Investigation into Early Steps of Actin Recognition by the Intrinsically Disordered N-WASP Domain V


Abstract
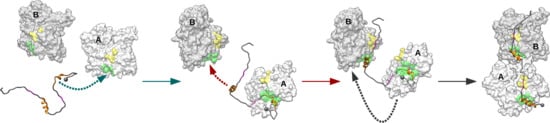
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
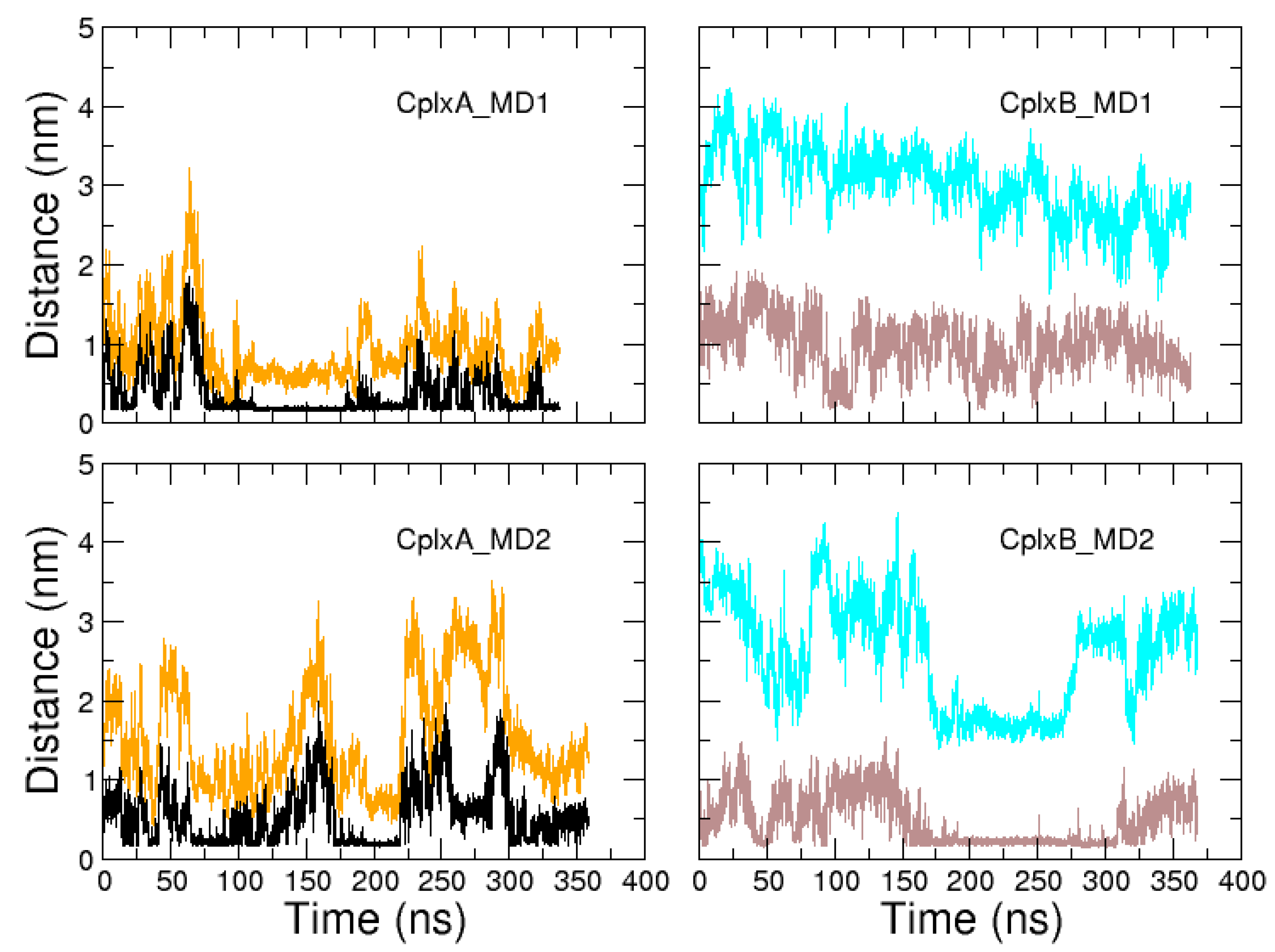{kind=link}
{kind=link}
{kind=link}
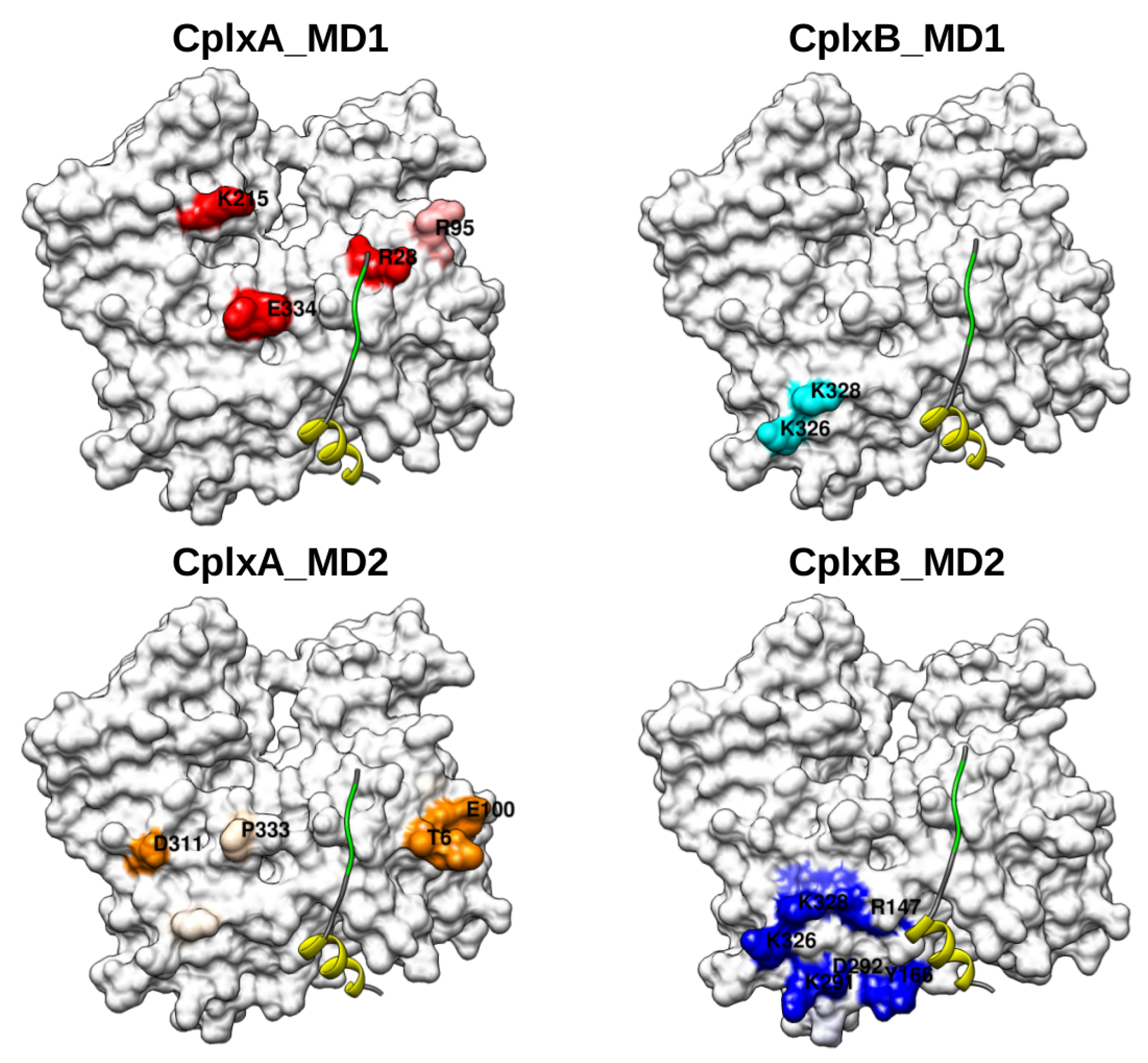{kind=link}
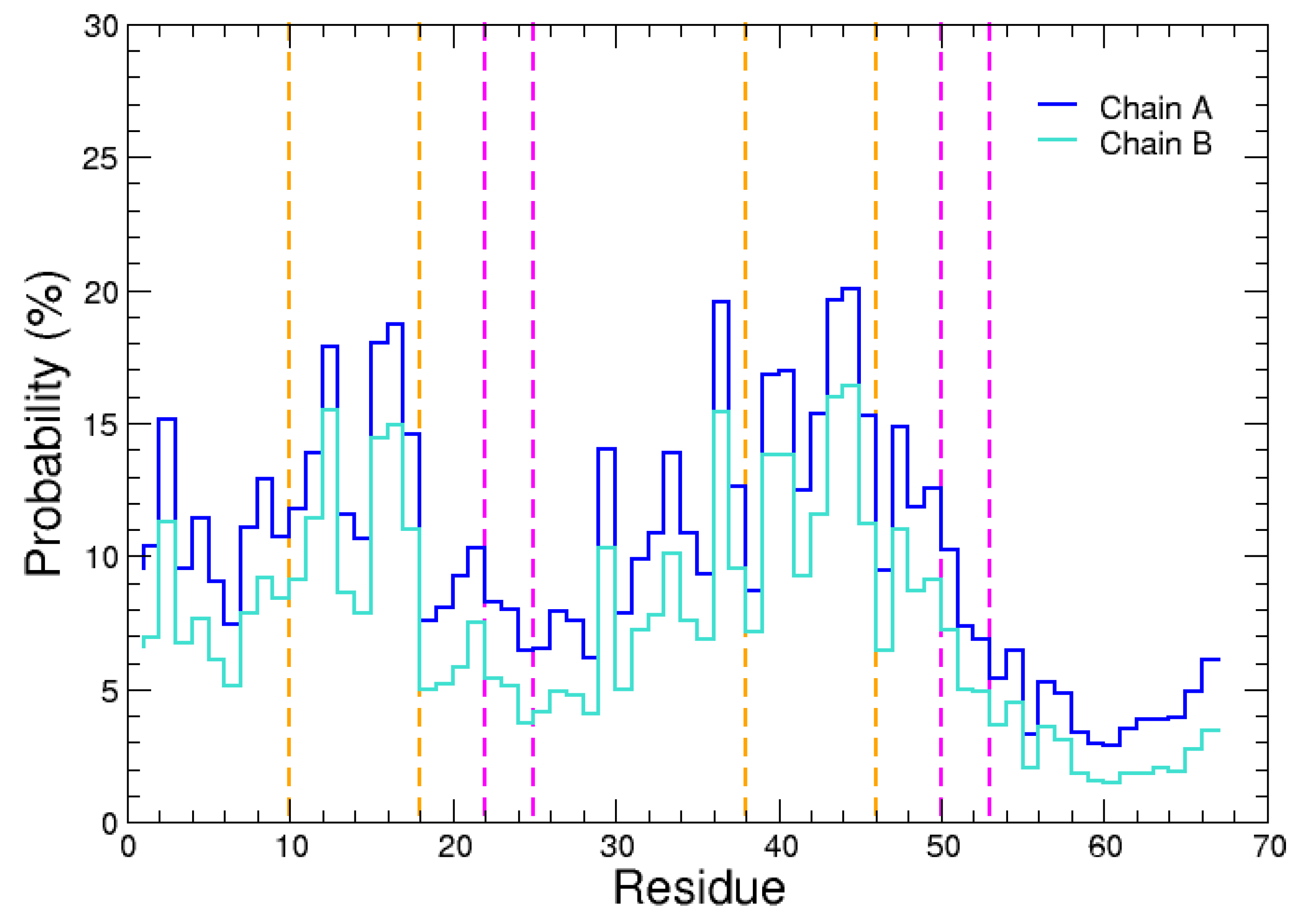{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
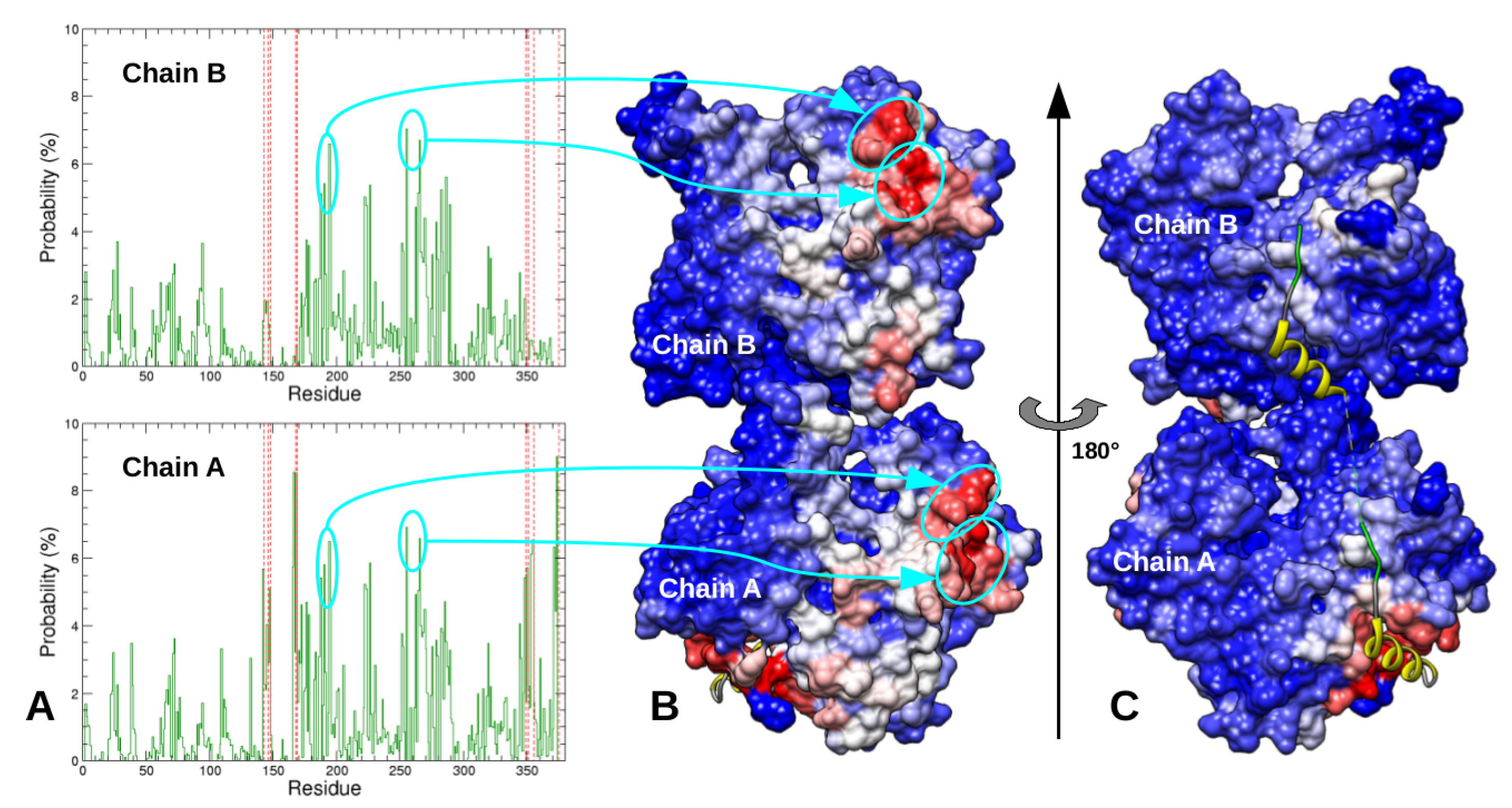
2.1. Monomeric Actin–Domain V Encounter Complexes Generated by Docking Calculations
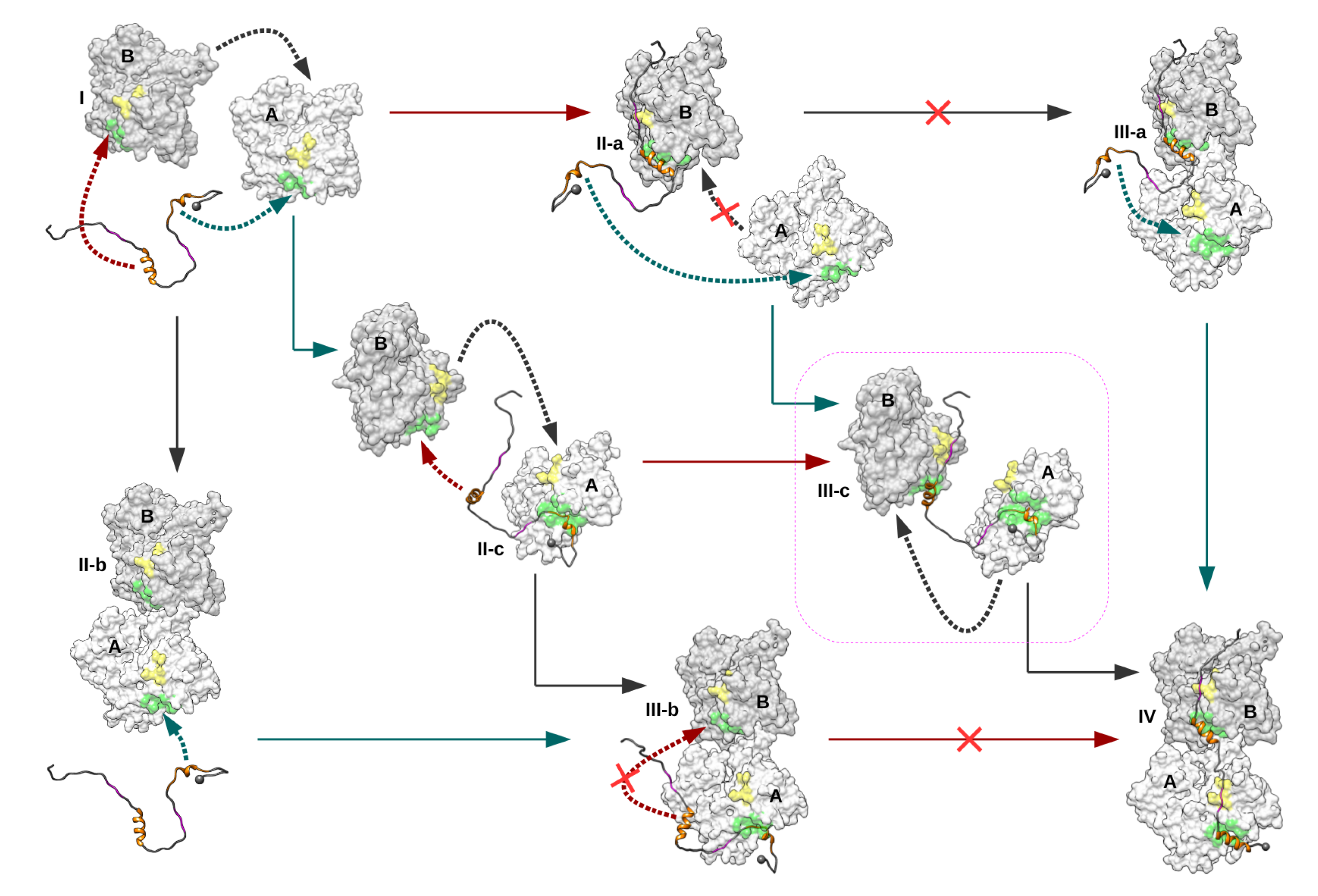
2.2. Identification and MD Simulations of Productive Actin–Domain V Encounter Complexes
2.3. Dimeric Actin–Domain V Encounter Complexes Generated by Docking Calculations
3. Discussion
4. Methods
4.1. Conformational Clustering
4.2. Protein–Protein Docking
4.3. MD Simulations
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| IDP | Intrinsically Disordered Protein |
| IDR | Intrinsically Disordered Region |
| PDB | Protein Data Bank |
| N-WASP | Neural Wiskott–Aldrich Syndrome Protein |
| MoRF | Molecular Recognition Feature |
| NMR | Nuclear Magnetic Resonance |
| SAXS | Small-Angle X-ray Scattering |
| MD | Molecular Dynamics |
| RMSD | Root Mean Square Deviation |
References
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008, 18, 756–764. [Google Scholar] [CrossRef]
- Zea, D.J.; Monzon, A.M.; Gonzalez, C.; Fornasari, M.S.; Tosatto, S.C.E.; Parisi, G. Disorder transitions and conformational diversity cooperatively modulate biological function in proteins. Protein Sci. 2016, 25, 1138–1146. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Romero, P.; Uversky, V.N.; Dunker, A.K. Coupled Folding and Binding with α-Helix-Forming Molecular Recognition Elements. Biochemistry 2005, 44, 12454–12470. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Oldfield, C.J.; Radivojac, P.; Vacic, V.; Cortese, M.S.; Dunker, A.K.; Uversky, V.N. Analysis of Molecular Recognition Features (MoRFs). J. Mol. Biol. 2006, 362, 1043–1059. [Google Scholar] [CrossRef]
- Vacic, V.; Oldfield, C.J.; Mohan, A.; Radivojac, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Characterization of Molecular Recognition Features, MoRFs, and Their Binding Partners. J. Proteome Res. 2007, 6, 2351–2366. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Oldfield, C.J.; Meng, J.; Romero, P.; Uversky, V.N.; Dunker, A.K. Mining α-helix-forming molecular recognition features with cross species sequence alignments. Biochemistry 2007, 46, 13468–13477. [Google Scholar] [CrossRef]
- Lee, C.; Kalmar, L.; Xue, B.; Tompa, P.; Daughdrill, G.W.; Uversky, V.N.; Han, K.H. Contribution of proline to the pre-structuring tendency of transient helical secondary structure elements in intrinsically disordered proteins. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Carlier, M.F.; Husson, C.; Renault, L.; Didry, D. Chapter Two–Control of Actin Assembly by the WH2 Domains and Their Multifunctional Tandem Repeats in Spire and Cordon-Bleu. In International Review of Cell and Molecular Biology; Jeon, K.W., Ed.; Academic Press: Cambridge, MA, USA, 2011; Volume 290, pp. 55–85. [Google Scholar]
- Derry, J.M.J.; Ochs, H.D.; Francke, U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. Cell 1994, 78, 635–644. [Google Scholar] [CrossRef]
- Palma, A.; Ortega, C.; Romero, P.; Garcia-V, A.; Roman, C.; Molina, I.; Santamaria, M. Wiskott-Aldrich syndrome protein (WASp) and relatives: A many-sided family. Immunologia 2004, 23, 217–230. [Google Scholar]
- Chereau, D.; Kerff, F.; Graceffa, P.; Grabarek, Z.; Langsetmo, K.; Dominguez, R. Actin-bound structures of Wiskott-Aldrich syndrome protein (WASP)-homology domain 2 and the implications for filament assembly. Proc. Natl. Acad. Sci. USA 2005, 102, 16644–16649. [Google Scholar] [CrossRef] [PubMed]
- Renault, L.; Deville, C.; van Heijenoort, C. Structural features and interfacial properties of WH2, β-thymosin domains and other intrinsically disordered domains in the regulation of actin cytoskeleton dynamics. Cytoskeleton 2013, 70, 686–705. [Google Scholar] [CrossRef][Green Version]
- Kiefhaber, T.; Bachmann, A.; Jensen, K.S. Dynamics and mechanisms of coupled protein folding and binding reactions. Curr. Opin. Struct. Biol. 2012, 22, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, J.; Chen, J. Residual Structure Accelerates Binding of Intrinsically Disordered ACTR by Promoting Efficient Folding upon Encounter. J. Mol. Biol. 2019, 431, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Li, K.; Hall, D.R.; Beglov, D.; Zheng, J.; Vakili, P.; Schueler-Furman, O.; Paschalidis, I.C.; Clore, G.M.; Vajda, S. Encounter complexes and dimensionality reduction in protein–protein association. eLife 2014, 3, e01370. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D.; Borisy, G.G. Cellular Motility Driven by Assembly and Disassembly of Actin Filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef]
- Takenawa, T.; Suetsugu, S. The WASP–WAVE protein network: connecting the membrane to the cytoskeleton. Nat. Rev. Mol. Cell Biol. 2007, 8, 37–48. [Google Scholar] [CrossRef]
- Miki, H.; Miura, K.; Takenawa, T. N-WASP, a novel actin-depolymerizing protein, regulates the cortical cytoskeletal rearrangement in a PIP2-dependent manner downstream of tyrosine kinases. Embo J. 1996, 15, 5326–5335. [Google Scholar] [CrossRef]
- Prehoda, K.E.; Scott, J.A.; Mullins, R.D.; Lim, W.A. Integration of Multiple Signals Through Cooperative Regulation of the N-WASP-Arp2/3 Complex. Science 2000, 290, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, J.; Pawson, T. N-WASP Regulation—The Sting in the Tail. Science 2000, 290, 725–726. [Google Scholar] [CrossRef] [PubMed]
- Luan, Q.; Zelter, A.; MacCoss, M.J.; Davis, T.N.; Nolen, B.J. Identification of Wiskott-Aldrich syndrome protein (WASP) binding sites on the branched actin filament nucleator Arp2/3 complex. Proc. Natl. Acad. Sci. USA 2018, 115, E1409–E1418. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, R. Actin filament nucleation and elongation factors— Structure–function relationships. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Rebowski, G.; Namgoong, S.; Boczkowska, M.; Leavis, P.C.; Navaza, J.; Dominguez, R. Structure of a Longitudinal Actin Dimer Assembled by Tandem W Domains: Implications for Actin Filament Nucleation. J. Mol. Biol. 2010, 403, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Gaucher, J.F.; Maugé, C.; Didry, D.; Guichard, B.; Renault, L.; Carlier, M.F. Interactions of Isolated C-terminal Fragments of Neural Wiskott-Aldrich Syndrome Protein (N-WASP) with Actin and Arp2/3 Complex. J. Biol. Chem. 2012, 287, 34646–34659. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Miki, H.; Suetsugu, S.; Ma, L.; Kirschner, M.W.; Takenawa, T. Two tandem verprolin homology domains are necessary for a strong activation of Arp2/3 complex-induced actin polymerization and induction of microspike formation by N-WASP. Proc. Natl. Acad. Sci. USA 2000, 97, 12631–12636. [Google Scholar] [CrossRef]
- Chan-Yao-Chong, M.; Deville, C.; Pinet, L.; van Heijenoort, C.; Durand, D.; Ha-Duong, T. Structural Characterization of N-WASP Domain V Using MD Simulations with NMR and SAXS Data. Biophys. J. 2019, 116, 1216–1227. [Google Scholar] [CrossRef]
- Andreani, J.; Faure, G.; Guerois, R. InterEvScore: a novel coarse-grained interface scoring function using a multi-body statistical potential coupled to evolution. Bioinformatics 2013, 29, 1742–1749. [Google Scholar] [CrossRef]
- Kollmar, M.; Lbik, D.; Enge, S. Evolution of the eukaryotic ARP2/3 activators of the WASP family: WASP, WAVE, WASH, and WHAMM, and the proposed new family members WAWH and WAML. BMC Res. Notes 2012, 5, 88. [Google Scholar] [CrossRef]
- Chen, X.; Ni, F.; Tian, X.; Kondrashkina, E.; Wang, Q.; Ma, J. Structural Basis of Actin Filament Nucleation by Tandem W Domains. Cell Rep. 2013, 3, 1910–1920. [Google Scholar] [CrossRef] [PubMed]
- Merino, F.; Pospich, S.; Funk, J.; Wagner, T.; Küllmer, F.; Arndt, H.D.; Bieling, P.; Raunser, S. Structural transitions of F-actin upon ATP hydrolysis at near-atomic resolution revealed by cryo-EM. Nat. Struct. Mol. Biol. 2018, 25, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Hertzog, M.; van Heijenoort, C.; Didry, D.; Gaudier, M.; Coutant, J.; Gigant, B.; Didelot, G.; Préat, T.; Knossow, M.; Guittet, E.; et al. The β-Thymosin/WH2 Domain: Structural Basis for the Switch from Inhibition to Promotion of Actin Assembly. Cell 2004, 117, 611–623. [Google Scholar] [CrossRef]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; van Gunsteren, W.F.; Mark, A.E. Peptide Folding: When Simulation Meets Experiment. Angew. Chem. Int. Ed. 1999, 38, 236–240. [Google Scholar] [CrossRef]
- Saladin, A.; Fiorucci, S.; Poulain, P.; Prévost, C.; Zacharias, M. PTools: An opensource molecular docking library. BMC Struct. Biol. 2009, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Basdevant, N.; Borgis, D.; Ha-Duong, T. A Coarse-Grained Protein–Protein Potential Derived from an All-Atom Force Field. J. Phys. Chem. B 2007, 111, 9390–9399. [Google Scholar] [CrossRef] [PubMed]
- Basdevant, N.; Borgis, D.; Ha-Duong, T. Modeling Protein–Protein Recognition in Solution Using the Coarse-Grained Force Field SCORPION. J. Chem. Theory Comput. 2013, 9, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Pall, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. SETTLE: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Best, R.B.; Mittal, J. Protein Simulations with an Optimized Water Model: Cooperative Helix Formation and Temperature-Induced Unfolded State Collapse. J. Phys. Chem. B 2010, 114, 14916–14923. [Google Scholar] [CrossRef]
- Best, R.B.; Zheng, W.; Mittal, J. Balanced Protein–Water Interactions Improve Properties of Disordered Proteins and Non-Specific Protein Association. J. Chem. Theory Comput. 2014, 10, 5113–5124. [Google Scholar] [CrossRef] [PubMed]
- Heinig, M.; Frishman, D. STRIDE: A web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004, 32, W500–W502. [Google Scholar] [CrossRef] [PubMed]












© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan-Yao-Chong, M.; Durand, D.; Ha-Duong, T. Investigation into Early Steps of Actin Recognition by the Intrinsically Disordered N-WASP Domain V. Int. J. Mol. Sci. 2019, 20, 4493. https://doi.org/10.3390/ijms20184493
Chan-Yao-Chong M, Durand D, Ha-Duong T. Investigation into Early Steps of Actin Recognition by the Intrinsically Disordered N-WASP Domain V. International Journal of Molecular Sciences. 2019; 20(18):4493. https://doi.org/10.3390/ijms20184493
Chicago/Turabian StyleChan-Yao-Chong, Maud, Dominique Durand, and Tâp Ha-Duong. 2019. "Investigation into Early Steps of Actin Recognition by the Intrinsically Disordered N-WASP Domain V" International Journal of Molecular Sciences 20, no. 18: 4493. https://doi.org/10.3390/ijms20184493
APA StyleChan-Yao-Chong, M., Durand, D., & Ha-Duong, T. (2019). Investigation into Early Steps of Actin Recognition by the Intrinsically Disordered N-WASP Domain V. International Journal of Molecular Sciences, 20(18), 4493. https://doi.org/10.3390/ijms20184493