MicroRNA Networks Modulate Oxidative Stress in Cancer

Abstract
1. Introduction
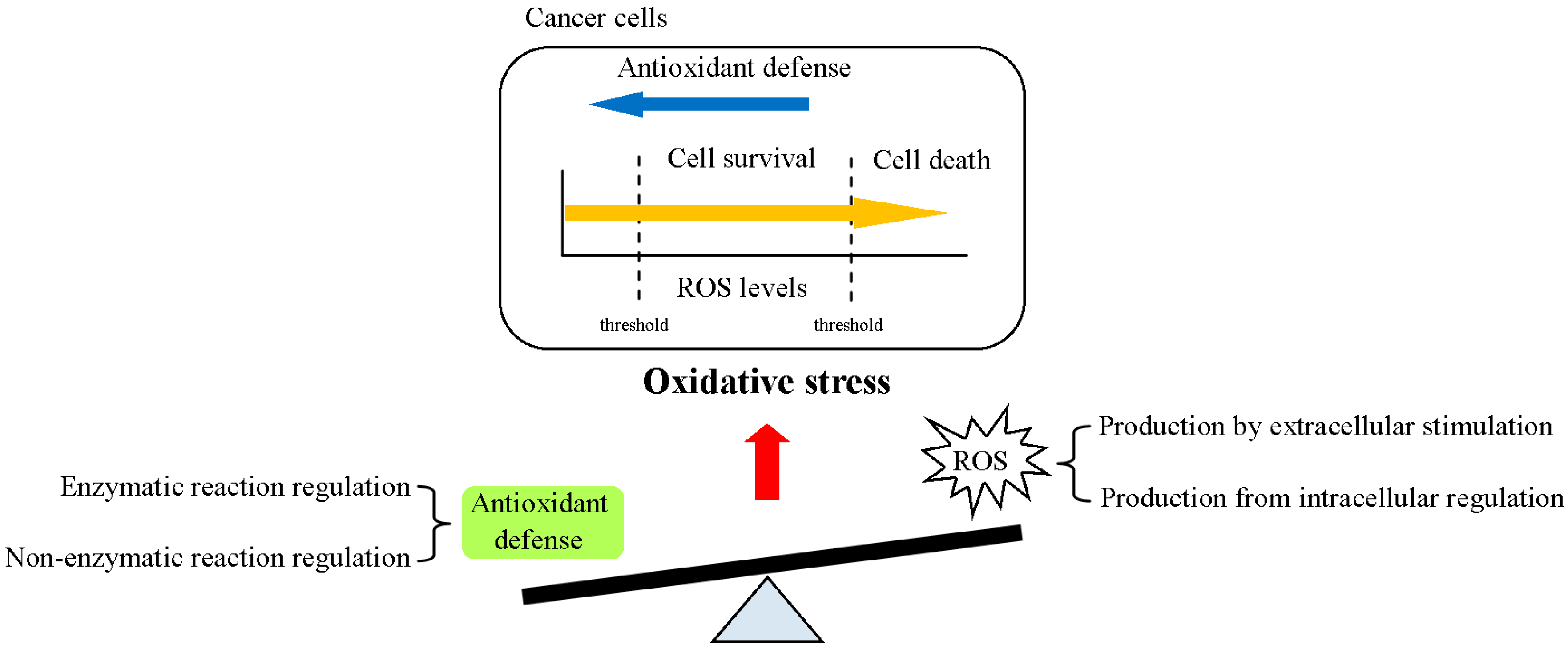
2. Regulation of ROS Homeostasis in Cells
3. MiRNAs and Their Roles in Oxidative Stress
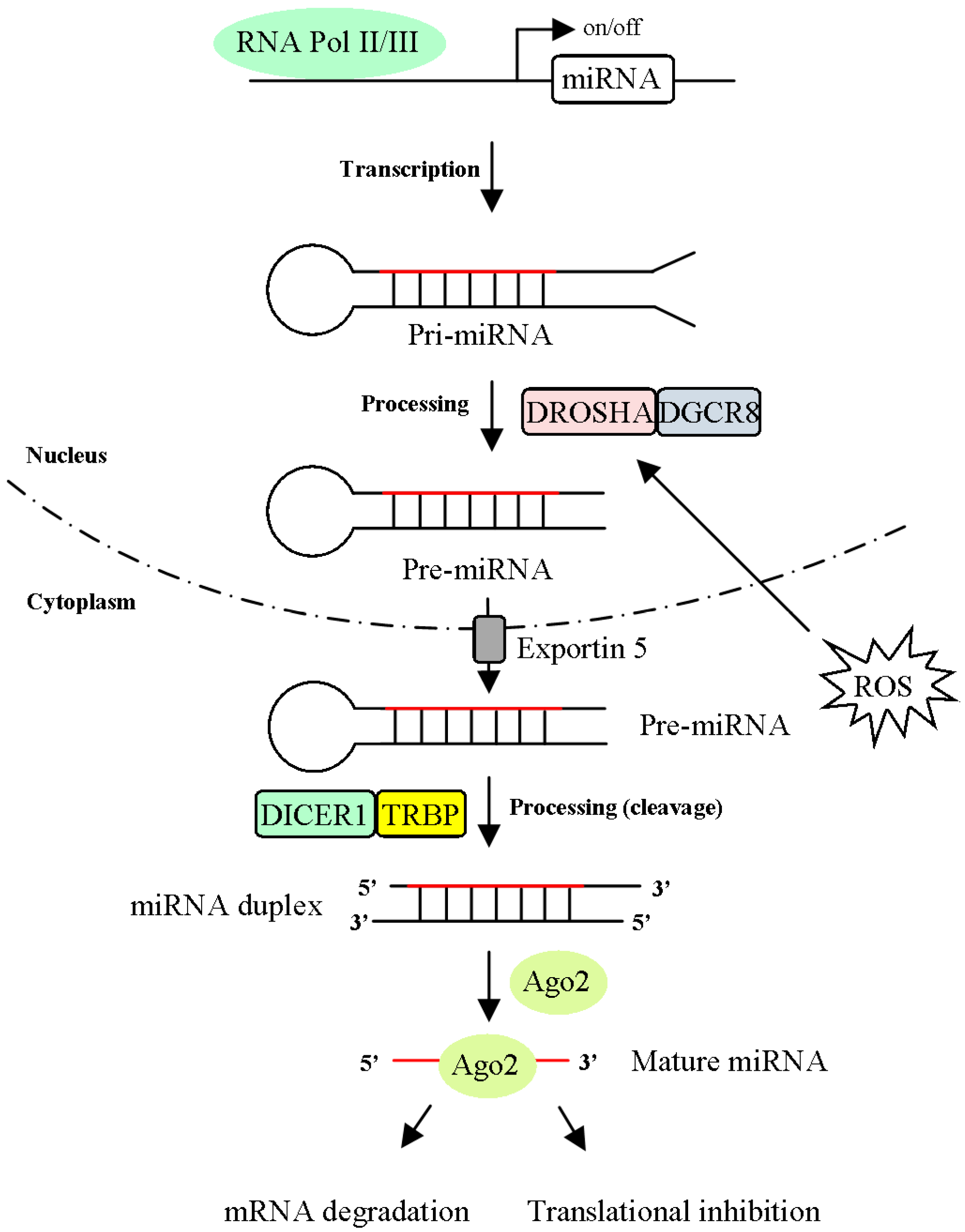
3.1. MiRNA Processing is Regulated by ROS
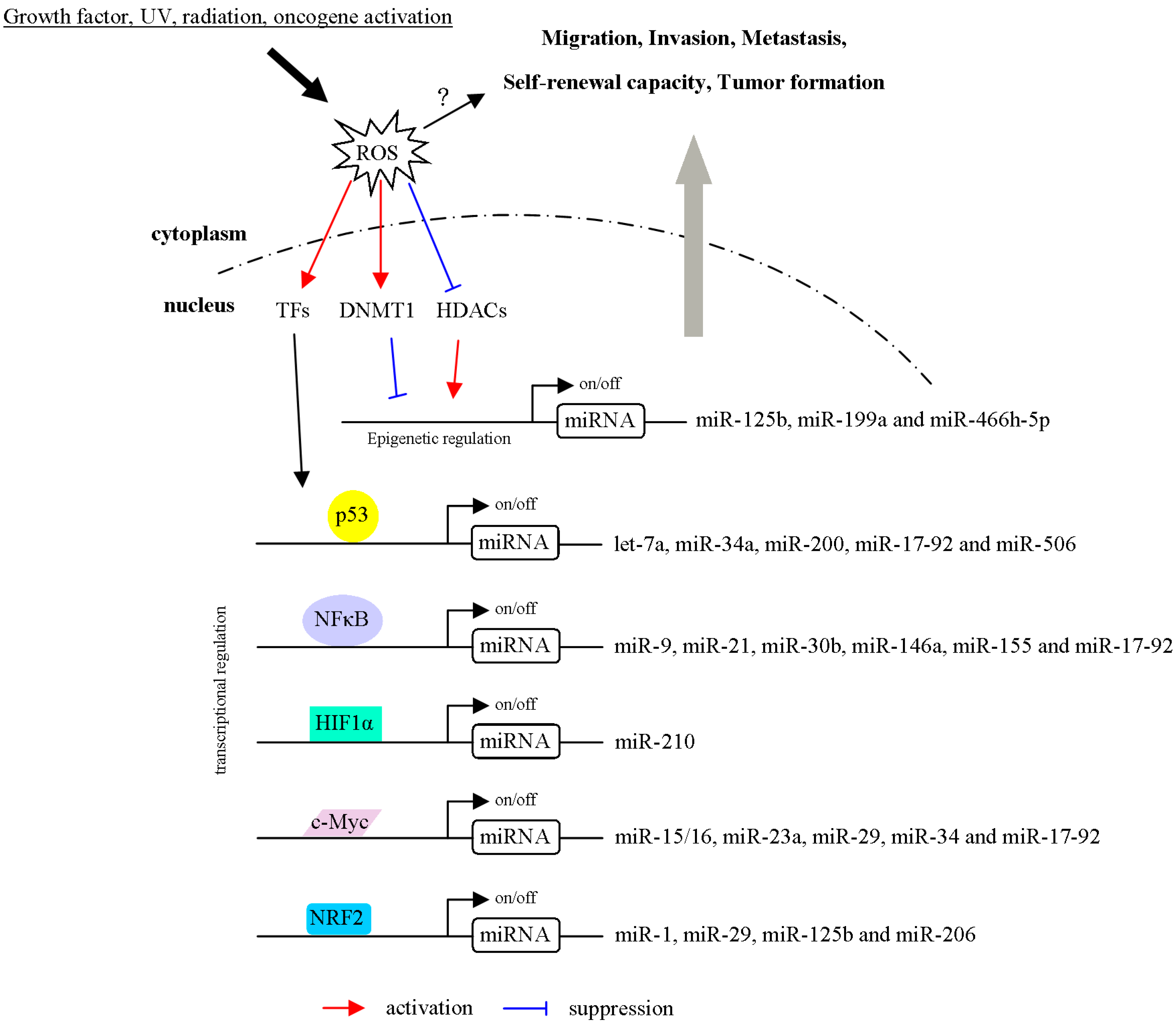
3.2. ROS Regulate miRNA Expression through the Modulation of Transcription Factors
3.3. ROS Regulate miRNA Expression via Epigenetic Regulation
4. Interplay between Oxidative Stress, miRNA and Cancer Development
4.1. Association between OS, miRNA and Hypoxia
4.2. Association between OS, miRNA and Angiogenesis
4.3. Association between OS, miRNA and Metastasis
4.4. Association between OS, miRNA and Metabolism
4.5. Association between OS, miRNA and Cancer Stem Cells
4.6. Association between OS, miRNA and Senescence
5. ROS-Mediated Therapeutic Strategies in Cancer
6. Conclusions
Funding
Conflicts of Interest
References
- Dong, Y.; Xu, W.; Liu, C.; Liu, P.; Li, P.; Wang, K. Reactive oxygen species related noncoding RNAs as regulators of cardiovascular diseases. Int. J. Biol. Sci. 2019, 15, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Engedal, N.; Zerovnik, E.; Rudov, A.; Galli, F.; Olivieri, F.; Procopio, A.D.; Rippo, M.R.; Monsurro, V.; Betti, M.; Albertini, M.C. From oxidative stress damage to pathways, networks, and autophagy via microRNAs. Oxid. Med. Cell. Longev. 2018, 2018, 4968321. [Google Scholar] [CrossRef] [PubMed]
- Castaneda-Arriaga, R.; Perez-Gonzalez, A.; Reina, M.; Alvarez-Idaboy, J.R.; Galano, A. Comprehensive investigation of the antioxidant and pro-oxidant effects of phenolic compounds: A double-edged sword in the context of oxidative stress? J. Phys. Chem. B 2018, 122, 6198–6214. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. (Lausanne) 2018, 9, 402. [Google Scholar] [CrossRef]
- Tan, W.; Liu, B.; Qu, S.; Liang, G.; Luo, W.; Gong, C. MicroRNAs and cancer: Key paradigms in molecular therapy. Oncol. Lett. 2018, 15, 2735–2742. [Google Scholar] [CrossRef]
- Liu, Y.; Qiang, W.; Xu, X.; Dong, R.; Karst, A.M.; Liu, Z.; Kong, B.; Drapkin, R.I.; Wei, J.J. Role of miR-182 in response to oxidative stress in the cell fate of human fallopian tube epithelial cells. Oncotarget 2015, 6, 38983–38998. [Google Scholar] [CrossRef]
- Fierro-Fernandez, M.; Miguel, V.; Lamas, S. Role of redoximiRs in fibrogenesis. Redox Biol. 2016, 7, 58–67. [Google Scholar] [CrossRef]
- Meseguer, S.; Martinez-Zamora, A.; Garcia-Arumi, E.; Andreu, A.L.; Armengod, M.E. The ROS-sensitive microRNA-9/9* controls the expression of mitochondrial tRNA-modifying enzymes and is involved in the molecular mechanism of MELAS syndrome. Hum. Mol. Genet. 2015, 24, 167–184. [Google Scholar] [CrossRef]
- La Sala, L.; Mrakic-Sposta, S.; Micheloni, S.; Prattichizzo, F.; Ceriello, A. Glucose-sensing microRNA-21 disrupts ROS homeostasis and impairs antioxidant responses in cellular glucose variability. Cardiovasc. Diabetol. 2018, 17, 105. [Google Scholar] [CrossRef]
- Saki, M.; Prakash, A. DNA damage related crosstalk between the nucleus and mitochondria. Free Radic. Biol. Med. 2017, 107, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Case, A.J. On the origin of superoxide dismutase: An evolutionary perspective of superoxide-mediated redox signaling. Antioxidants (Basel) 2017, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Meitzler, J.L.; Antony, S.; Wu, Y.; Juhasz, A.; Liu, H.; Jiang, G.; Lu, J.; Roy, K.; Doroshow, J.H. Nadph oxidases: A perspective on reactive oxygen species production in tumor biology. Antioxid. Redox Signal. 2014, 20, 2873–2889. [Google Scholar] [CrossRef] [PubMed]
- Collin, F. Chemical basis of reactive oxygen species reactivity and involvement in neurodegenerative diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef] [PubMed]
- Pylvas, M.; Puistola, U.; Laatio, L.; Kauppila, S.; Karihtala, P. Elevated serum 8-OHdG is associated with poor prognosis in epithelial ovarian cancer. Anticancer Res. 2011, 31, 1411–1415. [Google Scholar] [PubMed]
- Plachetka, A.; Adamek, B.; Strzelczyk, J.K.; Krakowczyk, L.; Migula, P.; Nowak, P.; Wiczkowski, A. 8-hydroxy-2’-deoxyguanosine in colorectal adenocarcinoma--is it a result of oxidative stress? Med. Sci. Monit. 2013, 19, 690–695. [Google Scholar] [PubMed]
- Ma-On, C.; Sanpavat, A.; Whongsiri, P.; Suwannasin, S.; Hirankarn, N.; Tangkijvanich, P.; Boonla, C. Oxidative stress indicated by elevated expression of NRF2 and 8-OHdG promotes hepatocellular carcinoma progression. Med. Oncol. 2017, 34, 57. [Google Scholar] [CrossRef]
- Tanaka, H.; Fujita, N.; Sugimoto, R.; Urawa, N.; Horiike, S.; Kobayashi, Y.; Iwasa, M.; Ma, N.; Kawanishi, S.; Watanabe, S.; et al. Hepatic oxidative DNA damage is associated with increased risk for hepatocellular carcinoma in chronic hepatitis c. Br. J. Cancer 2008, 98, 580–586. [Google Scholar] [CrossRef]
- Akhtar, M.J.; Ahamed, M.; Alhadlaq, H.A.; Alshamsan, A. Mechanism of ROS scavenging and antioxidant signalling by redox metallic and fullerene nanomaterials: Potential implications in ROS associated degenerative disorders. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 802–813. [Google Scholar] [CrossRef]
- He, J.; Jiang, B.H. Interplay between reactive oxygen species and microRNAs in cancer. Curr. Pharmacol. Rep. 2016, 2, 82–90. [Google Scholar] [CrossRef]
- Gong, Y.Y.; Luo, J.Y.; Wang, L.; Huang, Y. MicroRNAs regulating reactive oxygen species in cardiovascular diseases. Antioxid. Redox Signal. 2018, 29, 1092–1107. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Huang, Z.; Han, J.; Shao, J.; Huang, C. Redox regulation of microRNAs in cancer. Cancer Lett. 2018, 418, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Tucsek, Z.; Sosnowska, D.; Toth, P.; Gautam, T.; Podlutsky, A.; Csiszar, A.; Losonczy, G.; Valcarcel-Ares, M.N.; Sonntag, W.E.; et al. Aging-induced dysregulation of dicer1-dependent microRNA expression impairs angiogenic capacity of rat cerebromicrovascular endothelial cells. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 877–891. [Google Scholar] [CrossRef] [PubMed]
- Shilo, S.; Roy, S.; Khanna, S.; Sen, C.K. Evidence for the involvement of miRNA in redox regulated angiogenic response of human microvascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Kozakowska, M.; Ciesla, M.; Stefanska, A.; Skrzypek, K.; Was, H.; Jazwa, A.; Grochot-Przeczek, A.; Kotlinowski, J.; Szymula, A.; Bartelik, A.; et al. Heme oxygenase-1 inhibits myoblast differentiation by targeting myomirs. Antioxid. Redox Signal. 2012, 16, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Markopoulos, G.S.; Roupakia, E.; Tokamani, M.; Alabasi, G.; Sandaltzopoulos, R.; Marcu, K.B.; Kolettas, E. Roles of NF-kB signaling in the regulation of miRNAs impacting on inflammation in cancer. Biomedicines 2018, 6, 40. [Google Scholar] [CrossRef]
- Graves, J.A.; Metukuri, M.; Scott, D.; Rothermund, K.; Prochownik, E.V. Regulation of reactive oxygen species homeostasis by peroxiredoxins and c-myc. J. Biol. Chem. 2009, 284, 6520–6529. [Google Scholar] [CrossRef]
- Ferro, E.; Goitre, L.; Retta, S.F.; Trabalzini, L. The interplay between ROS and Ras GTPases: Physiological and pathological implications. J. Signal. Transduct. 2012, 2012, 365769. [Google Scholar] [CrossRef]
- Jackstadt, R.; Hermeking, H. MicroRNAs as regulators and mediators of c-Myc function. Biochim. Biophys. Acta 2015, 1849, 544–553. [Google Scholar] [CrossRef]
- Xue, G.; Yan, H.L.; Zhang, Y.; Hao, L.Q.; Zhu, X.T.; Mei, Q.; Sun, S.H. c-MYC-mediated repression of miR-15-16 in hypoxia is induced by increased HIF-2alpha and promotes tumor angiogenesis and metastasis by upregulating FGF2. Oncogene 2015, 34, 1393–1406. [Google Scholar] [CrossRef]
- Li, S.G.; Shi, Q.W.; Yuan, L.Y.; Qin, L.P.; Wang, Y.; Miao, Y.Q.; Chen, Z.; Ling, C.Q.; Qin, W.X. c-Myc-dependent repression of two oncogenic miRNA clusters contributes to triptolide-induced cell death in hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. 2018, 37, 51. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, A.; Falcone, E.; Garibaldi, F.; Piaggio, G. Dysregulation of microRNA biogenesis in cancer: The impact of mutant p53 on drosha complex activity. J. Exp. Clin. Cancer Res. 2016, 35, 45. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.R.; Vuppusetty, C.; Colley, T.; Papaioannou, A.I.; Fenwick, P.; Donnelly, L.; Ito, K.; Barnes, P.J. Oxidative stress dependent microRNA-34a activation via pi3kalpha reduces the expression of sirtuin-1 and sirtuin-6 in epithelial cells. Sci. Rep. 2016, 6, 35871. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Yan, W.; Lu, L.; Wang, Y.; Lu, W.; Cao, Y.; Cai, W. P38/p53/miR-200a-3p feedback loop promotes oxidative stress-mediated liver cell death. Cell Cycle 2015, 14, 1548–1558. [Google Scholar] [CrossRef] [PubMed]
- Magenta, A.; Cencioni, C.; Fasanaro, P.; Zaccagnini, G.; Greco, S.; Sarra-Ferraris, G.; Antonini, A.; Martelli, F.; Capogrossi, M.C. MiR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via zeb1 inhibition. Cell Death Differ. 2011, 18, 1628–1639. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ding, J.; Yang, J.; Guo, X.; Zheng, Y. MicroRNA roles in the nuclear factor kappa b signaling pathway in cancer. Front. Immunol. 2018, 9, 546. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Becker Buscaglia, L.E.; Barker, J.R.; Li, Y. MicroRNAs in NF-kappaB signaling. J. Mol. Cell Biol. 2011, 3, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, F.; Rossato, M.; Fabbri, M.; Gaudiosi, D.; Mirolo, M.; Mori, L.; Tamassia, N.; Mantovani, A.; Cassatella, M.A.; Locati, M. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc. Natl. Acad. Sci. USA 2009, 106, 5282–5287. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Shi, Y.; Tan, G.; Yang, C.H.; Fan, M.; Pfeffer, L.M.; Wu, Z.H. DNA damage induces NF-kappaB-dependent microRNA-21 up-regulation and promotes breast cancer cell invasion. J. Biol. Chem. 2012, 287, 21783–21795. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Guo, Y.; Li, P.; Chao, L. Role of kallistatin treatment in aging and cancer by modulating miR-34a and miR-21 expression. Oxid. Med. Cell. Longev. 2017, 2017, 5025610. [Google Scholar] [CrossRef]
- Pratheeshkumar, P.; Son, Y.O.; Divya, S.P.; Wang, L.; Zhang, Z.; Shi, X. Oncogenic transformation of human lung bronchial epithelial cells induced by arsenic involves ROS-dependent activation of STAT3-miR-21-PDCD4 mechanism. Sci. Rep. 2016, 6, 37227. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Hu, G.; Gong, A.Y.; Chen, X.M. Binding of NF-kappaB p65 subunit to the promoter elements is involved in LPS-induced transactivation of miRNA genes in human biliary epithelial cells. Nucleic Acids Res. 2010, 38, 3222–3232. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Tan, W.; Cheng, S.; Wang, H.; Ye, S.; Yu, C.; He, Y.; Zeng, J.; Cen, J.; Hu, J.; et al. Upregulation of microRNA-126 in hepatic stellate cells may affect pathogenesis of liver fibrosis through the NF-kappaB pathway. DNA Cell Biol. 2015, 34, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Ren, X.; Zhang, X.; Luo, Y.; Wang, G.; Huang, K.; Feng, S.; Bao, X.; Huang, K.; He, X.; et al. Selective killing of lung cancer cells by miRNA-506 molecule through inhibiting NF-kappaB p65 to evoke reactive oxygen species generation and p53 activation. Oncogene 2015, 34, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Sykiotis, G.P.; Bohmann, D. Stress-activated cap’n’collar transcription factors in aging and human disease. Sci. Signal. 2010, 3, re3. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Zhang, C.; Shu, L.; Kong, A.N. MicroRNAs: New players in cancer prevention targeting Nrf2, oxidative stress and inflammatory pathways. Curr. Pharmacol. Rep. 2015, 1, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.H.; Ku, C.H.; Siow, R.C.M. Regulation of the Nrf2 antioxidant pathway by microRNAs: New players in micromanaging redox homeostasis. Free Radic. Biol. Med. 2013, 64, 4–11. [Google Scholar] [CrossRef]
- Singh, A.; Happel, C.; Manna, S.K.; Acquaah-Mensah, G.; Carrerero, J.; Kumar, S.; Nasipuri, P.; Krausz, K.W.; Wakabayashi, N.; Dewi, R.; et al. Transcription factor Nrf2 regulates miR-1 and miR-206 to drive tumorigenesis. J. Clin. Investig. 2013, 123, 2921–2934. [Google Scholar] [CrossRef]
- Kurinna, S.; Schafer, M.; Ostano, P.; Karouzakis, E.; Chiorino, G.; Bloch, W.; Bachmann, A.; Gay, S.; Garrod, D.; Lefort, K.; et al. A novel Nrf2-miR-29-desmocollin-2 axis regulates desmosome function in keratinocytes. Nat. Commun. 2014, 5, 5099. [Google Scholar] [CrossRef]
- Joo, M.S.; Lee, C.G.; Koo, J.H.; Kim, S.G. Mir-125b transcriptionally increased by nrf2 inhibits AHR repressor, which protects kidney from cisplatin-induced injury. Cell Death Dis. 2013, 4, e899. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Yao, Y.; Eades, G.; Zhang, Y.; Zhou, Q. Mir-28 regulates Nrf2 expression through a Keap1-independent mechanism. Breast Cancer Res. Treat. 2011, 129, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Gao, Y.; Qin, J.; Lu, S. The role of mir-34a in the hepatoprotective effect of hydrogen sulfide on ischemia/reperfusion injury in young and old rats. PLoS ONE 2014, 9, e113305. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Ronghe, A.M.; Chatterjee, A.; Bhat, N.K.; Bhat, H.K. MicroRNA-93 regulates Nrf2 expression and is associated with breast carcinogenesis. Carcinogenesis 2013, 34, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Eades, G.; Yang, M.; Yao, Y.; Zhang, Y.; Zhou, Q. Mir-200a regulates Nrf2 activation by targeting Keap1 mRNA in breast cancer cells. J. Biol. Chem. 2011, 286, 40725–40733. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, A.; Tambaro, F.P.; Dell’Aversana, C.; Altucci, L. Cancer epigenetics: Moving forward. PLoS Genet. 2018, 14, e1007362. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Xu, Q.; Jing, Y.; Agani, F.; Qian, X.; Carpenter, R.; Li, Q.; Wang, X.R.; Peiper, S.S.; Lu, Z.; et al. Reactive oxygen species regulate ERBB2 and ERBB3 expression via miR-199a/125b and DNA methylation. EMBO Rep. 2012, 13, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Druz, A.; Betenbaugh, M.; Shiloach, J. Glucose depletion activates mmu-miR-466h-5p expression through oxidative stress and inhibition of histone deacetylation. Nucleic Acids Res. 2012, 40, 7291–7302. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef]
- Azimi, I.; Petersen, R.M.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Hypoxia-induced reactive oxygen species mediate n-cadherin and serpine1 expression, EGFR signalling and motility in mda-mb-468 breast cancer cells. Sci. Rep. 2017, 7, 15140. [Google Scholar] [CrossRef]
- Cardin, R.; Piciocchi, M.; Sinigaglia, A.; Lavezzo, E.; Bortolami, M.; Kotsafti, A.; Cillo, U.; Zanus, G.; Mescoli, C.; Rugge, M.; et al. Oxidative DNA damage correlates with cell immortalization and miR-92 expression in hepatocellular carcinoma. BMC Cancer 2012, 12, 177. [Google Scholar] [CrossRef]
- Yatabe, N.; Kyo, S.; Maida, Y.; Nishi, H.; Nakamura, M.; Kanaya, T.; Tanaka, M.; Isaka, K.; Ogawa, S.; Inoue, M. HIF-1-mediated activation of telomerase in cervical cancer cells. Oncogene 2004, 23, 3708–3715. [Google Scholar] [CrossRef] [PubMed]
- Jung, N.; Kwon, H.J.; Jung, H.J. Downregulation of mitochondrial UQCRB inhibits cancer stem cell-like properties in glioblastoma. Int. J. Oncol. 2018, 52, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, S.E.; O’Neill, L.A. HIF1alpha and metabolic reprogramming in inflammation. J. Clin. Investig. 2016, 126, 3699–3707. [Google Scholar] [CrossRef] [PubMed]
- Bavelloni, A.; Ramazzotti, G.; Poli, A.; Piazzi, M.; Focaccia, E.; Blalock, W.; Faenza, I. Mirna-210: A current overview. Anticancer Res. 2017, 37, 6511–6521. [Google Scholar]
- Fallah, A.; Sadeghinia, A.; Kahroba, H.; Samadi, A.; Heidari, H.R.; Bradaran, B.; Zeinali, S.; Molavi, O. Therapeutic targeting of angiogenesis molecular pathways in angiogenesis-dependent diseases. Biomed. Pharmacother. 2019, 110, 775–785. [Google Scholar] [CrossRef]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; Van Oosterom, A.T.; De Bruijn, E.A. Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev. 2004, 56, 549–580. [Google Scholar] [CrossRef]
- Ferrari, G.; Cook, B.D.; Terushkin, V.; Pintucci, G.; Mignatti, P. Transforming growth factor-beta 1 (TGF-beta1) induces angiogenesis through vascular endothelial growth factor (VEGF)-mediated apoptosis. J. Cell. Physiol. 2009, 219, 449–458. [Google Scholar] [CrossRef]
- Fagiani, E.; Christofori, G. Angiopoietins in angiogenesis. Cancer Lett. 2013, 328, 18–26. [Google Scholar] [CrossRef]
- Raica, M.; Cimpean, A.M. Platelet-derived growth factor (PDGF)/PDGFreceptors (PDGFR) axis as target for antitumor and antiangiogenic therapy. Pharmaceuticals 2010, 3, 572–599. [Google Scholar] [CrossRef]
- Espinosa-Diez, C.; Wilson, R.; Chatterjee, N.; Hudson, C.; Ruhl, R.; Hipfinger, C.; Helms, E.; Khan, O.F.; Anderson, D.G.; Anand, S. MicroRNA regulation of the MRN complex impacts DNA damage, cellular senescence, and angiogenic signaling. Cell Death Dis. 2018, 9, 632. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.Y.; Ramakrishnan, D.P.; Silverstein, R.L. Thrombospondin-1 modulates VEGF signaling via CD36 by recruiting SHP-1 to VEGFR2 complex in microvascular endothelial cells. Blood 2013, 122, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. Atm activation by oxidative stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Okuno, Y.; Nakamura-Ishizu, A.; Otsu, K.; Suda, T.; Kubota, Y. Pathological neoangiogenesis depends on oxidative stress regulation by atm. Nat. Med. 2012, 18, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Economopoulou, M.; Langer, H.F.; Celeste, A.; Orlova, V.V.; Choi, E.Y.; Ma, M.; Vassilopoulos, A.; Callen, E.; Deng, C.; Bassing, C.H.; et al. Histone h2ax is integral to hypoxia-driven neovascularization. Nat. Med. 2009, 15, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.; Espinosa-Diez, C.; Kanner, N.; Chatterjee, N.; Ruhl, R.; Hipfinger, C.; Advani, S.J.; Li, J.; Khan, O.F.; Franovic, A.; et al. MicroRNA regulation of endothelial TREX1 reprograms the tumour microenvironment. Nat. Commun. 2016, 7, 13597. [Google Scholar] [CrossRef]
- Yang, Z.; Wa, Q.D.; Lu, C.; Pan, W.; Lu, Z.; Ao, J. Mir3283p enhances the radiosensitivity of osteosarcoma and regulates apoptosis and cell viability via H2AX. Oncol. Rep. 2018, 39, 545–553. [Google Scholar] [PubMed]
- Marampon, F.; Codenotti, S.; Megiorni, F.; Del Fattore, A.; Camero, S.; Gravina, G.L.; Festuccia, C.; Musio, D.; De Felice, F.; Nardone, V.; et al. NRF2 orchestrates the redox regulation induced by radiation therapy, sustaining embryonal and alveolar rhabdomyosarcoma cells radioresistance. J. Cancer Res. Clin. Oncol. 2019, 145, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Crosstalk between oxidative stress and SIRT1: Impact on the aging process. Int. J. Mol. Sci. 2013, 14, 3834–3859. [Google Scholar] [CrossRef]
- Luo, J.; Chen, P.; Xie, W.; Wu, F. MicroRNA-138 inhibits cell proliferation in hepatocellular carcinoma by targeting SIRT1. Oncol. Rep. 2017, 38, 1067–1074. [Google Scholar] [CrossRef][Green Version]
- Zhou, B.; Li, C.; Qi, W.; Zhang, Y.; Zhang, F.; Wu, J.X.; Hu, Y.N.; Wu, D.M.; Liu, Y.; Yan, T.T.; et al. Downregulation of miR-181a upregulates sirtuin-1 (SIRT1) and improves hepatic insulin sensitivity. Diabetologia 2012, 55, 2032–2043. [Google Scholar] [CrossRef] [PubMed]
- Yamakuchi, M. MicroRNA regulation of SIRT1. Front. Physiol. 2012, 3, 68. [Google Scholar] [CrossRef] [PubMed]
- Rane, S.; He, M.; Sayed, D.; Vashistha, H.; Malhotra, A.; Sadoshima, J.; Vatner, D.E.; Vatner, S.F.; Abdellatif, M. Downregulation of miR-199a derepresses hypoxia-inducible factor-1alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ. Res. 2009, 104, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging biological principles of metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed]
- Peiris-Pages, M.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Metastasis and oxidative stress: Are antioxidants a metabolic driver of progression? Cell Metab. 2015, 22, 956–958. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.G.; Piskounova, E.; Morrison, S.J. Cancer, oxidative stress, and metastasis. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.L.; Liu, C.M.; Chen, H.A.; Yang, S.T.; Shigemura, K.; Kitagawa, K.; Yamamichi, F.; Fujisawa, M.; Liu, Y.R.; Lee, W.H.; et al. Reactive oxygen species-mediated switching expression of MMP-3 in stromal fibroblasts and cancer cells during prostate cancer progression. Sci. Rep. 2017, 7, 9065. [Google Scholar] [CrossRef] [PubMed]
- Cichon, M.A.; Radisky, D.C. ROS-induced epithelial-mesenchymal transition in mammary epithelial cells is mediated by NF-kB-dependent activation of snail. Oncotarget 2014, 5, 2827–2838. [Google Scholar] [CrossRef]
- Zeller, K.S.; Riaz, A.; Sarve, H.; Li, J.; Tengholm, A.; Johansson, S. The role of mechanical force and ROS in integrin-dependent signals. PLoS ONE 2013, 8, e64897. [Google Scholar] [CrossRef]
- Mori, K.; Shibanuma, M.; Nose, K. Invasive potential induced under long-term oxidative stress in mammary epithelial cells. Cancer Res. 2004, 64, 7464–7472. [Google Scholar] [CrossRef]
- Sawicki, G. Intracellular regulation of matrix metalloproteinase-2 activity: New strategies in treatment and protection of heart subjected to oxidative stress. Scientifica 2013, 2013, 130451. [Google Scholar] [CrossRef] [PubMed]
- Dezerega, A.; Madrid, S.; Mundi, V.; Valenzuela, M.A.; Garrido, M.; Paredes, R.; Garcia-Sesnich, J.; Ortega, A.V.; Gamonal, J.; Hernandez, M. Pro-oxidant status and matrix metalloproteinases in apical lesions and gingival crevicular fluid as potential biomarkers for asymptomatic apical periodontitis and endodontic treatment response. J. Inflamm. 2012, 9, 8. [Google Scholar] [CrossRef]
- Yoon, S.O.; Park, S.J.; Yoon, S.Y.; Yun, C.H.; Chung, A.S. Sustained production of H(2)O(2) activates pro-matrix metalloproteinase-2 through receptor tyrosine kinases/phosphatidylinositol 3-kinase/NF-kappa B pathway. J. Biol. Chem. 2002, 277, 30271–30282. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.H.; Wang, J.; Nie, G.; Chen, Y.J.; Li, X.; Jiang, X.; Cao, W.H. MicroRNA-509-5p functions as an anti-oncogene in breast cancer via targeting sod2. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3617–3625. [Google Scholar] [PubMed]
- Yu, L.; Chen, X.; Sun, X.; Wang, L.; Chen, S. The glycolytic switch in tumors: How many players are involved? J. Cancer 2017, 8, 3430–3440. [Google Scholar] [CrossRef] [PubMed]
- Pacella, I.; Procaccini, C.; Focaccetti, C.; Miacci, S.; Timperi, E.; Faicchia, D.; Severa, M.; Rizzo, F.; Coccia, E.M.; Bonacina, F.; et al. Fatty acid metabolism complements glycolysis in the selective regulatory t cell expansion during tumor growth. Proc. Natl. Acad. Sci. USA 2018, 115, E6546–E6555. [Google Scholar] [CrossRef]
- Schwartz, L.; Supuran, C.T.; Alfarouk, K.O. The warburg effect and the hallmarks of cancer. Anticancer Agents Med. Chem. 2017, 17, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, J.; Bae, J.S. ROS homeostasis and metabolism: A critical liaison for cancer therapy. Exp. Mol. Med. 2016, 48, e269. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef]
- Budanov, A.V. The role of tumor suppressor p53 in the antioxidant defense and metabolism. Subcell. Biochem. 2014, 85, 337–358. [Google Scholar]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed]
- Mello, T.; Simeone, I.; Galli, A. Mito-nuclear communication in hepatocellular carcinoma metabolic rewiring. Cells 2019, 8, 417. [Google Scholar] [CrossRef] [PubMed]
- Horie, T.; Nishino, T.; Baba, O.; Kuwabara, Y.; Nakao, T.; Nishiga, M.; Usami, S.; Izuhara, M.; Sowa, N.; Yahagi, N.; et al. MicroRNA-33 regulates sterol regulatory element-binding protein 1 expression in mice. Nat. Commun. 2013, 4, 2883. [Google Scholar] [CrossRef] [PubMed]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.D.; Savage, J.E.; Cao, L.; Soule, B.P.; Ly, D.; DeGraff, W.; Harris, C.C.; Mitchell, J.B.; Simone, N.L. Cellular stress induced alterations in microRNA let-7a and let-7b expression are dependent on p53. PLoS ONE 2011, 6, e24429. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ng, W.L.; Wang, P.; Tian, L.; Werner, E.; Wang, H.; Doetsch, P.; Wang, Y. MicroRNA-21 modulates the levels of reactive oxygen species by targeting sod3 and tnfalpha. Cancer Res. 2012, 72, 4707–4713. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Ren, Z.J.; Tang, J.H. MicroRNA-34a: A potential therapeutic target in human cancer. Cell Death Dis. 2014, 5, e1327. [Google Scholar] [CrossRef]
- Balzano, F.; Cruciani, S.; Basoli, V.; Santaniello, S.; Facchin, F.; Ventura, C.; Maioli, M. Mir200 and miR302: Two big families influencing stem cell behavior. Molecules 2018, 23, 282. [Google Scholar] [CrossRef]
- Kim, J.H.; Park, S.G.; Song, S.Y.; Kim, J.K.; Sung, J.H. Reactive oxygen species-responsive miR-210 regulates proliferation and migration of adipose-derived stem cells via ptpn2. Cell Death Dis. 2013, 4, e588. [Google Scholar] [CrossRef]
- Mei, Y.; Bian, C.; Li, J.; Du, Z.; Zhou, H.; Yang, Z.; Zhao, R.C. Mir-21 modulates the erk-mapk signaling pathway by regulating spry2 expression during human mesenchymal stem cell differentiation. J. Cell. Biochem. 2013, 114, 1374–1384. [Google Scholar] [CrossRef]
- Yu, G.; Yao, W.; Xiao, W.; Li, H.; Xu, H.; Lang, B. MicroRNA-34a functions as an anti-metastatic microRNA and suppresses angiogenesis in bladder cancer by directly targeting cd44. J. Exp. Clin. Cancer Res. 2014, 33, 779. [Google Scholar] [CrossRef] [PubMed]
- Peter, M.E. Let-7 and miR-200 microRNAs: Guardians against pluripotency and cancer progression. Cell Cycle 2009, 8, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Yang, Z.; Zhu, Q.; Wu, Y.; Sun, K.; Alahdal, M.; Zhang, Y.; Xing, Y.; Shen, Y.; Xia, T.; et al. Up-regulation of miR-210 induced by a hypoxic microenvironment promotes breast cancer stem cells metastasis, proliferation, and self-renewal by targeting e-cadherin. FASEB J. 2018, 32, fj201801013R. [Google Scholar] [CrossRef] [PubMed]
- Maciel-Baron, L.A.; Moreno-Blas, D.; Morales-Rosales, S.L.; Gonzalez-Puertos, V.Y.; Lopez-Diazguerrero, N.E.; Torres, C.; Castro-Obregon, S.; Konigsberg, M. Cellular senescence, neurological function, and redox state. Antioxid. Redox Signal. 2018, 28, 1704–1723. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Xu, Y. P53, oxidative stress, and aging. Antioxid. Redox Signal. 2011, 15, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Dellago, H.; Preschitz-Kammerhofer, B.; Terlecki-Zaniewicz, L.; Schreiner, C.; Fortschegger, K.; Chang, M.W.; Hackl, M.; Monteforte, R.; Kuhnel, H.; Schosserer, M.; et al. High levels of oncomiR-21 contribute to the senescence-induced growth arrest in normal human cells and its knock-down increases the replicative lifespan. Aging Cell 2013, 12, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Takeshita, F.; Hino, Y.; Fukunaga, S.; Kudo, Y.; Tamaki, A.; Matsunaga, J.; Takahashi, R.U.; Takata, T.; Shimamoto, A.; et al. Mir-22 represses cancer progression by inducing cellular senescence. J. Cell Biol. 2011, 193, 409–424. [Google Scholar] [CrossRef]
- Hu, Z.; Klein, J.D.; Mitch, W.E.; Zhang, L.; Martinez, I.; Wang, X.H. MicroRNA-29 induces cellular senescence in aging muscle through multiple signaling pathways. Aging 2014, 6, 160–175. [Google Scholar] [CrossRef]
- He, X.; Yang, A.; McDonald, D.G.; Riemer, E.C.; Vanek, K.N.; Schulte, B.A.; Wang, G.Y. Mir-34a modulates ionizing radiation-induced senescence in lung cancer cells. Oncotarget 2017, 8, 69797–69807. [Google Scholar] [CrossRef]
- Hong, L.; Lai, M.; Chen, M.; Xie, C.; Liao, R.; Kang, Y.J.; Xiao, C.; Hu, W.Y.; Han, J.; Sun, P. The miR-17-92 cluster of microRNAs confers tumorigenicity by inhibiting oncogene-induced senescence. Cancer Res. 2010, 70, 8547–8557. [Google Scholar] [CrossRef]
- Nyholm, A.M.; Lerche, C.M.; Manfe, V.; Biskup, E.; Johansen, P.; Morling, N.; Thomsen, B.M.; Glud, M.; Gniadecki, R. Mir-125b induces cellular senescence in malignant melanoma. BMC Dermatol. 2014, 14, 8. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, F.; Lazzarini, R.; Recchioni, R.; Marcheselli, F.; Rippo, M.R.; Di Nuzzo, S.; Albertini, M.C.; Graciotti, L.; Babini, L.; Mariotti, S.; et al. Mir-146a as marker of senescence-associated pro-inflammatory status in cells involved in vascular remodelling. Age 2013, 35, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Menghini, R.; Casagrande, V.; Cardellini, M.; Martelli, E.; Terrinoni, A.; Amati, F.; Vasa-Nicotera, M.; Ippoliti, A.; Novelli, G.; Melino, G.; et al. MicroRNA 217 modulates endothelial cell senescence via silent information regulator 1. Circulation 2009, 120, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Shang, F.; Wang, S.C.; Hsu, C.Y.; Miao, Y.; Martin, M.; Yin, Y.; Wu, C.C.; Wang, Y.T.; Wu, G.; Chien, S.; et al. MicroRNA-92a mediates endothelial dysfunction in ckd. J. Am. Soc. Nephrol. 2017, 28, 3251–3261. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wu, H.Y.; Wang, W.Y.; Zhao, Z.L.; Liu, X.Y.; Wang, L.Y. Regulation of miR-92a on vascular endothelial aging via mediating nrf2-keap1-are signal pathway. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 2734–2742. [Google Scholar] [PubMed]
- Sanchez-Sanchez, B.; Gutierrez-Herrero, S.; Lopez-Ruano, G.; Prieto-Bermejo, R.; Romo-Gonzalez, M.; Llanillo, M.; Pandiella, A.; Guerrero, C.; Miguel, J.F.; Sanchez-Guijo, F.; et al. Nadph oxidases as therapeutic targets in chronic myelogenous leukemia. Clin. Cancer Res. 2014, 20, 4014–4025. [Google Scholar] [CrossRef]
- Altenhofer, S.; Radermacher, K.A.; Kleikers, P.W.; Wingler, K.; Schmidt, H.H. Evolution of nadph oxidase inhibitors: Selectivity and mechanisms for target engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar] [CrossRef]
- Suzuki, S.; Pitchakarn, P.; Sato, S.; Shirai, T.; Takahashi, S. Apocynin, an nadph oxidase inhibitor, suppresses progression of prostate cancer via rac1 dephosphorylation. Exp. Toxicol. Pathol. 2013, 65, 1035–1041. [Google Scholar] [CrossRef]
- Fuji, S.; Suzuki, S.; Naiki-Ito, A.; Kato, H.; Hayakawa, M.; Yamashita, Y.; Kuno, T.; Takahashi, S. The nadph oxidase inhibitor apocynin suppresses preneoplastic liver foci of rats. Toxicol. Pathol. 2017, 45, 544–550. [Google Scholar] [CrossRef]
- Kumari, S.; Badana, A.K.; G, M.M.; G, S.; Malla, R. Reactive oxygen species: A key constituent in cancer survival. Biomark. Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, T.W.; Zhou, Y.; Peng, H.; Liu, T.; Zandi, E.; Martinez-Chantar, M.L.; Mato, J.M.; Lu, S.C. Activation of a novel c-Myc-miR27-prohibitin 1 circuitry in cholestatic liver injury inhibits glutathione synthesis in mice. Antioxid. Redox Signal. 2015, 22, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Diez, C.; Fierro-Fernandez, M.; Sanchez-Gomez, F.; Rodriguez-Pascual, F.; Alique, M.; Ruiz-Ortega, M.; Beraza, N.; Martinez-Chantar, M.L.; Fernandez-Hernando, C.; Lamas, S. Targeting of gamma-glutamyl-cysteine ligase by miR-433 reduces glutathione biosynthesis and promotes tgf-beta-dependent fibrogenesis. Antioxid. Redox Signal. 2015, 23, 1092–1105. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhou, M.; Zhou, W. MicroRNA-30e regulates tgf-beta-mediated nadph oxidase 4-dependent oxidative stress by snai1 in atherosclerosis. Int. J. Mol. Med. 2019, 43, 1806–1816. [Google Scholar] [PubMed]
- Cho, K.J.; Song, J.; Oh, Y.; Lee, J.E. MicroRNA-let-7a regulates the function of microglia in inflammation. Mol. Cell. Neurosci. 2015, 68, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Fulciniti, M.; Amodio, N.; Bandi, R.L.; Cagnetta, A.; Samur, M.K.; Acharya, C.; Prabhala, R.; D’Aquila, P.; Bellizzi, D.; Passarino, G.; et al. Mir-23b/sp1/c-myc forms a feed-forward loop supporting multiple myeloma cell growth. Blood Cancer J. 2016, 6, e380. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, F.; Wang, Y.; Liu, Z.; Sun, A.; Zen, K.; Zhang, C.Y.; Zhang, Q. The transcription factor c-myc suppresses miR-23b and miR-27b transcription during fetal distress and increases the sensitivity of neurons to hypoxia-induced apoptosis. PLoS ONE 2015, 10, e0120217. [Google Scholar] [CrossRef][Green Version]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. C-myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef]
- Mott, J.L.; Kurita, S.; Cazanave, S.C.; Bronk, S.F.; Werneburg, N.W.; Fernandez-Zapico, M.E. Transcriptional suppression of miR-29b-1/miR-29a promoter by c-myc, hedgehog, and nf-kappab. J. Cell. Biochem. 2010, 110, 1155–1164. [Google Scholar] [CrossRef]
- Hou, M.; Zuo, X.; Li, C.; Zhang, Y.; Teng, Y. Mir-29b regulates oxidative stress by targeting sirt1 in ovarian cancer cells. Cell. Physiol. Biochem. 2017, 43, 1767–1776. [Google Scholar] [CrossRef]
- Mazzoccoli, L.; Robaina, M.C.; Apa, A.G.; Bonamino, M.; Pinto, L.W.; Queiroga, E.; Bacchi, C.E.; Klumb, C.E. Mir-29 silencing modulates the expression of target genes related to proliferation, apoptosis and methylation in burkitt lymphoma cells. J. Cancer Res. Clin. Oncol. 2018, 144, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.W.; Hsu, N.Y.; Wang, Y.C.; Lee, M.C.; Cheng, Y.W.; Chen, C.Y.; Lee, H. C-myc suppresses microRNA-29b to promote tumor aggressiveness and poor outcomes in non-small cell lung cancer by targeting fhit. Oncogene 2015, 34, 2072–2082. [Google Scholar] [CrossRef] [PubMed]
- Okada, N.; Lin, C.P.; Ribeiro, M.C.; Biton, A.; Lai, G.; He, X.; Bu, P.; Vogel, H.; Jablons, D.M.; Keller, A.C.; et al. A positive feedback between p53 and miR-34 miRNAs mediates tumor suppression. Genes Dev. 2014, 28, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Mihailovich, M.; Bremang, M.; Spadotto, V.; Musiani, D.; Vitale, E.; Varano, G.; Zambelli, F.; Mancuso, F.M.; Cairns, D.A.; Pavesi, G.; et al. Mir-17-92 fine-tunes myc expression and function to ensure optimal b cell lymphoma growth. Nat. Commun. 2015, 6, 8725. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.L.; Xue, G.; Mei, Q.; Wang, Y.Z.; Ding, F.X.; Liu, M.F.; Lu, M.H.; Tang, Y.; Yu, H.Y.; Sun, S.H. Repression of the miR-17-92 cluster by p53 has an important function in hypoxia-induced apoptosis. EMBO J. 2009, 28, 2719–2732. [Google Scholar] [CrossRef] [PubMed]
- Borkowski, R.; Du, L.; Zhao, Z.; McMillan, E.; Kosti, A.; Yang, C.R.; Suraokar, M.; Wistuba, I.I.; Gazdar, A.F.; Minna, J.D.; et al. Genetic mutation of p53 and suppression of the miR-17 approximately 92 cluster are synthetic lethal in non-small cell lung cancer due to upregulation of vitamin d signaling. Cancer Res. 2015, 75, 666–675. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| miRNA | Regulation Mechanism a | ROS Production b | Expression in Cancer c | Cell/Cancer Types | Molecules, Cellular Processes and Signaling Pathways Involved d | References |
|---|---|---|---|---|---|---|
| Let-7a | OS, p53 | ✓ | Down | CSC, prostate cancer, pancreatic cancer | PTEN, LIN28b | [105,135] |
| miR-1 | NRF2, HDAC4 | ✓ | Down | Non-small cell lung cancer | NRF2, KEAP1, glucose metabolism, tumor growth | [49] |
| miR-15/16 | c-Myc | ✓ | Down | Skin, colon cancer | FGF2, HIF-2α, senescence-like phenotype, angiogenesis, metastasis | [30] |
| miR-21 | Glucose, NFκB, STAT3 | ✓ | Up | CSCs, lung cancer, liver cancer, colorectal cancer | MAPK pathway, cell migration, invasion and EMT phenotype, self-renewal ability | [41,110] |
| miR-23a | c-Myc | ✓ | - | Cardiac disease, myeloma | Glutaminase, MnSOD, apoptosis, cell growth | [136,137,138] |
| miR-29 | c-Myc, H2O2, NRF2 | ✓ | Dual role | Ovarian cancer, lung cancer, lymphoma | SIRT1, senescence, proliferation, apoptosis | [50,139,140,141,142] |
| miR-33a/b | - | ✓ | Down | Liver | HDL biosynthesis, apoptosis, OS resistance | [103] |
| miR-34 | OS, c-Myc, p53 | ✓ | Down | Stromal cells, CSC, bladder cancer, lung cancer | CD44, EMT markers, SIRT1, senescence, metastasis | [33,111,143] |
| miR-17-92 | c-Myc, p53, NFκB | ✓ | Up | Lung cancer, | Vitamin D, Senescence, apoptosis | [120,144,145,146] |
| miR-92a | - | ✓ | Up | Endothelial cells | SIRT1, KLF2, KLF4 | [124,125] |
| miR-125b | DNMT1, H2O2, NRF2 | ✓ | Dual role | Ovarian cancer, liver | Epigenetic regulation | [51,57] |
| miR-181 | - | ✓ | Up | Macrophagy, HCC | SIRT1, insulin sensitivity, NFκB activity, apoptosis | [80] |
| miR-199a | DNMT1, H2O2 | ✓ | Down, (hypermethylation) | Ovarian cancer | HIF1α, SIRT1, Epigenetic regulation | [57,83] |
| miR-200 | P53, H2O2 | ✓ | Down | CSC, breast cancer, liver cancer | Bmil-1, Suz12, Notch-1, self-renewal capacity, EMT markers, senescence | [34,35] |
| miR-210 | Hypoxia | ✓ | Up | CSCs | E-cadherin, Hypoxia, proliferation, self-renewal capacity, migration and invasion, senescence | [65,109] [113] |
| miR-217 | - | - | Dual role | Endothelial cells | SIRT1, Angiogenesis, premature senescence-like phenotype | [123] |
| miR-466h-5p | ROS, HDAC2 | - | - | Mouse ovarian epithelial | BCL2L2, apoptosis | [58] |
| MiR-506 | P53 | ✓ | Down | Lung cancer | NFκB signaling pathway | [44] |
| miR-509 | - | ✓ | Down | Breast cancer | SOD2, Cell growth, migration and invasion | [94] |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Y.-H. MicroRNA Networks Modulate Oxidative Stress in Cancer. Int. J. Mol. Sci. 2019, 20, 4497. https://doi.org/10.3390/ijms20184497
Lin Y-H. MicroRNA Networks Modulate Oxidative Stress in Cancer. International Journal of Molecular Sciences. 2019; 20(18):4497. https://doi.org/10.3390/ijms20184497
Chicago/Turabian StyleLin, Yang-Hsiang. 2019. "MicroRNA Networks Modulate Oxidative Stress in Cancer" International Journal of Molecular Sciences 20, no. 18: 4497. https://doi.org/10.3390/ijms20184497
APA StyleLin, Y.-H. (2019). MicroRNA Networks Modulate Oxidative Stress in Cancer. International Journal of Molecular Sciences, 20(18), 4497. https://doi.org/10.3390/ijms20184497