Mechanism of Action of the Tumor Vessel Targeting Agent NGR-hTNF: Role of Both NGR Peptide and hTNF in Cell Binding and Signaling

 , , ,
, , ,
Abstract
1. Introduction
2. Results
2.1. Crystal Structure of NGR-hTNF
2.2. In Vitro Characterization of the Binding Properties of the CNGRC Peptide
2.3. In Vitro Characterization of the Binding Properties of NGR-hTNF
2.4. In Vivo Characterization of the Binding Properties of the CNGRC Peptide
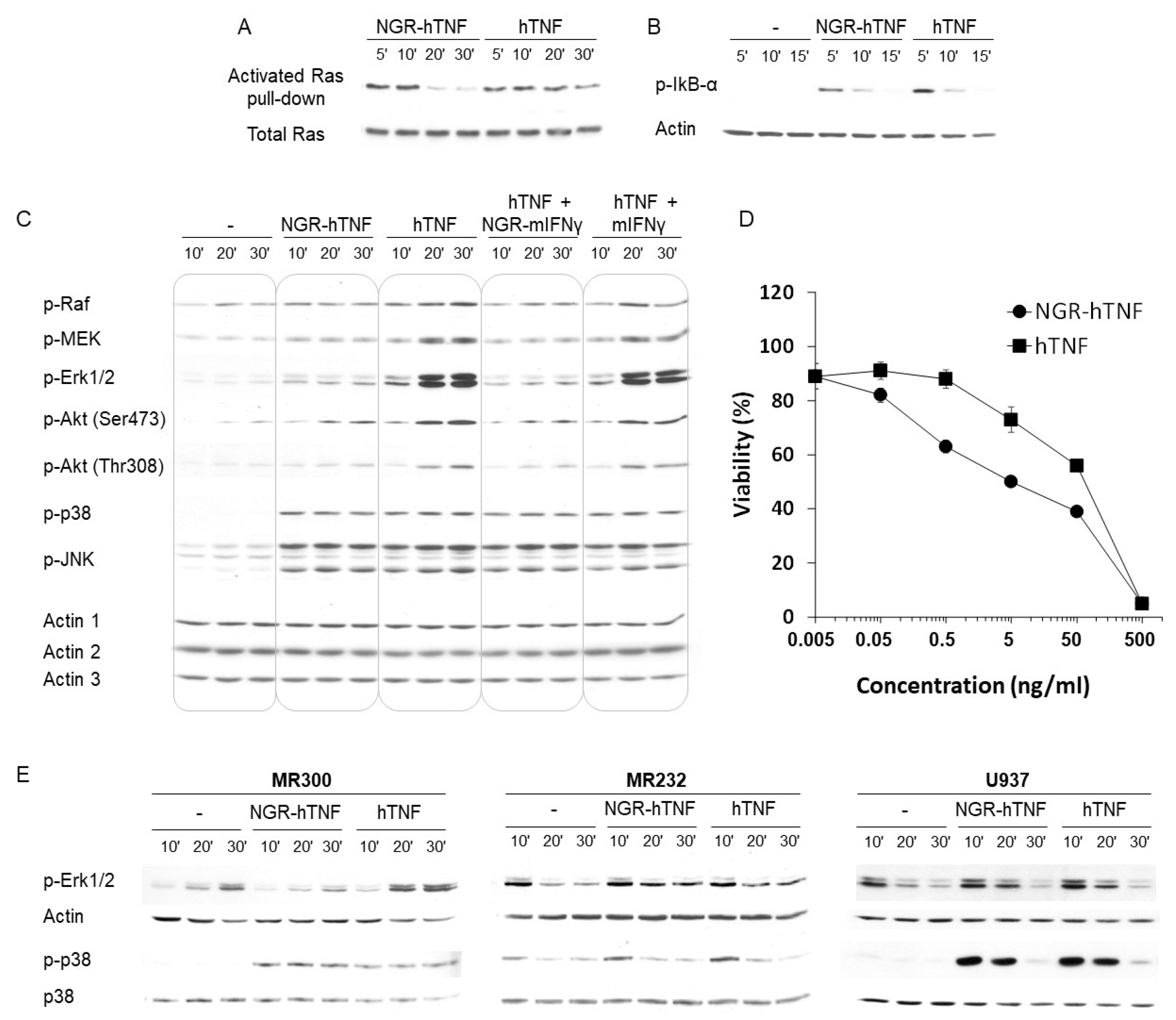
2.5. Characterization of the Signaling Properties of NGR-hTNF
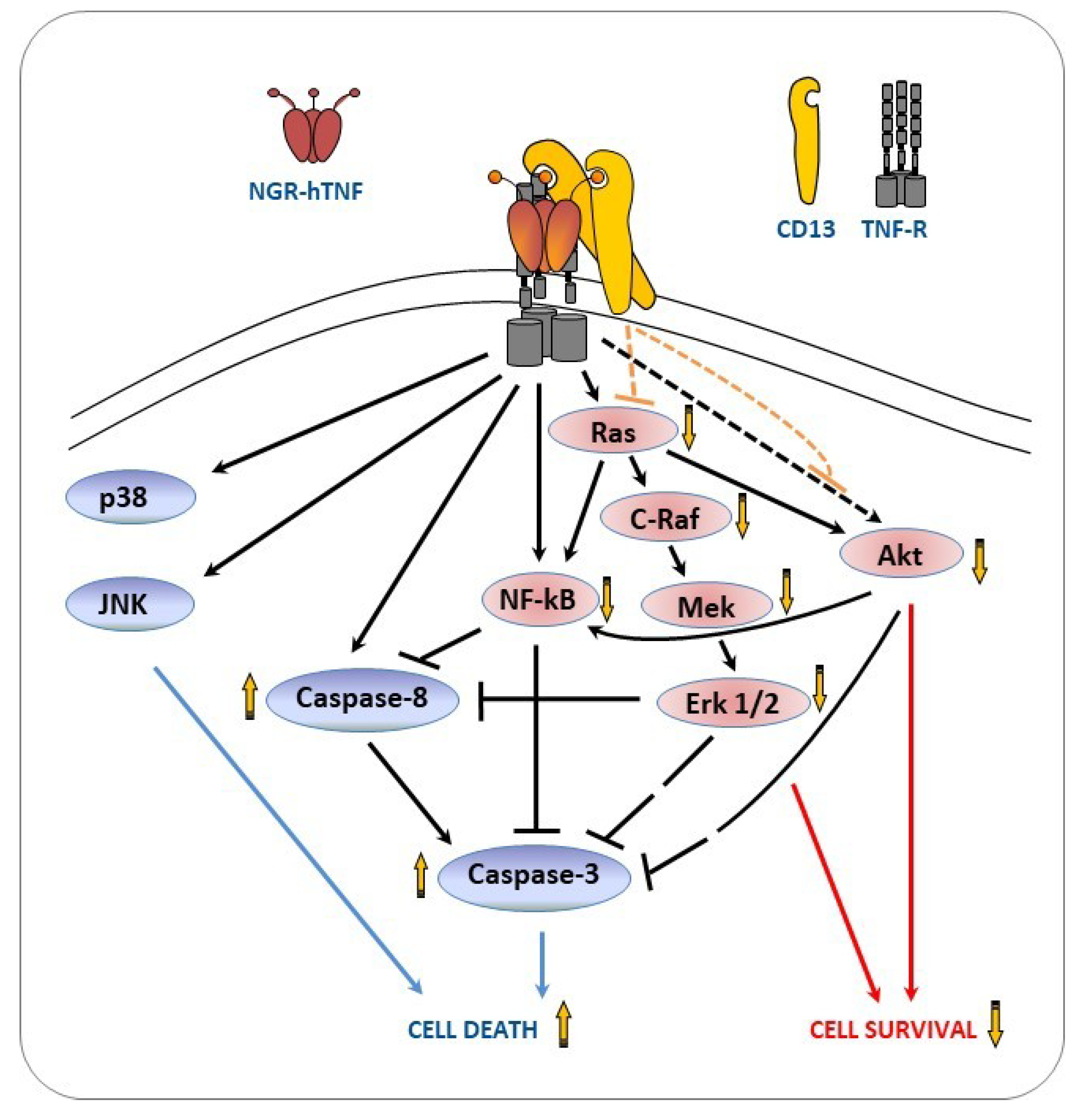
2.6. NGR-hTNF Signal Transduction Pathways and Biological Effects in HUVEC
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Animal Models
4.3. Cytokines
4.4. Peptide Binding Assay
4.5. Conjugation of Peptide to Nanoparticles (Qdots)
4.6. In Vivo Administration of Peptid-Qdots and Whole Mount Analysis
4.7. TNF Binding Assays and Competition
4.8. Flow Cytometry
4.9. X-ray Diffraction
4.10. 2D TR-NOE Spectroscopy (TR-NOESY) Experiments by NMR Technique
4.11. Surface Plasmon Resonance Analysis
4.12. Analysis of Signal Transduction
4.13. Caspase Activation and Cytotoxicity Assay
4.14. Tubule Formation Assay
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Arap, W.; Pasqualini, R.; Ruoslahti, E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 1998, 279, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Curnis, F.; Sacchi, A.; Borgna, L.; Magni, F.; Gasparri, A.; Corti, A. Enhancement of tumor necrosis factor alpha antitumor immunotherapeutic properties by targeted delivery to aminopeptidase n (cd13). Nat. Biotechnol. 2000, 18, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Corti, A.; Pastorino, F.; Curnis, F.; Arap, W.; Ponzoni, M.; Pasqualini, R. Targeted drug delivery and penetration into solid tumors. Med. Res. Rev. 2011. [Google Scholar] [CrossRef] [PubMed]
- Curnis, F.; Sacchi, A.; Corti, A. Improving chemotherapeutic drug penetration in tumors by vascular targeting and barrier alteration. J. Clin. Investig. 2002, 110, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Ferreri, A.J.M.; Calimeri, T.; Conte, G.M.; Cattaneo, D.; Fallanca, F.; Ponzoni, M.; Scarano, E.; Curnis, F.; Nonis, A.; Lopedote, P.; et al. R-chop preceded by blood-brain barrier permeabilization with engineered tumor necrosis factor-alpha in primary cns lymphoma. Blood 2019. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Grioni, M.; Jachetti, E.; Curnis, F.; Mondino, A.; Parmiani, G.; Corti, A.; Bellone, M. Targeting tnf-alpha to neoangiogenic vessels enhances lymphocyte infiltration in tumors and increases the therapeutic potential of immunotherapy. J. Immunol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Di Matteo, P.; Hackl, C.; Jedeszko, C.; Valentinis, B.; Bordignon, C.; Traversari, C.; Kerbel, R.S.; Rizzardi, G.P. Ngr-tnf, a novel vascular-targeting agent, does not induce cytokine recruitment of proangiogenic bone marrow-derived cells. Br. J. Cancer 2013, 109, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Porcellini, S.; Asperti, C.; Valentinis, B.; Tiziano, E.; Mangia, P.; Bordignon, C.; Rizzardi, G.P.; Traversari, C. The tumor vessel targeting agent ngr-tnf controls the different stages of the tumorigenic process in transgenic mice by distinct mechanisms. Oncoimmunology 2015, 4, e1041700. [Google Scholar] [CrossRef]
- Corti, A.; Curnis, F.; Rossoni, G.; Marcucci, F.; Gregorc, V. Peptide-mediated targeting of cytokines to tumor vasculature: The ngr-htnf example. BioDrugs 2013, 27, 591–603. [Google Scholar] [CrossRef]
- Gregorc, V.; Cavina, R.; Novello, S.; Grossi, F.; Lazzari, C.; Capelletto, E.; Genova, C.; Salini, G.; Lambiase, A.; Santoro, A. Ngr-htnf and doxorubicin as second-line treatment of patients with small cell lung cancer. Oncologist 2018, 23, 1133-e112. [Google Scholar] [CrossRef]
- Gregorc, V.; Gaafar, R.M.; Favaretto, A.; Grossi, F.; Jassem, J.; Polychronis, A.; Bidoli, P.; Tiseo, M.; Shah, R.; Taylor, P.; et al. Ngr-htnf in combination with best investigator choice in previously treated malignant pleural mesothelioma (ngr015): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2018, 19, 799–811. [Google Scholar] [CrossRef]
- Parmiani, G.; Pilla, L.; Corti, A.; Doglioni, C.; Cimminiello, C.; Bellone, M.; Parolini, D.; Russo, V.; Capocefalo, F.; Maccalli, C. A pilot phase i study combining peptide-based vaccination and ngr-htnf vessel targeting therapy in metastatic melanoma. Oncoimmunology 2014, 3, e963406. [Google Scholar] [CrossRef] [PubMed]
- Elia, A.R.; Grioni, M.; Basso, V.; Curnis, F.; Freschi, M.; Corti, A.; Mondino, A.; Bellone, M. Targeting tumor vasculature with tnf leads effector t cells to the tumor and enhances therapeutic efficacy of immune checkpoint blockers in combination with adoptive cell therapy. Clin. Cancer Res. 2018, 24, 2171–2181. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B. Signalling pathways of the tnf superfamily: A double-edged sword. Nat. Rev. Immunol. 2003, 3, 745–756. [Google Scholar] [CrossRef]
- Brenner, D.; Blaser, H.; Mak, T.W. Regulation of tumour necrosis factor signalling: Live or let die. Nat. Rev. Immunol. 2015, 15, 362–374. [Google Scholar] [CrossRef]
- Modur, V.; Zimmerman, G.A.; Prescott, S.M.; McIntyre, T.M. Endothelial cell inflammatory responses to tumor necrosis factor alpha. Ceramide-dependent and -independent mitogen-activated protein kinase cascades. J. Biol. Chem. 1996, 271, 13094–13102. [Google Scholar] [CrossRef]
- Madge, L.A.; Pober, J.S. Tnf signaling in vascular endothelial cells. Exp. Mol. Pathol. 2001, 70, 317–325. [Google Scholar] [CrossRef]
- Sainson, R.C.; Johnston, D.A.; Chu, H.C.; Holderfield, M.T.; Nakatsu, M.N.; Crampton, S.P.; Davis, J.; Conn, E.; Hughes, C.C. Tnf primes endothelial cells for angiogenic sprouting by inducing a tip cell phenotype. Blood 2008, 111, 4997–5007. [Google Scholar] [CrossRef]
- Bieler, G.; Hasmim, M.; Monnier, Y.; Imaizumi, N.; Ameyar, M.; Bamat, J.; Ponsonnet, L.; Chouaib, S.; Grell, M.; Goodman, S.L.; et al. Distinctive role of integrin-mediated adhesion in tnf-induced pkb/akt and nf-kappab activation and endothelial cell survival. Oncogene 2007, 26, 5722–5732. [Google Scholar] [CrossRef]
- Ihnatko, R.; Kubes, M. Tnf signaling: Early events and phosphorylation. Gen. Physiol. Biophys. 2007, 26, 159–167. [Google Scholar]
- Jin, Z.; Li, Y.; Pitti, R.; Lawrence, D.; Pham, V.C.; Lill, J.R.; Ashkenazi, A. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell 2009, 137, 721–735. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Parrish, A.B.; Freel, C.D.; Kornbluth, S. Cellular mechanisms controlling caspase activation and function. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Allan, L.A.; Morrice, N.; Brady, S.; Magee, G.; Pathak, S.; Clarke, P.R. Inhibition of caspase-9 through phosphorylation at thr 125 by erk mapk. Nat. Cell Biol. 2003, 5, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Tran, S.E.; Holmstrom, T.H.; Ahonen, M.; Kahari, V.M.; Eriksson, J.E. Mapk/erk overrides the apoptotic signaling from fas, tnf, and trail receptors. J. Biol. Chem. 2001, 276, 16484–16490. [Google Scholar] [CrossRef] [PubMed]
- Stehlik, C.; de Martin, R.; Kumabashiri, I.; Schmid, J.A.; Binder, B.R.; Lipp, J. Nuclear factor (nf)-kappab-regulated x-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor alpha-induced apoptosis. J. Exp. Med. 1998, 188, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Mayo, M.W.; Korneluk, R.G.; Goeddel, D.V.; Baldwin, A.S., Jr. Nf-kappab antiapoptosis: Induction of traf1 and traf2 and c-iap1 and c-iap2 to suppress caspase-8 activation. Science 1998, 281, 1680–1683. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Lens, S.; Gaide, O.; Alevizopoulos, K.; Tschopp, J. Nf-kappab signals induce the expression of c-flip. Mol. Cell Biol. 2001, 21, 5299–5305. [Google Scholar] [CrossRef]
- Pasqualini, R.; Koivunen, E.; Kain, R.; Lahdenranta, J.; Sakamoto, M.; Stryhn, A.; Ashmun, R.A.; Shapiro, L.H.; Arap, W.; Ruoslahti, E. Aminopeptidase n is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res. 2000, 60, 722–727. [Google Scholar]
- Curnis, F.; Arrigoni, G.; Sacchi, A.; Fischetti, L.; Arap, W.; Pasqualini, R.; Corti, A. Differential binding of drugs containing the ngr motif to cd13 isoforms in tumor vessels, epithelia, and myeloid cells. Cancer Res. 2002, 62, 867–874. [Google Scholar]
- Di Matteo, P.; Arrigoni, G.L.; Alberici, L.; Corti, A.; Gallo-Stampino, C.; Traversari, C.; Doglioni, C.; Rizzardi, G.P. Enhanced expression of cd13 in vessels of inflammatory and neoplastic tissues. J. Histochem. Cytochem. 2011, 59, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Mina-Osorio, P. The moonlighting enzyme cd13: Old and new functions to target. Trends Mol. Med. 2008, 14, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.N.; Langner, J.; Herrmann, M.; Riemann, D. Aminopeptidase n/cd13 is directly linked to signal transduction pathways in monocytes. Cell Immunol. 2000, 201, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Mina-Osorio, P.; Shapiro, L.H.; Ortega, E. Cd13 in cell adhesion: Aminopeptidase n (cd13) mediates homotypic aggregation of monocytic cells. J. Leukoc. Biol. 2006, 79, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Mina-Osorio, P.; Ortega, E. Aminopeptidase n (cd13) functionally interacts with fcgammars in human monocytes. J. Leukoc. Biol. 2005, 77, 1008–1017. [Google Scholar] [CrossRef]
- Petrovic, N.; Schacke, W.; Gahagan, J.R.; O’Conor, C.A.; Winnicka, B.; Conway, R.E.; Mina-Osorio, P.; Shapiro, L.H. Cd13/apn regulates endothelial invasion and filopodia formation. Blood 2007, 110, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Subramani, J.; Ghosh, M.; Rahman, M.M.; Caromile, L.A.; Gerber, C.; Rezaul, K.; Han, D.K.; Shapiro, L.H. Tyrosine phosphorylation of cd13 regulates inflammatory cell-cell adhesion and monocyte trafficking. J. Immunol. 2013, 191, 3905–3912. [Google Scholar] [CrossRef] [PubMed]
- Eck, M.J.; Sprang, S.R. The structure of tumor necrosis factor-alpha at 2.6 a resolution. Implications for receptor binding. J. Biol. Chem. 1989, 264, 17595–17605. [Google Scholar]
- Banner, D.W.; D’Arcy, A.; Janes, W.; Gentz, R.; Schoenfeld, H.J.; Broger, C.; Loetscher, H.; Lesslauer, W. Crystal structure of the soluble human 55 kd tnf receptor-human tnf beta complex: Implications for tnf receptor activation. Cell 1993, 73, 431–445. [Google Scholar] [CrossRef]
- Buehler, A.; van Zandvoort, M.A.; Stelt, B.J.; Hackeng, T.M.; Schrans-Stassen, B.H.; Bennaghmouch, A.; Hofstra, L.; Cleutjens, J.P.; Duijvestijn, A.; Smeets, M.B.; et al. Cngr: A novel homing sequence for cd13/apn targeted molecular imaging of murine cardiac angiogenesis in vivo. Arterioscler Thromb. Vasc. Biol. 2006, 26, 2681–2687. [Google Scholar] [CrossRef]
- Curnis, F.; Cattaneo, A.; Longhi, R.; Sacchi, A.; Gasparri, A.M.; Pastorino, F.; Di Matteo, P.; Traversari, C.; Bachi, A.; Ponzoni, M.; et al. Critical role of flanking residues in ngr-to-isodgr transition and cd13/integrin receptor switching. J. Biol. Chem. 2010, 285, 9114–9123. [Google Scholar] [CrossRef] [PubMed]
- Tonlorenzi, R.; Dellavalle, A.; Schnapp, E.; Cossu, G.; Sampaolesi, M. Isolation and characterization of mesoangioblasts from mouse, dog, and human tissues. Curr. Protoc. Stem. Cell Biol. 2007, 2, 1. [Google Scholar]
- Mari, S.; Invernizzi, C.; Spitaleri, A.; Alberici, L.; Ghitti, M.; Bordignon, C.; Traversari, C.; Rizzardi, G.P.; Musco, G. 2d tr-noesy experiments interrogate and rank ligand-receptor interactions in living human cancer cells. Angew. Chem. Int. Ed. Engl. 2010, 49, 1071–1074. [Google Scholar] [CrossRef] [PubMed]
- Curnis, F.; Sacchi, A.; Gasparri, A.; Longhi, R.; Bachi, A.; Doglioni, C.; Bordignon, C.; Traversari, C.; Rizzardi, G.P.; Corti, A. Isoaspartate-glycine-arginine: A new tumor vasculature-targeting motif. Cancer Res. 2008, 68, 7073–7082. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, S.V.; Lahdenranta, J.; Giordano, R.; Arap, W.; Pasqualini, R.; Shapiro, L.H. Cd13/apn is activated by angiogenic signals and is essential for capillary tube formation. Blood 2001, 97, 652–659. [Google Scholar] [CrossRef]
- Hemmi, S.; Peghini, P.; Metzler, M.; Merlin, G.; Dembic, Z.; Aguet, M. Cloning of murine interferon gamma receptor cdna: Expression in human cells mediates high-affinity binding but is not sufficient to confer sensitivity to murine interferon gamma. Proc. Natl. Acad. Sci. USA 1989, 86, 9901–9905. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.J. Specificity of receptor tyrosine kinase signaling: Transient versus sustained extracellular signal-regulated kinase activation. Cell 1995, 80, 179–185. [Google Scholar] [CrossRef]
- Sharrocks, A.D. Cell cycle: Sustained erk signalling represses the inhibitors. Curr. Biol. 2006, 16, R540–R542. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; Gerstberger, S.; Carlson, L.; Franzoso, G.; Siebenlist, U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science 1995, 267, 1485–1488. [Google Scholar] [CrossRef]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Ruoslahti, E.; Bhatia, S.N.; Sailor, M.J. Targeting of drugs and nanoparticles to tumors. J. Cell Biol. 2010, 188, 759–768. [Google Scholar] [CrossRef]
- Curnis, F.; Gasparri, A.; Sacchi, A.; Cattaneo, A.; Magni, F.; Corti, A. Targeted delivery of ifngamma to tumor vessels uncouples antitumor from counterregulatory mechanisms. Cancer Res. 2005, 65, 2906–2913. [Google Scholar] [CrossRef]
- Corti, A.; Fiocchi, M.; Curnis, F. Targeting cd13 with asn-gly-arg (ngr) peptide-drug conjugates. In Next-Generation Therapies and Technologies for Immune-Mediated Inflammatory Diseases; Mina-Osorio, P., Ed.; Springer International Publishing: Cham, Switzerland, 2017. [Google Scholar]
- Moujalled, D.M.; Cook, W.D.; Lluis, J.M.; Khan, N.R.; Ahmed, A.U.; Callus, B.A.; Vaux, D.L. In mouse embryonic fibroblasts, neither caspase-8 nor cellular flice-inhibitory protein (flip) is necessary for tnf to activate nf-kappab, but caspase-8 is required for tnf to cause cell death, and induction of flip by nf-kappab is required to prevent it. Cell Death Differ. 2012, 19, 808–815. [Google Scholar] [CrossRef]
- Ozes, O.N.; Mayo, L.D.; Gustin, J.A.; Pfeffer, S.R.; Pfeffer, L.M.; Donner, D.B. Nf-kappab activation by tumour necrosis factor requires the akt serine-threonine kinase. Nature 1999, 401, 82–85. [Google Scholar] [CrossRef]
- Duronio, V. The life of a cell: Apoptosis regulation by the pi3k/pkb pathway. Biochem. J. 2008, 415, 333–344. [Google Scholar] [CrossRef]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef]
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies within. Science 1998, 281, 1312–1316. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Reddy, E.P. Jnk signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef]
- Donovan, D.; Brown, N.J.; Bishop, E.T.; Lewis, C.E. Comparison of three in vitro human ‘angiogenesis’ assays with capillaries formed in vivo. Angiogenesis 2001, 4, 113–121. [Google Scholar] [CrossRef]
- Bulotta, A.; Gregorc, V.; Rossoni, G.; Todisco, G.; Viganò, M.G.; Ammannati, C.; Mazzola, G.; Lambiase, A.; Bordignon, C. Relationships of peripheral blood lymphocyte counts (pblc) with antitumor activity of ngr-htnf given in combination with chemotherapy (ct). J. Clin. Oncol. 2013, 31, 3038. [Google Scholar]
- Lionello, I.; Mangia, P.; Gattinoni, L.; Pende, D.; Cippone, A.; Sensi, M.; Rigatti, P.; Traversari, C. Cd8(+) t lymphocytes isolated from renal cancer patients recognize tumour cells through an hla- and tcr/cd3-independent pathway. Cancer Immunol. Immunother. 2007, 56, 1065–1076. [Google Scholar] [CrossRef]
- Tanzarella, S.; Russo, V.; Lionello, I.; Dalerba, P.; Rigatti, D.; Bordignon, C.; Traversari, C. Identification of a promiscuous t-cell epitope encoded by multiple members of the mage family. Cancer Res. 1999, 59, 2668–2674. [Google Scholar]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. Phenix: A comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. Molprobity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Karplus, P.A.; Diederichs, K. Linking crystallographic model and data quality. Science 2012, 336, 1030–1033. [Google Scholar] [CrossRef]
- Papeo, G.; Avanzi, N.; Bettoni, S.; Leone, A.; Paolucci, M.; Perego, R.; Quartieri, F.; Riccardi-Sirtori, F.; Thieffine, S.; Montagnoli, A.; et al. Insights into parp inhibitors’ selectivity using fluorescence polarization and surface plasmon resonance binding assays. J. Biomol. Screen 2014, 19, 1212–1219. [Google Scholar] [CrossRef][Green Version]
- Karlsson, R.; Katsamba, P.S.; Nordin, H.; Pol, E.; Myszka, D.G. Analyzing a kinetic titration series using affinity biosensors. Anal. Biochem. 2006, 349, 136–147. [Google Scholar] [CrossRef]
- Reis, C.R.; van Assen, A.H.; Quax, W.J.; Cool, R.H. Unraveling the binding mechanism of trivalent tumor necrosis factor ligands and their receptors. Mol. Cell Proteom. 2011, 10, M110-002808. [Google Scholar] [CrossRef]
- Lortat-Jacob, H.; Chouin, E.; Cusack, S.; van Raaij, M.J. Kinetic analysis of adenovirus fiber binding to its receptor reveals an avidity mechanism for trimeric receptor-ligand interactions. J. Biol. Chem. 2001, 276, 9009–9015. [Google Scholar] [CrossRef]
- Beyaert, R.; Vanhaesebroeck, B.; Suffys, P.; Van Roy, F.; Fiers, W. Lithium chloride potentiates tumor necrosis factor-mediated cytotoxicity in vitro and in vivo. Proc. Natl. Acad. Sci. USA 1989, 86, 9494–9498. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | |
| Crystal parameters | a = b = 92.011 Å; c = 116.879 Å α = β = γ = 90° |
| Space group | P41212 |
| Wavelength (Å) | 0.931 |
| Resolution range (Å) | 49.33–2.65 (2.78–2.65) |
| Reflections observed | 241,457 (32,201) |
| Reflections unique | 15,203 (1973) |
| Completeness (%) | 100.0 (100.0) |
| Multiplicity | 15.9 (16.3) |
| <I/σ(I)> | 15.6 (1.0) |
| CC1/2 | 0.999 (0.469) |
| Rpim | 0.035 (0.905) |
| Refinement Statistics | |
| Reflections used | 15138 |
| Rcrys | 0.209 |
| Rfree | 0.252 |
| rmsd bonds (Å) | 0.003 |
| rmsd angles (°) | 0.626 |
| <B> (Å2) | |
| Protein | 89.8 |
| Solvent | 63.4 |
| Ramachandran Statistics (% of Residues) | |
| Favoured | 96.7 |
| Allowed | 3.3 |
| Disallowed | 0 |
| kon1 × 105 (M−1·s−1) | koff1 × 10−4 (s−1) | t1/2 1 (min) | KD (nM) | kon2 × 10−3 (RU−1·s−1) | koff2 × 10−3 (s−1) | t1/2 2 (min) | ||
|---|---|---|---|---|---|---|---|---|
| TNFR1 | NGR-hTNF | 4.68 (1.40) | 15.8 (1.8) | 7.4 (0.8) | 3.6 (1.4) | 4.29 (0.55) | 1.19 (0.19) | 9.9 (1.6) |
| hTNF | 2.09 (0.03) | 9.2 (0.8) | 12.7 (1.0) | 4.4 (0.4) | 2.70 (0.35) | 1.16 (0.04) | 10.0 (0.4) | |
| TNFR2 | NGR-hTNF | 5.88 (0.04) | 336 (1.4) | 0.3 (0.0) | 57 (0.2) | 0.54 (0.00) | 0.39 (0.00) | 29.4 (0.1) |
| hTNF | 2.74 (0.23) | 204 (46.7) | 0.6 (0.1) | 74 (1.1) | 0.55 (0.01) | 0.30 (0.02) | 38.5 (2.3) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valentinis, B.; Porcellini, S.; Asperti, C.; Cota, M.; Zhou, D.; Di Matteo, P.; Garau, G.; Zucchelli, C.; Avanzi, N.R.; Rizzardi, G.P.; et al. Mechanism of Action of the Tumor Vessel Targeting Agent NGR-hTNF: Role of Both NGR Peptide and hTNF in Cell Binding and Signaling. Int. J. Mol. Sci. 2019, 20, 4511. https://doi.org/10.3390/ijms20184511
Valentinis B, Porcellini S, Asperti C, Cota M, Zhou D, Di Matteo P, Garau G, Zucchelli C, Avanzi NR, Rizzardi GP, et al. Mechanism of Action of the Tumor Vessel Targeting Agent NGR-hTNF: Role of Both NGR Peptide and hTNF in Cell Binding and Signaling. International Journal of Molecular Sciences. 2019; 20(18):4511. https://doi.org/10.3390/ijms20184511
Chicago/Turabian StyleValentinis, Barbara, Simona Porcellini, Claudia Asperti, Manuela Cota, Dan Zhou, Paola Di Matteo, Gianpiero Garau, Chiara Zucchelli, Nilla Roberta Avanzi, Gian Paolo Rizzardi, and et al. 2019. "Mechanism of Action of the Tumor Vessel Targeting Agent NGR-hTNF: Role of Both NGR Peptide and hTNF in Cell Binding and Signaling" International Journal of Molecular Sciences 20, no. 18: 4511. https://doi.org/10.3390/ijms20184511
APA StyleValentinis, B., Porcellini, S., Asperti, C., Cota, M., Zhou, D., Di Matteo, P., Garau, G., Zucchelli, C., Avanzi, N. R., Rizzardi, G. P., Degano, M., Musco, G., & Traversari, C. (2019). Mechanism of Action of the Tumor Vessel Targeting Agent NGR-hTNF: Role of Both NGR Peptide and hTNF in Cell Binding and Signaling. International Journal of Molecular Sciences, 20(18), 4511. https://doi.org/10.3390/ijms20184511