-Secretase (BACE-1). PLoS ONE 2012, 7, e38086. [Google Scholar] [CrossRef] [PubMed]
- Mokrani, E.H.; Bensegueni, A.; Chaput, L.; Beauvineau, C.; Djeghim, H.; Mouawad, L. Identification of New Potent Acetylcholinesterase Inhibitors Using Virtual Screening and In Vitro Approaches. Mol. Inform. 2019, 38, 1800118. [Google Scholar] [CrossRef]
- Russo Spena, C.; De Stefano, L.; Poli, G.; Granchi, C.; El Boustani, M.; Ecca, F.; Grassi, G.; Grassi, M.; Canzonieri, V.; Giordano, A.; et al. Virtual screening identifies a PIN1 inhibitor with possible antiovarian cancer effects. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Mouawad, N.; Jha, V.; Poli, G.; Granchi, C.; Rizzolio, F.; Caligiuri, I.; Minutolo, F.; Lapillo, M.; Tuccinardi, T.; Macchia, M. Computationally driven discovery of phenyl(piperazin-1-yl) methanone derivatives as reversible monoacylglycerol lipase (MAGL) inhibitors. J. Enzyme Inhib. Med. Chem. 2019, 34, 589–596. [Google Scholar]
- Damm-Ganamet, K.L.; Dunbar, J.B.; Ahmed, A.; Esposito, E.X.; Stuckey, J.A.; Gestwicki, J.E.; Chinnaswamy, K.; Delproposto, J.; Smith, R.D.; Carlson, H.A.; et al. CSAR Data Set Release 2012: Ligands, Affinities, Complexes, and Docking Decoys. J. Chem. Inf. Model. 2013, 53, 1842–1852. [Google Scholar]
- Walters, W.P.; Liu, S.; Chiu, M.; Shao, C.; Rudolph, M.G.; Burley, S.K.; Gilson, M.K.; Feher, V.A.; Gaieb, Z.; Kuhn, B.; et al. D3R Grand Challenge 2: Blind prediction of protein–ligand poses, affinity rankings, and relative binding free energies. J. Comput. Aided Mol. Des. 2017, 32, 1–20. [Google Scholar]
- Nevins, N.; Yang, H.; Walters, W.P.; Ameriks, M.K.; Parks, C.D.; Gilson, M.K.; Gaieb, Z.; Lambert, M.H.; Shao, C.; Chiu, M.; et al. D3R Grand Challenge 3: Blind prediction of protein–ligand poses and affinity rankings. J. Comput. Aided Mol. Des. 2019, 33, 1–18. [Google Scholar]
- Shuker, S.B.; Hajduk, P.J.; Meadows, R.P.; Fesik, S.W. Discovering High-Affinity Ligands for Proteins: SAR by NMR. Science 1996, 274, 1531–1534. [Google Scholar] [CrossRef] [PubMed]
- Romasanta, A.K.S.; van der Sijde, P.; Hellsten, I.; Hubbard, R.E.; Keseru, G.M.; van Muijlwijk-Koezen, J.; de Esch, I.J.P. When fragments link: A bibliometric perspective on the development of fragment-based drug discovery. Drug Discov. Today 2018, 23, 1596–1609. [Google Scholar] [CrossRef]
- Erlanson, D.A. Introduction to fragment-based drug discovery. Top. Curr. Chem. 2012, 317, 1–32. [Google Scholar]
- Hann, M.M.; Leach, A.R.; Harper, G. Molecular Complexity and Its Impact on the Probability of Finding Leads for Drug Discovery. J. Chem. Inf. Comput. Sci. 2001, 41, 856–864. [Google Scholar] [CrossRef]
- Leach, A.R.; Hann, M.M. Molecular complexity and fragment-based drug discovery: Ten years on. Curr. Opin. Chem. Biol. 2011, 15, 489–496. [Google Scholar] [CrossRef]
- Fink, T.; Raymond, J.L. Virtual exploration of the chemical universe up to 11 atoms of C, N, O, F: Assembly of 26.4 million structures (110.9 million stereoisomers) and analysis for new ring systems, stereochemistry, physicochemical properties, compound classes, and drug discovery. J. Chem. Inf. Model. 2007, 47, 342–353. [Google Scholar]
- Lyu, J.; Irwin, J.J.; Roth, B.L.; Shoichet, B.K.; Levit, A.; Wang, S.; Tolmachova, K.; Singh, I.; Tolmachev, A.A.; Che, T.; et al. Ultra-large library docking for discovering new chemotypes. Nature 2019, 566, 224. [Google Scholar] [CrossRef]
- Scott, D.E.; Coyne, A.G.; Hudson, S.A.; Abell, C. Fragment-based approaches in drug discovery and chemical biology. Biochemistry 2012, 51, 4990–5003. [Google Scholar] [CrossRef] [PubMed]
- Blundell, T.L.; Jhoti, H.; Abell, C. High-throughput crystallography for lead discovery in drug design. Nat. Rev. Drug Discov. 2002, 1, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Abad-Zapatero, C.; Metz, J.T. Ligand efficiency indices as guideposts for drug discovery. Drug Discov. Today 2005, 10, 464–469. [Google Scholar] [CrossRef]
- Reynolds, C.H.; Bembenek, S.D.; Tounge, B.A. The role of molecular size in ligand efficiency. Bioorg. Med. Chem. Lett. 2007, 17, 4258–4261. [Google Scholar] [CrossRef] [PubMed]
- Schultes, S.; De Graaf, C.; Haaksma, E.E.J.; De Esch, I.J.P.; Leurs, R.; Krämer, O. Ligand efficiency as a guide in fragment hit selection and optimization. Drug Discov. Today Technol. 2010, 7, 157–162. [Google Scholar] [CrossRef]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A ‘Rule of Three’ for fragment-based lead discovery? Recent. Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- Jhoti, H.; Williams, G.; Rees, D.C.; Murray, C.W. The “rule of three” for fragment-based drug discovery: Where are we now? Nat. Rev. Drug Discov. 2013, 12, 644. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Morley, A.D.; Pugliese, A.; Birchall, K.; Bower, J.; Brennan, P.; Brown, N.; Chapman, T.; Drysdale, M.; Gilbert, I.H.; Hoelder, S.; et al. Fragment-based hit identification: Thinking in 3D. Drug Discov. Today 2013, 18, 1221–1227. [Google Scholar] [CrossRef]
- Verheij, H.J. Leadlikeness and structural diversity of synthetic screening libraries. Mol. Divers. 2006, 10, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Hubbard, R.E. Fragment-based ligand discovery. Mol. Interv. 2009, 9, 22–30. [Google Scholar] [CrossRef]
- Schuffenhauer, A.; Ruedisser, S.; Jahnke, W.; Marzinzik, A.; Selzer, P.; Jacoby, E. Library Design for Fragment Based Screening. Curr. Top. Med. Chem. 2005, 5, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Lewell, X.Q.; Judd, D.B.; Watson, S.P.; Hann, M.M. RECAP—Retrosynthetic Combinatorial Analysis Procedure: A powerful new technique for identifying privileged molecular fragments with useful applications in combinatorial chemistry. J. Chem. Inf. Comput. Sci. 1998, 38, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Prescher, H.; Koch, G.; Schuhmann, T.; Ertl, P.; Bussenault, A.; Glick, M.; Dix, I.; Petersen, F.; Lizos, D.E. Construction of a 3D-shaped, natural product like fragment library by fragmentation and diversification of natural products. Bioorg. Med. Chem. 2017, 25, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shoichet, B.K. Molecular docking and ligand specificity in fragment-based inhibitor discovery. Nat. Chem. Biol. 2009, 5, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Fjellström, O.; Akkaya, S.; Beisel, H.G.; Eriksson, P.O.; Erixon, K.; Gustafsson, D.; Jurva, U.; Kang, D.; Karis, D.; Knecht, W.; et al. Creating novel activated factor XI inhibitors through fragment based lead generation and structure aided drug design. PLoS ONE 2015, 10, e0113705. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Shin, Y.; Kim, J.; Hong, S. Application of Fragment-Based de Novo Design to the Discovery of Selective Picomolar Inhibitors of Glycogen Synthase Kinase-3 Beta. J. Med. Chem. 2016, 59, 9018–9034. [Google Scholar] [CrossRef]
- Wang, R.; Gao, Y.; Lai, L. LigBuilder: A Multi-Purpose Program for Structure-Based Drug Design. J. Mol. Model. 2004, 6, 498–516. [Google Scholar] [CrossRef]
- Zhao, H.; Gartenmann, L.; Dong, J.; Spiliotopoulos, D.; Caflisch, A. Discovery of BRD4 bromodomain inhibitors by fragment-based high-throughput docking. Bioorg. Med. Chem. Lett. 2014, 24, 2493–2496. [Google Scholar] [CrossRef]
- Rudling, A.; Gustafsson, R.; Almlöf, I.; Homan, E.; Scobie, M.; Warpman Berglund, U.; Helleday, T.; Stenmark, P.; Carlsson, J. Fragment-Based Discovery and Optimization of Enzyme Inhibitors by Docking of Commercial Chemical Space. J. Med. Chem. 2017, 60, 8160–8169. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, J.; Hoffer, L.; Coutard, B.; Querat, G.; Roche, P.; Morelli, X.; Decroly, E.; Barral, K. Optimization of a fragment linking hit toward Dengue and Zika virus NS5 methyltransferases inhibitors. Eur. J. Med. Chem. 2019, 161, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Akabayov, S.R.; Richardson, C.C.; Arthanari, H.; Akabayov, B.; Ilic, S.; Wagner, G. Identification of DNA primase inhibitors via a combined fragment-based and virtual screening. Sci. Rep. 2016, 6, 36322. [Google Scholar]
- Amaning, K.; Lowinski, M.; Vallee, F.; Steier, V.; Marcireau, C.; Ugolini, A.; Delorme, C.; Foucalt, F.; McCort, G.; Derimay, N.; et al. The use of virtual screening and differential scanning fluorimetry for the rapid identification of fragments active against MEK1. Bioorg. Med. Chem. Lett. 2013, 23, 3620–3626. [Google Scholar] [CrossRef] [PubMed]
- Barelier, S.; Eidam, O.; Fish, I.; Hollander, J.; Figaroa, F.; Nachane, R.; Irwin, J.J.; Shoichet, B.K.; Siegal, G. Increasing chemical space coverage by combining empirical and computational fragment screens. ACS Chem. Biol. 2014, 9, 1528–1535. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.; Kobayashi, T.; Lawson, J.D.; Saitoh, M.; Shimokawa, K.; Bigi, S.V.; Hixon, M.S.; Smith, C.R.; Tatamiya, T.; Goto, M.; et al. Fragment-based drug discovery of potent and selective MKK3/6 inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 1086–1089. [Google Scholar] [CrossRef]
- Darras, F.H.; Pockes, S.; Huang, G.; Wehle, S.; Strasser, A.; Wittmann, H.J.; Nimczick, M.; Sotriffer, C.A.; Decker, M. Synthesis, biological evaluation, and computational studies of Tri- and tetracyclic nitrogen-bridgehead compounds as potent dual-acting AChE inhibitors and h H3 receptor antagonists. ACS Chem. Neurosci. 2014, 5, 225–242. [Google Scholar] [CrossRef]
- He, Y.; Guo, X.; Yu, Z.H.; Wu, L.; Gunawan, A.M.; Zhang, Y.; Dixon, J.E.; Zhang, Z.Y. A potent and selective inhibitor for the UBLCP1 proteasome phosphatase. Bioorg. Med. Chem. 2015, 23, 2798–2809. [Google Scholar] [CrossRef]
- Bishop, C.M. Pattern Recognition and Machine Learning; Springer: New York, NY, USA, 2007; ISBN 978-0-387-31073-2. [Google Scholar]
- Ashtawy, H.M.; Mahapatra, N.R. A comparative assessment of predictive accuracies of conventional and machine learning scoring functions for protein-ligand binding affinity prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2015, 12, 335–347. [Google Scholar] [CrossRef]
- Ashtawy, H.M.; Mahapatra, N.R. Machine-learning scoring functions for identifying native poses of ligands docked to known and novel proteins. BMC Bioinform. 2015, 16. [Google Scholar] [CrossRef]
- Hassan, M.; Mogollón, D.C.; Fuentes, O. DLSCORE: A Deep Learning Model for Predicting Protein-Ligand Binding Affinities. ChemRxiv 2018, 13, 53. [Google Scholar]
- Ouyang, X.; Handoko, S.D.; Kwoh, C.K. Cscore: A Simple Yet Effective Scoring Function for Protein–Ligand Binding Affinity Prediction Using Modified Cmac Learning Architecture. J. Bioinform. Comput. Biol. 2011, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kinnings, S.L.; Liu, N.; Tonge, P.J.; Jackson, R.M.; Xie, L.; Bourne, P.E. A machine learning-based method to improve docking scoring functions and its application to drug repurposing. J. Chem. Inf. Model. 2011, 51, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Hsin, K.Y.; Ghosh, S.; Kitano, H. Combining machine learning systems and multiple docking simulation packages to improve docking prediction reliability for network pharmacology. PLoS ONE 2013, 8, e83922. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.C.; Caffarena, E.R.; Dos Santos, C.N. Boosting Docking-Based Virtual Screening with Deep Learning. J. Chem. Inf. Model. 2016, 56, 2495–2506. [Google Scholar] [CrossRef] [PubMed]
- Pason, L.P.; Sotriffer, C.A. Empirical Scoring Functions for Affinity Prediction of Protein-ligand Complexes. Mol. Inform. 2016, 35, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.G.; Simoes, C.J.V.; Carreiras, P.; Brito, R.M.M. Enhancing Scoring Performance of Docking-Based Virtual Screening Through Machine Learning. Curr. Bioinform. 2016, 11, 408–420. [Google Scholar] [CrossRef]
- Korkmaz, S.; Zararsiz, G.; Goksuluk, D. MLViS: A web tool for machine learning-based virtual screening in early-phase of drug discovery and development. PLoS ONE 2015, 10, e0124600. [Google Scholar] [CrossRef]
- Springer, C.; Adalsteinsson, H.; Young, M.M.; Kegelmeyer, P.W.; Roe, D.C. PostDOCK: A Structural, Empirical Approach to Scoring Protein Ligand Complexes. J. Med. Chem. 2005, 48, 6821–6831. [Google Scholar] [CrossRef]
- Ashtawy, H.M.; Mahapatra, N.R. Task-Specific Scoring Functions for Predicting Ligand Binding Poses and Affinity and for Screening Enrichment. J. Chem. Inf. Model. 2018, 58, 119–133. [Google Scholar] [CrossRef]
- Imrie, F.; Bradley, A.R.; Van Der Schaar, M.; Deane, C.M. Protein Family-Specific Models Using Deep Neural Networks and Transfer Learning Improve Virtual Screening and Highlight the Need for More Data. J. Chem. Inf. Model. 2018, 58, 2319–2330. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guo, Y.; Kuang, Q.; Pu, X.; Ji, Y.; Zhang, Z.; Li, M. A comparative study of family-specific protein-ligand complex affinity prediction based on random forest approach. J. Comput. Aided Mol. Des. 2015, 29, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Wójcikowski, M.; Ballester, P.J.; Siedlecki, P. Performance of machine-learning scoring functions in structure-based virtual screening. Sci. Rep. 2017, 7, 46710. [Google Scholar] [CrossRef]
- Cao, Y.; Li, L. Improved protein-ligand binding affinity prediction by using a curvature-dependent surface-area model. Bioinformatics 2014, 30, 1674–1680. [Google Scholar] [CrossRef] [PubMed]
- Yuriev, E.; Ramsland, P.A. Latest developments in molecular docking: 2010–2011 in review. J. Mol. Recognit. 2013, 26, 215–239. [Google Scholar] [CrossRef] [PubMed]
- Guedes, I.A.; Pereira, F.S.S.; Dardenne, L.E. Empirical scoring functions for structure-based virtual screening: Applications, critical aspects, and challenges. Front. Pharmacol. 2018, 9, 1089. [Google Scholar] [CrossRef]
- Li, L.; Wang, B.; Meroueh, S.O. Support Vector Regression Scoring of Receptor–Ligand Complexes for Rank-Ordering and Virtual Screening of Chemical Libraries. J. Chem. Inf. Model. 2011, 51, 2132–2138. [Google Scholar] [CrossRef]
- Guyon, I.; Elisseeff, A. An Introduction to Variable and Feature Selection. J. Mach. Learn. Res. 2011, 3, 1157–1182. [Google Scholar]
- Koppisetty, C.A.K.; Frank, M.; Kemp, G.J.L.; Nyholm, P.G. Computation of binding energies including their enthalpy and entropy components for protein-ligand complexes using support vector machines. J. Chem. Inf. Model. 2013, 53, 2559–2570. [Google Scholar] [CrossRef]
- Liu, Q.; Kwoh, C.K.; Li, J. Binding affinity prediction for protein-ligand complexes based on β contacts and B factor. J. Chem. Inf. Model. 2013, 53, 3076–3085. [Google Scholar] [CrossRef]
- Ballester, P.J.; Schreyer, A.; Blundell, T.L. Does a More Precise Chemical Description of Protein—Ligand Complexes Lead to More Accurate Prediction of Binding Affinity? J. Chem. Inf. Model. 2014, 54, 944–955. [Google Scholar] [CrossRef] [PubMed]
- Kundu, I.; Paul, G.; Banerjee, R. A machine learning approach towards the prediction of protein–ligand binding affinity based on fundamental molecular properties. RSC Adv. 2018, 8, 12127–12137. [Google Scholar] [CrossRef]
- Srinivas, R.; Klimovich, P.V.; Larson, E.C. Implicit-descriptor ligand-based virtual screening by means of collaborative filtering. J. Cheminform. 2018, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Ragoza, M.; Hochuli, J.; Idrobo, E.; Sunseri, J.; Koes, D.R. Protein-Ligand Scoring with Convolutional Neural Networks. J. Chem. Inf. Model. 2017, 57, 942–957. [Google Scholar] [CrossRef] [PubMed]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. Convolutional Neural Networks ImageNet Classification with Deep Convolutional Neural Network. Commun. ACM 2017, 60. [Google Scholar] [CrossRef]
- Khamis, M.A.; Gomaa, W.; Ahmed, W.F. Machine learning in computational docking. Artif. Intell. Med. 2015, 63, 135–152. [Google Scholar] [CrossRef]
- Sieg, J.; Flachsenberg, F.; Rarey, M. In the need of bias control: Evaluation of chemical data for Machine Learning Methods in Virtual Screening. J. Chem. Inf. Model. 2019, 59, 947–961. [Google Scholar] [CrossRef]
- Durrant, J.D.; Carlson, K.E.; Martin, T.A.; Offutt, T.L.; Mayne, C.G.; Katzenellenbogen, J.A.; Amaro, R.E. Neural-Network Scoring Functions Identify Structurally Novel Estrogen-Receptor Ligands. J. Chem. Inf. Model. 2015, 55, 1953–1961. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Ascher, D.B. CSM-lig: A web server for assessing and comparing protein-small molecule affinities. Nucleic Acids Res. 2016, 44, W557–W561. [Google Scholar] [CrossRef]
- Zilian, D.; Sotriffer, C.A. SFCscore RF: A Random Forest-Based Scoring Function for Improved Affinity Prediction of Protein–Ligand Complexes. J. Chem. Inf. Model. 2013, 53, 1923–1933. [Google Scholar] [CrossRef]
- Li, G.-B.; Yang, L.-L.; Wang, W.-J.; Li, L.-L.; Yang, S.-Y. ID-Score: A New Empirical Scoring Function Based on a Comprehensive Set of Descriptors Related to Protein–Ligand Interactions. J. Chem. Inf. Model. 2013, 53, 592–600. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Software | Posing | Scoring | Availability | Reference |
|---|---|---|---|---|
| Vina | Iterated Local Search + BFGS Local Optimiser | Empirical/Knowledge-Based | Free (Apache License) | Trott, 2010 [3] |
| AutoDock4 | Lamarckian Genetic Algorithm, Genetic Algorithm or Simulated Annealing | Semiempirical | Free (GNU License) | Morris, 2009; Huey, 2007 [31,32] |
| Molegro/MolDock | Differential Evolution (Alternatively Simplex Evolution and Iterated Simplex) | Semiempirical | Commercial | Thomsen, 2006 [9] |
| Smina | Monte Carlo stochastic sampling + local optimisation | Empirical (customisable) | Free (GNU License) | Koes, 2013 [33] |
| Plants | Ant Colony Optimisation | Empirical | Academic License | Korb, 2007; Korb, 2009 [34,35] |
| ICM | Biased Probability Monte Carlo + Local Optimisation | Physics-Based | Commercial | Abagyan, 1993; Abagyan, 1994 [36,37] |
| Glide | Systematic search + Optimisation (XP mode also uses anchor-and-grow) | Empirical | Commercial | Friesner, 2004 [38] |
| Surflex | Fragmentation and alignment to idealised molecule (Protomol) + BFGS optimisation | Empirical | Commercial | Jain, 2003; Jain 2007 [39,40] |
| GOLD | Genetic Algorithm | Physics-based (GoldScore), Empirical (ChemScore, ChemPLP) and Knowledge-based (ASP) | Commercial | Jones, 1997; Verdonk 2003 [6,7] |
| GEMDOCK | Generic Evolutionary Algorithm | Empirical (includes pharmacophore potential) | Free (for non-commercial research) | Yang, 2004 [41] |
| Dock6 | Anchor-and-grow incremental construction | Physics-based (several other options) | Academic License | Allen, 2015 [42] |
| GAsDock | Entropy-based multi-population genetic algorithm | Physics-based | * | Li, 2004 [43] |
| FlexX | Fragment-Based Pattern-recognition (Pose Clustering) + Incremental Growth | Empirical | Commercial | Rarey, 1996; Rarey, 1996b [8,44] |
| Fred | Conformer generation + Systematic rigid body search | Empirical (defaults to Chemgauss3) | Commercial | McGann, 2011 [45] |
| DockThor | Steady-state genetic algorithm (with Dynamic Modified Restricted Tournament Selection method) | Physics-based + Empirical | Free (Webserver) | De Magalhães, 2014 [4,25] |
| Source | T a | Posing b | F c | Consensus Strategy | Analysis | Ref. |
|---|---|---|---|---|---|---|
| DUD-E/ PDB | 102/3 | 4 | 4 | Standard Deviation Consensus (SDC), Variable SDC (vSDC) | Rank/Score curves Hit recovery count | Chaput, 2016 [121] |
| DUD-E | 21 | 8 | 8 | Gradient Boosting | EF, ROCAUC | Ericksen, 2017 [124] |
| PDBBind DUD | 228/1 | Vina, AutoDock | 2 | Compound rejection if pose RMSD > 2.0 Å | Success rate | Houston, 2013 [114] |
| PDB | 3 | GAsDock | 2 | Multi-Objective Scoring Function Optimisation | EF | Kang, 2019 [108] |
| mTOR d Inhibitors | 1 | Glide | 26 | Linear Combination | BEI Correlation | Li, 2018 [119] |
| PDB | 220 | FlexX | 9 | Several e | Compression and Accuracy | Oda, 2006 [120] |
| DUD-E | 102 | Dock 3.6 | 15 | Genetic Algorithm used to combine SF components | EF, BEDROC | Perez-Castillo, 2019 [116] |
| PDBBind | 1300 | 7 | 7 | RMSD-based pose consensus, multivariate linear regression | Success rate | Plewczynski, 2011 [115] |
| DUD | 35 | 10 | 10 | Compound rejection based on RMSD consensus level | EF | Poli, 2016 [112] |
| PDBBind | 3535 | 11 | 11 | Selection of representative pose with minimum RMSD | Success rate | Ren, 2018 [111] |
| PDB | 100 | AutoDock | 11 | Supervised Learning (Random Forests), Rank-by-rank | Average RMSD, Success rate | Teramoto, 2007 [125] |
| PDB DUD | 130/3 | 10 | 10 | Compound rejection based on RMSD consensus level | EF, ROCAUC | Tuccinardi (2014) [113] |
| PDBBind CSAR | 421 | Glide | 7 | Support Vector Rank Regression | Top pose /Top Rank | Wang, 2013 [126] |
| PDB | 4 | GEMDOCK GOLD | 2 | Rank-by-rank, Rank-by-score | Rank/Score curve, GH Score, CS index | Yang, 2005 [127] |
| Target | Lig. | Posing | F a | Consensus Strategy | Hits/Test | Best Activity (IC50) | Ref. |
|---|---|---|---|---|---|---|---|
| EBOV Glycoprotein | 3.57 × 107 | VINA, FlexX | 2 | Sequential Docking | - | - | Onawole, 2018 [117] |
| β-secretase (BACE1) | 1.13 × 105 | Surflex | 12 | Z-scaled rank-by-number Principal Component Analysis | 2/20 | 51.6 μM | Liu, 2012 [128] |
| c-Met Kinase | 738 | 2 | 2 | Sequential Docking Compound rejection if pose RMSD > 2.0 Å | - | - | Aliebrahimi, 2017 [118] |
| Acetylcholinesterase | 14,758 | 4 | 4 | vSDC [121] | 12/14 | 47.3 nM | Mokrani, 2019 [129] |
| PIN1 | 32,500 | 10 | 10 | Compound rejection based on RMSD consensus level | 1/10 | 13.4 μM 53.9 µM c | Spena, 2019 [130] |
| Akt1 | 47 | LigandFit | 5 | Support Vector Regression | 6/6 b | 7.7 nM | Zhan, 2014 [123] |
| Monoacylglycerol Lipase (MAGL) | 4.80 × 105 | 4 | 4 | Compound rejection based on RMSD consensus level | 1/3 | 6.1 µM | Mouawad, 2019 [131] |
| SF Name | ML Algorithm | Training Database | Best Performance | Generic or Family Specific | Type of Docking Study | Reference |
|---|---|---|---|---|---|---|
| RF-Score | RF a | PDBbind | Rp b = 0.776 | Generic | BAP c | Ballester 2010 [77] |
| B2BScore | RF | PDBbind | Rp = 0.746 | Generic | BAP | Liu 2013 [192] |
| SFCScoreRF | RF | PDBbind | Rp = 0.779 | Generic | BAP | Zilian, 2013 [202] |
| PostDOCK | RF | Constructed from PDB | 92% accuracy | Generic | VS d | Springer, 2005 [181] |
| - | SVM e | DUD | - | Both | VS | Kinnings, 2011 [175] |
| ID-Score | SVR f | PDBbind | Rp = 0.85 | Generic | BAP | Li, 2013 [203] |
| NNScore | NN g | PDB; MOAD; PDBbind-CN | EF = 10.3 | Generic | VS | Durrant, 2010 [79] |
| CScore | NN | PDBbind | Rp = 0.7668 (gen.) Rp = 0.8237 (fam. spec.) | Both | BAP | Ouyang, 2011 [174] |
| - | Deep NN | CSAR, DUD-E | ROCAUC = 0.868 | Generic | VS | Ragoza, 2017 [196] |
| - | Deep NN | DUD-E | ROCAUC = 0.92 | Both | VS | Imrie, 2018 [183] |
| DLScore | Deep NN | PDBbind | Rp = 0.82 | Generic | BAP | Hassan, 2018 [173] |
| DeepVS | Deep NN | DUD | ROCAUC = 0.81 | Generic | VS | Pereira, 2016 [177] |
| Kdeep | Deep NN | PDBbind | Rp = 0.82 | Generic | BAP | Jiménez, 2018 [78] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres, P.H.M.; Sodero, A.C.R.; Jofily, P.; Silva-Jr, F.P. Key Topics in Molecular Docking for Drug Design. Int. J. Mol. Sci. 2019, 20, 4574. https://doi.org/10.3390/ijms20184574
Torres PHM, Sodero ACR, Jofily P, Silva-Jr FP. Key Topics in Molecular Docking for Drug Design. International Journal of Molecular Sciences. 2019; 20(18):4574. https://doi.org/10.3390/ijms20184574
Chicago/Turabian StyleTorres, Pedro H. M., Ana C. R. Sodero, Paula Jofily, and Floriano P. Silva-Jr. 2019. "Key Topics in Molecular Docking for Drug Design" International Journal of Molecular Sciences 20, no. 18: 4574. https://doi.org/10.3390/ijms20184574
APA StyleTorres, P. H. M., Sodero, A. C. R., Jofily, P., & Silva-Jr, F. P. (2019). Key Topics in Molecular Docking for Drug Design. International Journal of Molecular Sciences, 20(18), 4574. https://doi.org/10.3390/ijms20184574