Identification of Long Non-Coding RNAs and the Regulatory Network Responsive to Arbuscular Mycorrhizal Fungi Colonization in Maize Roots

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Phenotypic Responses of Maize Seedlings to AM Fungus
2.2. Ultra-Deep RNA Sequencing and Mapping onto Reference Genome
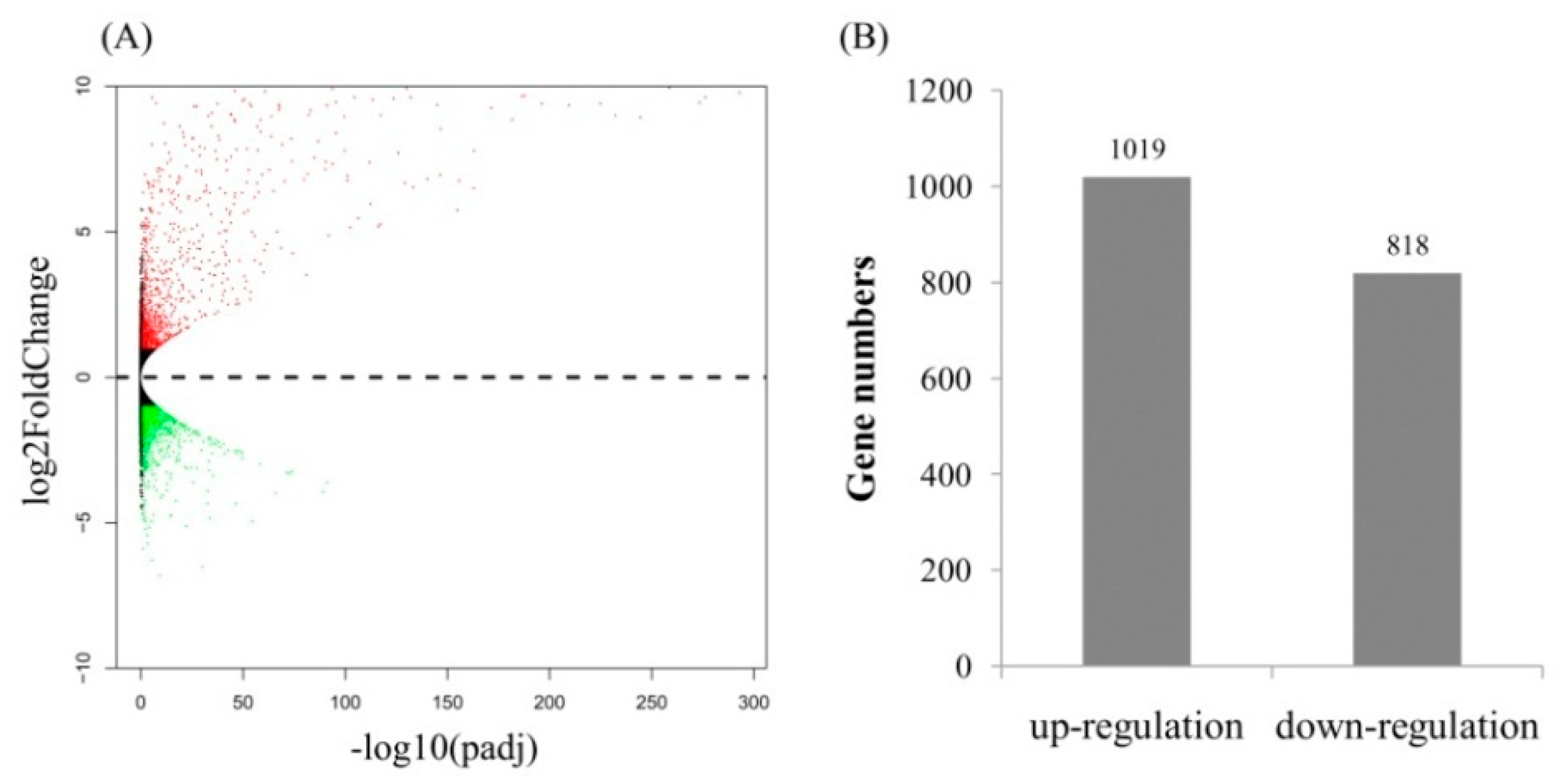
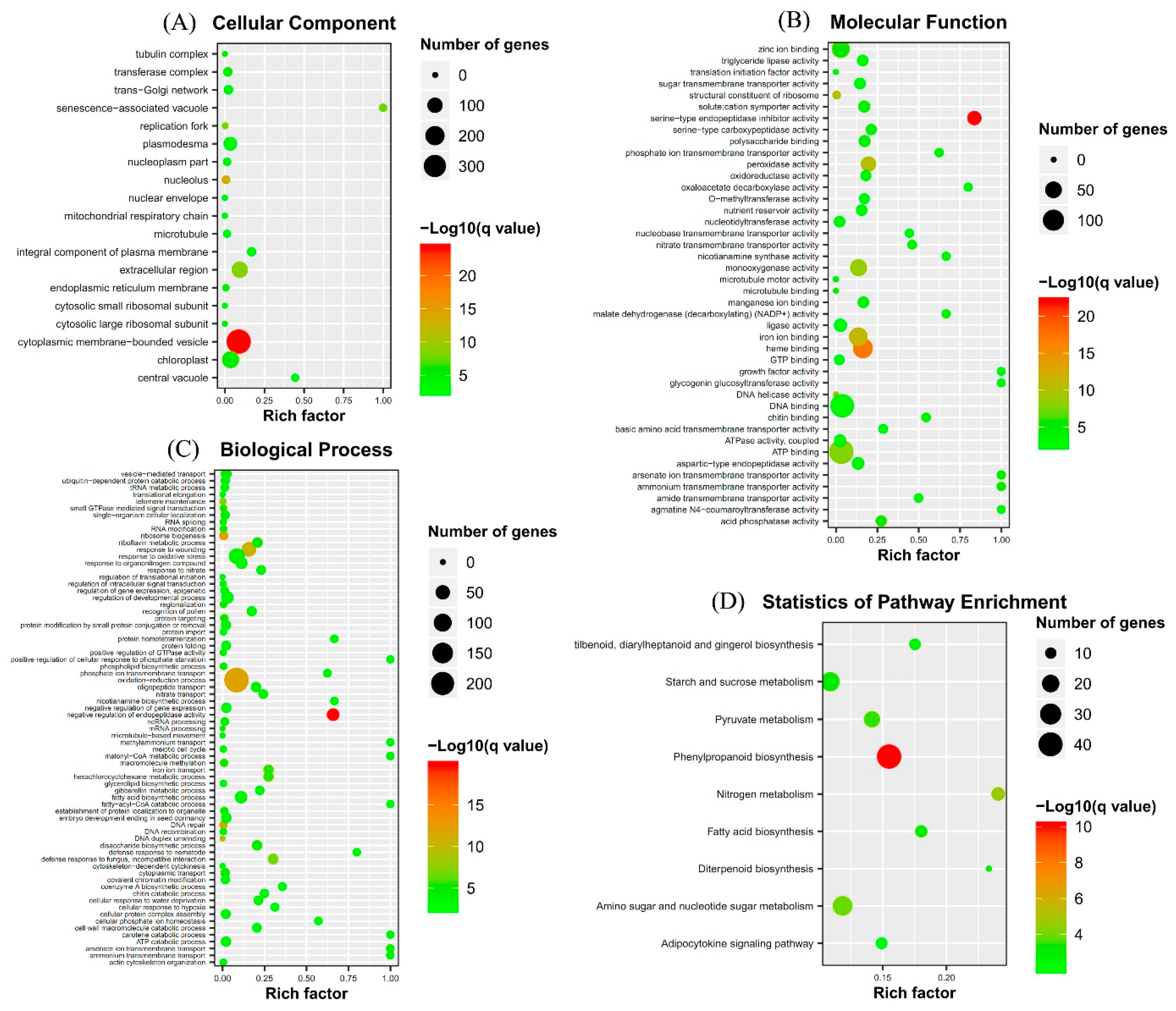
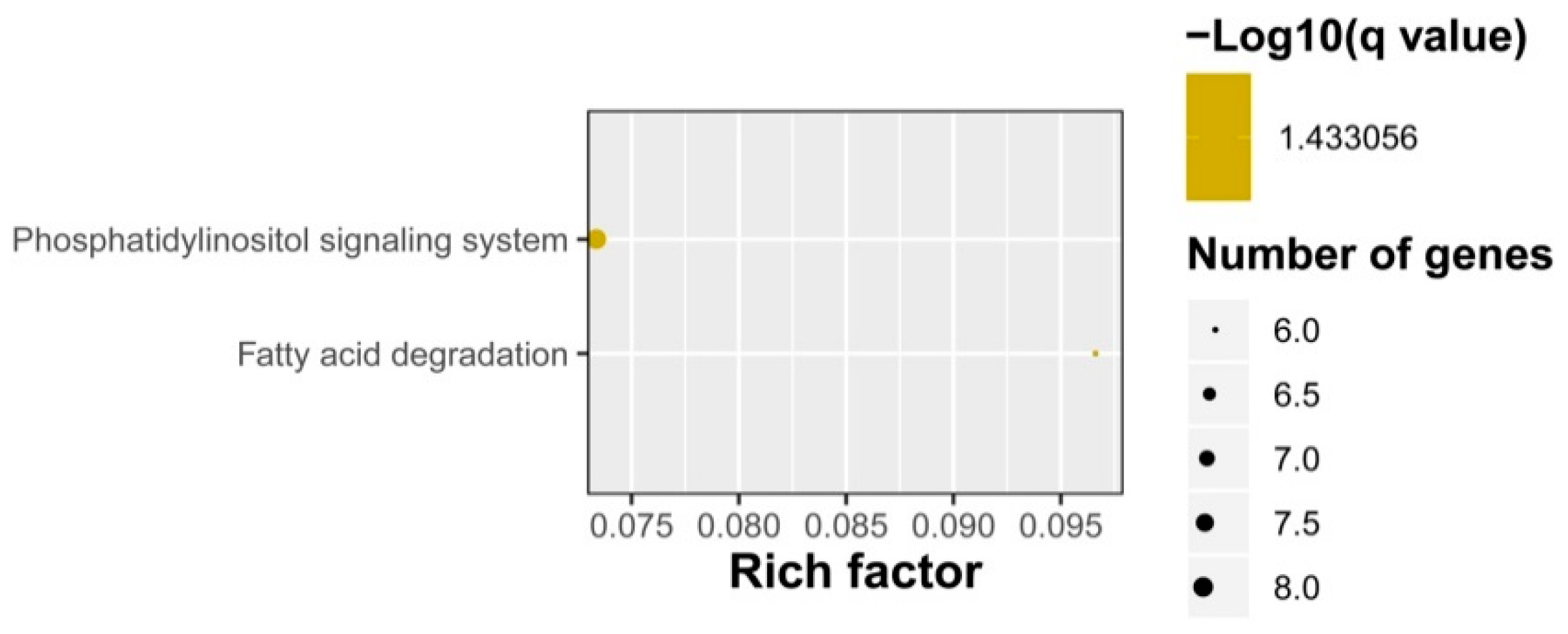
2.3. Gene Ontology (GO) Term and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Enrichment Analyses of Identified DEGs
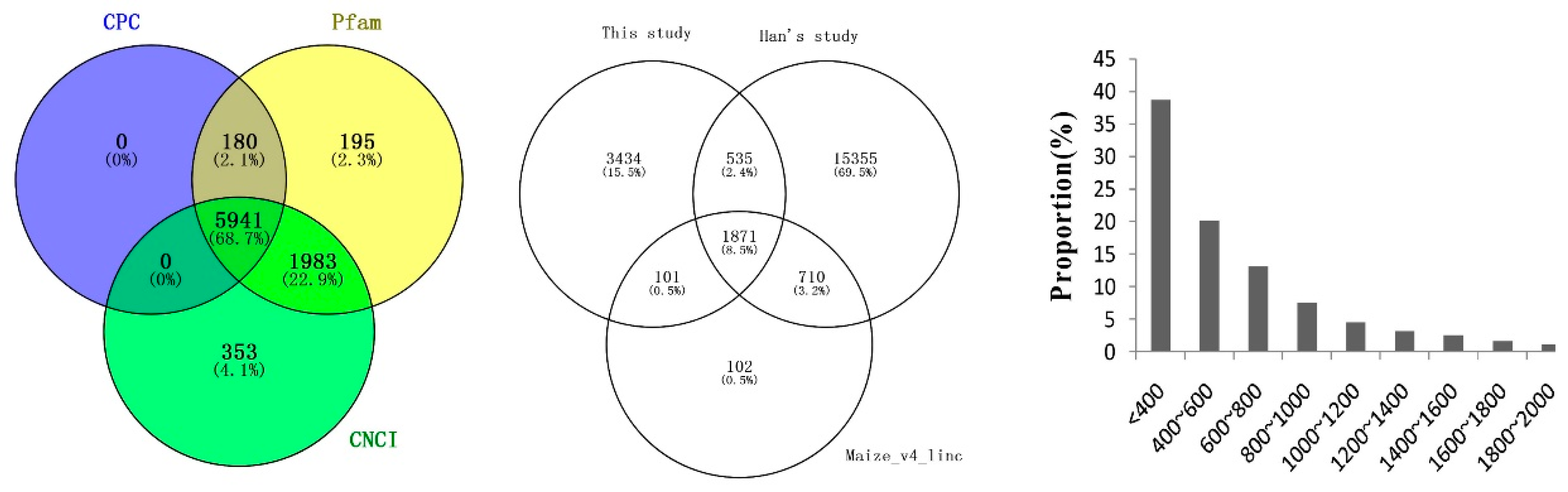
2.4. Identification and Characterization of Novel LncRNAs
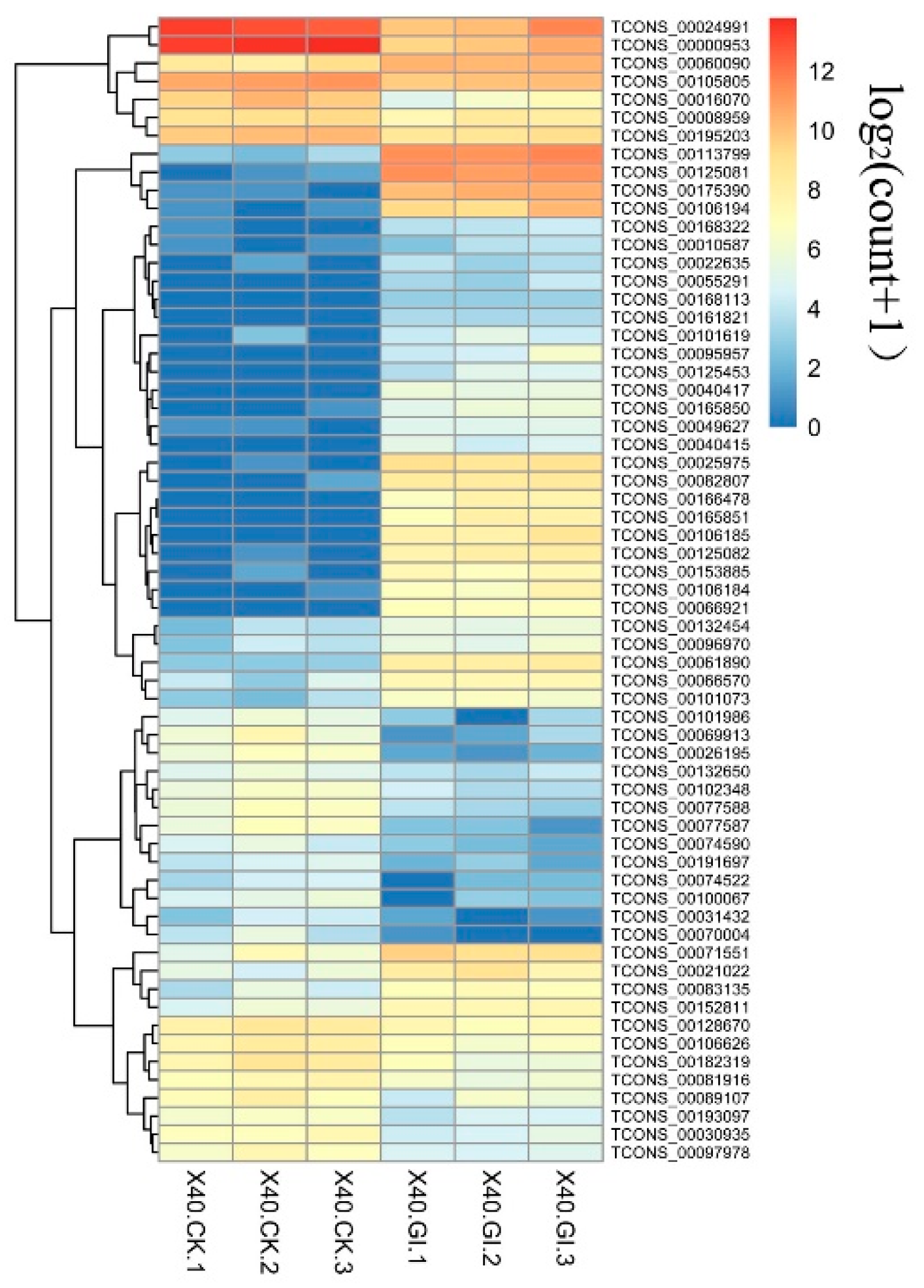
2.5. Putative Target Genes of Differentially Expressed lncRNAs (DELs)
2.6. Regulatory Network of DELs
2.7. Putative Vital DELs
2.8. Real-Time PCR Verification of Five DEGs and DELs
3. Discussion
4. Materials and Methods
4.1. Plant Symbiosis with AM Fungus
4.2. RNA Extraction and Total RNA-Seq Sequencing
4.3. Pipelines for Differentially Expressed Genes (DEGs) and Novel LncRNAS Identification
4.4. Prediction of Putative Cis- and Trans-targets of DELs
4.5. GO and KEGG Pathway Enrichment Analyses
4.6. Construction of the Regulatory Network
4.7. Validation of DEGs and DELs by Quantitative RT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lv, Y.; Liang, Z.; Ge, M.; Qi, W.; Zhang, T.; Lin, F.; Peng, Z.; Zhao, H. Genome-wide identification and functional prediction of nitrogen-responsive intergenic and intronic long non-coding RNAs in maize (Zea mays L.). BMC Genom. 2016, 17, 350. [Google Scholar] [CrossRef] [PubMed]
- Hurni, S.; Scheuermann, D.; Krattinger, S.G.; Kessel, B.; Wicker, T.; Herren, G.; Fitze, M.N.; Breen, J.; Presterl, T.; Ouzunova, M.; et al. The maize disease resistance gene Htn1 against northern corn leaf blight encodes a wall-associated receptor-like kinase. Proc. Natl. Acad. Sci. USA 2015, 112, 8780–8785. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Balint-Kurti, P.; Xu, M. Quantitative Disease Resistance: Dissection and Adoption in Maize. Mol. Plant 2017, 10, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; He, Y.; Kabahuma, M.; Chaya, T.; Kelly, A.; Borrego, E.; Bian, Y.; El Kasmi, F.; Yang, L.; Teixeira, P.; et al. A gene encoding maize caffeoyl-CoA O-methyltransferase confers quantitative resistance to multiple pathogens. Nat. Genet. 2017, 49, 1364–1372. [Google Scholar] [CrossRef]
- Li, W.; Xiang, F.; Zhong, M.; Zhou, L.; Liu, H.; Li, S.; Wang, X. Transcriptome and metabolite analysis identifies nitrogen utilization genes in tea plant (Camellia sinensis). Sci. Rep. 2017, 7, 1693. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Chen, X.; Wang, X.; Xu, Z.; Gao, X.; Jiang, C.; Deng, R.; Han, G. The Algicidal Fungus Trametes versicolor F21a Eliminating Blue Algae via Genes Encoding Degradation Enzymes and Metabolic Pathways Revealed by Transcriptomic Analysis. Front. Microbiol. 2018, 9, 826. [Google Scholar] [CrossRef]
- Gao, X.; Wang, C.; Dai, W.; Ren, S.; Tao, F.; He, X.; Han, G.; Wang, W. Proteomic analysis reveals large amounts of decomposition enzymes and major metabolic pathways involved in algicidal process of Trametes versicolor F21a. Sci. Rep. 2017, 7, 3907. [Google Scholar] [CrossRef]
- Plett, J.M.; Martin, F.M. Know your enemy, embrace your friend: Using omics to understand how plants respond differently to pathogenic and mutualistic microorganisms. Plant J. 2018, 93, 729–746. [Google Scholar] [CrossRef]
- van Velzen, R.; Holmer, R.; Bu, F.J.; Rutten, L.; van Zeijl, A.; Liu, W.; Santuari, L.; Cao, Q.Q.; Sharma, T.; Shen, D.F.; et al. Comparative genomics of the nonlegume Parasponia reveals insights into evolution of nitrogen-fixing rhizobium symbioses. Proc. Natl. Acad. Sci. USA 2018, 115, E4700–E4709. [Google Scholar] [CrossRef]
- Jiang, Y.N.; Xie, Q.J.; Wang, W.X.; Yang, J.; Zhang, X.W.; Yu, N.; Zhou, Y.; Wang, E.T. Medicago AP2-Domain Transcription Factor WRI5a Is a Master Regulator of Lipid Biosynthesis and Transfer during Mycorrhizal Symbiosis. Mol. Plant 2018, 11, 1344–1359. [Google Scholar] [CrossRef]
- Ferguson, B.J.; Mens, C.; Hastwell, A.H.; Zhang, M.; Su, H.; Jones, C.H.; Chu, X.; Gresshoff, P.M. Legume nodulation: The host controls the party. Plant Cell Environ. 2019, 42, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Hoysted, G.A.; Kowal, J.; Jacob, A.; Rimington, W.R.; Duckett, J.G.; Pressel, S.; Orchard, S.; Ryan, M.H.; Field, K.J.; Bidartondo, M.I. A mycorrhizal revolution. Curr. Opin. Plant Biol. 2018, 44, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Shi, J.; Xie, Q.; Jiang, Y.; Yu, N.; Wang, E. Nutrient Exchange and Regulation in Arbuscular Mycorrhizal Symbiosis. Mol. Plant 2017, 10, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Nadal, M.; Sawers, R.; Naseem, S.; Bassin, B.; Kulicke, C.; Sharman, A.; An, G.; An, K.; Ahern, K.R.; Romag, A.; et al. An N-acetylglucosamine transporter required for arbuscular mycorrhizal symbioses in rice and maize. Nat. Plants 2017, 3, 17073. [Google Scholar] [CrossRef] [PubMed]
- MacLean, A.M.; Bravo, A.; Harrison, M.J. Plant Signaling and Metabolic Pathways Enabling Arbuscular Mycorrhizal Symbiosis. Plant Cell 2017, 29, 23192–23335. [Google Scholar] [CrossRef]
- Oldroyd, G.E. Speak, friend, and enter: Signalling systems that promote beneficial symbiotic associations in plants. Nat. Rev. Microbiol. 2013, 11, 252–263. [Google Scholar] [CrossRef]
- Charpentier, M.; Sun, J.; Vaz Martins, T.; Radhakrishnan, G.V.; Findlay, K.; Soumpourou, E.; Thouin, J.; Very, A.A.; Sanders, D.; Morris, R.J.; et al. Nuclear-localized cyclic nucleotide-gated channels mediate symbiotic calcium oscillations. Science 2016, 352, 1102–1105. [Google Scholar] [CrossRef]
- Kanamori, N.; Madsen, L.H.; Radutoiu, S.; Frantescu, M.; Quistgaard, E.M.; Miwa, H.; Downie, J.A.; James, E.K.; Felle, H.H.; Haaning, L.L.; et al. A nucleoporin is required for induction of Ca2+ spiking in legume nodule development and essential for rhizobial and fungal symbiosis. Proc. Natl. Acad. Sci. USA 2006, 103, 359–364. [Google Scholar] [CrossRef]
- Saito, K.; Yoshikawa, M.; Yano, K.; Miwa, H.; Uchida, H.; Asamizu, E.; Sato, S.; Tabata, S.; Imaizumi-Anraku, H.; Umehara, Y.; et al. NUCLEOPORIN85 is required for calcium spiking, fungal and bacterial symbioses, and seed production in Lotus japonicus. Plant Cell 2007, 19, 610–624. [Google Scholar] [CrossRef]
- Groth, M.; Takeda, N.; Perry, J.; Uchida, H.; Draxl, S.; Brachmann, A.; Sato, S.; Tabata, S.; Kawaguchi, M.; Wang, T.L.; et al. NENA, a Lotus japonicus homolog of Sec13, is required for rhizodermal infection by arbuscular mycorrhiza fungi and rhizobia but dispensable for cortical endosymbiotic development. Plant Cell 2010, 22, 2509–2526. [Google Scholar] [CrossRef]
- Imaizumi-Anraku, H.; Takeda, N.; Charpentier, M.; Perry, J.; Miwa, H.; Umehara, Y.; Kouchi, H.; Murakami, Y.; Mulder, L.; Vickers, K.; et al. Plastid proteins crucial for symbiotic fungal and bacterial entry into plant roots. Nature 2005, 433, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Hogg, B.V.; Cullimore, J.V.; Ranjeva, R.; Bono, J.J. The DMI1 and DMI2 early symbiotic genes of medicago truncatula are required for a high-affinity nodulation factor-binding site associated to a particulate fraction of roots. Plant Physiol. 2006, 140, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Limpens, E.; Geurts, R. Transcriptional Regulation of Nutrient Exchange in Arbuscular Mycorrhizal Symbiosis. Mol. Plant 2018, 11, 14211–14423. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Klinnawee, L.; Zhou, Y.; Saridis, G.; Vijayakumar, V.; Brands, M.; Dormann, P.; Gigolashvili, T.; Turck, F.; Bucher, M. AP2 transcription factor CBX1 with a specific function in symbiotic exchange of nutrients in mycorrhizal Lotus japonicus. Proc. Natl. Acad. Sci. USA 2018, 115, E9239–E9246. [Google Scholar] [CrossRef] [PubMed]
- Wekesa, J.S.; Luan, Y.; Chen, M.; Meng, J. A Hybrid Prediction Method for Plant lncRNA-Protein Interaction. Cells 2019, 8, 521. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhao, M.; Zhang, X.; Liu, M.; Yang, C.; Chen, Y.; Chen, R.; Wen, J.; Mysore, K.S.; Zhang, W.H. Novel phosphate deficiency-responsive long non-coding RNAs in the legume model plant Medicago truncatula. J. Exp. Bot. 2017, 68, 59375–59948. [Google Scholar] [CrossRef] [PubMed]
- Nejat, N.; Mantri, N. Emerging roles of long non-coding RNAs in plant response to biotic and abiotic stresses. Crit. Rev. Biotechnol. 2018, 38, 93–105. [Google Scholar] [CrossRef]
- Yamamura, S.; Imai-Sumida, M.; Tanaka, Y.; Dahiya, R. Interaction and cross-talk between non-coding RNAs. Cell Mol. Life Sci. 2018, 75, 4674–4684. [Google Scholar] [CrossRef]
- Munschauer, M.; Nguyen, C.T.; Sirokman, K.; Hartigan, C.R.; Hogstrom, L.; Engreitz, J.M.; Ulirsch, J.C.; Fulco, C.P.; Subramanian, V.; Chen, J.; et al. The NORAD lncRNA assembles a topoisomerase complex critical for genome stability. Nature 2018, 561, 1321–1336. [Google Scholar] [CrossRef]
- Sethuraman, S.; Thomas, M.; Gay, L.A.; Renne, R. Computational analysis of ribonomics datasets identifies long non-coding RNA targets of gamma-herpesviral miRNAs. Nucleic Acids Res. 2018, 46, 8574–8589. [Google Scholar] [CrossRef]
- Li, W.Q.; Jia, Y.L.; Liu, F.Q.; Wang, F.Q.; Fan, F.J.; Wang, J.; Zhu, J.Y.; Xu, Y.; Zhong, W.G.; Yang, J. Genome-wide identification and characterization of long non-coding RNAs responsive to Dickeya zeae in rice. Rsc. Adv. 2018, 8, 34408–34417. [Google Scholar] [CrossRef]
- Liu, W.; Cheng, C.; Lin, Y.; XuHan, X.; Lai, Z. Genome-wide identification and characterization of mRNAs and lncRNAs involved in cold stress in the wild banana (Musa itinerans). PLoS ONE 2018, 13, e0200002. [Google Scholar] [CrossRef] [PubMed]
- Shen, E.; Zhu, X.; Hua, S.; Chen, H.; Ye, C.; Zhou, L.; Liu, Q.; Zhu, Q.H.; Fan, L.; Chen, X. Genome-wide identification of oil biosynthesis-related long non-coding RNAs in allopolyploid Brassica napus. BMC Genom. 2018, 19, 745. [Google Scholar] [CrossRef] [PubMed]
- Subburaj, S.; Jeon, Y.; Tu, L.; Jin, Y.-T.; Kumari, S.; Lee, G.J. Genome-wide identification, functional prediction and expression profiling of long non-coding RNAs in Camelina sativa. Plant Growth Regul. 2018, 86, 49–63. [Google Scholar] [CrossRef]
- Wang, T.Z.; Liu, M.; Zhao, M.G.; Chen, R.; Zhang, W.H. Identification and characterization of long non-coding RNAs involved in osmotic and salt stress in Medicago truncatula using genome-wide high-throughput sequencing. BMC Plant Biol. 2015, 15, 131. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Zhang, X.; Ma, X.; Zhao, J. Spatio-Temporal Transcriptional Dynamics of Maize Long Non-Coding RNAs Responsive to Drought Stress. Genes (Basel) 2019, 10, 138. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Liu, P.; Irwanto, N.; Loh, R.; Wong, S.M. Upregulation of LINC-AP2 is negatively correlated with AP2 gene expression with Turnip crinkle virus infection in Arabidopsis thaliana. Plant Cell Rep. 2016, 35, 2257–2267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Hu, W.; Hao, J.; Lv, S.; Wang, C.; Tong, W.; Wang, Y.; Wang, Y.; Liu, X.; Ji, W. Genome-wide identification and functional prediction of novel and fungi-responsive lincRNAs in Triticum aestivum. BMC Genom. 2016, 17, 238. [Google Scholar] [CrossRef]
- Jiang, N.; Cui, J.; Shi, Y.; Yang, G.; Zhou, X.; Hou, X.; Meng, J.; Luan, Y. Tomato lncRNA23468 functions as a competing endogenous RNA to modulate NBS-LRR genes by decoying miR482b in the tomato-Phytophthora infestans interaction. Hortic. Res. 2019, 6, 28. [Google Scholar] [CrossRef]
- Muthusamy, M.; Uma, S.; Suthanthiram, B.; Saraswathi, M.S.; Chandrasekar, A. Genome-wide identification of novel, long non-coding RNAs responsive to Mycosphaerella eumusae and Pratylenchus coffeae infections and their differential expression patterns in disease-resistant and sensitive banana cultivars. Plant Biotechnol. Rep. 2019, 13, 73–83. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, T.; Shen, D.; Wang, J.; Ling, X.; Hu, Z.; Chen, T.; Hu, J.; Huang, J.; Yu, W.; et al. Tomato yellow leaf curl virus intergenic siRNAs target a host long noncoding RNA to modulate disease symptoms. PLoS Pathog. 2019, 15, e1007534. [Google Scholar] [CrossRef] [PubMed]
- Runtsch, M.C.; O’Neill, L.A. GOTcha: lncRNA-ACOD1 targets metabolism during viral infection. Cell Res. 2018, 28, 137–138. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Jiang, N.; Meng, J.; Yang, G.; Liu, W.; Zhou, X.; Ma, N.; Hou, X.; Luan, Y. LncRNA33732-respiratory burst oxidase module associated with WRKY1 in tomato-Phytophthora infestans interactions. Plant J. cell Mol. Biol. 2019, 97, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Wang, K.; Zou, C.; Xu, C.; Li, W.X. The PILNCR1-miR399 Regulatory Module Is Important for Low Phosphate Tolerance in Maize. Plant Physiol. 2018, 177, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Mu, Z.; Luo, Z.; Pan, Q.; Li, L. New lncRNA annotation reveals extensive functional divergence of the transcriptome in maize. J. Integr. Plant Biol. 2019, 61, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.J.; Zhu, S.W.; Liu, F.; Wang, W.; Wang, X.W.; Han, G.M.; Cheng, B.J. Identification of Arbuscular Mycorrhiza Fungi Responsive microRNAs and Their Regulatory Network in Maize. Int. J. Mol. Sci. 2018, 19, 3201. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wang, W.; Xie, Q.; Liu, N.; Liu, L.; Wang, D.; Zhang, X.; Yang, C.; Chen, X.; Tang, D.; et al. Plants transfer lipids to sustain colonization by mutualistic mycorrhizal and parasitic fungi. Science 2017, 356, 1172–1175. [Google Scholar] [CrossRef]
- Luginbuehl, L.H.; Menard, G.N.; Kurup, S.; Van Erp, H.; Radhakrishnan, G.V.; Breakspear, A.; Oldroyd, G.E.D.; Eastmond, P.J. Fatty acids in arbuscular mycorrhizal fungi are synthesized by the host plant. Science 2017, 356, 1175–1178. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dong, J.; Bennetzen, J.L.; Zhong, M.; Yang, J.; Zhang, J.; Li, S.; Hao, X.; Zhang, Z.; Wang, X. Integrating transcriptome and microRNA analysis identifies genes and microRNAs for AHO-induced systemic acquired resistance in N. tabacum. Sci. Rep. 2017, 7, 12504. [Google Scholar] [CrossRef]
- Zhao, S.; Wang, X.; Yan, X.; Guo, L.; Mi, X.; Xu, Q.; Zhu, J.; Wu, A.; Liu, L.; Wei, C. Revealing of MicroRNA Involved Regulatory Gene Networks on Terpenoid Biosynthesis in Camellia sinensis in Different Growing Time Points. J. Agric. Food Chem. 2018, 66, 12604–12616. [Google Scholar] [CrossRef]
- Alonso, C.; Ramos-Cruz, D.; Becker, C. The role of plant epigenetics in biotic interactions. New Phytol. 2019, 221, 7317–7337. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Pfeifer, K.; Park, K.S. Circular RNAs and competing endogenous RNA (ceRNA) networks. Transl. Cancer Res. 2018, 7, S624–S628. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Peluso, P.; Shi, J.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.; Chin, C.S.; et al. Improved maize reference genome with single-molecule technologies. Nature 2017, 546, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Zhao, J.; Hu, G. Genome-wide methods for investigating long noncoding RNAs. Biomed. Pharm. 2019, 111, 395–401. [Google Scholar] [CrossRef]
- Dreyer, I.; Spitz, O.; Kanonenberg, K.; Montag, K.; Handrich, M.R.; Ahmad, S.; Schott-Verdugo, S.; Navarro-Retamal, C.; Rubio-Melendez, M.E.; Gomez-Porras, J.L.; et al. Nutrient exchange in arbuscular mycorrhizal symbiosis from a thermodynamic point of view. New Phytol. 2018, 222, 1043–1053. [Google Scholar] [CrossRef]
- Lanfranco, L.; Fiorilli, V.; Venice, F.; Bonfante, P. Strigolactones cross the kingdoms: Plants, fungi, and bacteria in the arbuscular mycorrhizal symbiosis. J. Exp. Bot. 2018, 69, 2175–2188. [Google Scholar] [CrossRef]
- Jalali, S.; Bhartiya, D.; Lalwani, M.K.; Sivasubbu, S.; Scaria, V. Systematic transcriptome wide analysis of lncRNA-miRNA interactions. PLoS ONE 2013, 8, e53823. [Google Scholar] [CrossRef]
- Zhu, Q.H.; Stephen, S.; Taylor, J.; Helliwell, C.A.; Wang, M.B. Long noncoding RNAs responsive to Fusarium oxysporum infection in Arabidopsis thaliana. New Phytol. 2014, 201, 574–584. [Google Scholar] [CrossRef]
- Branscheid, A.; Sieh, D.; Pant, B.D.; May, P.; Devers, E.A.; Elkrog, A.; Schauser, L.; Scheible, W.R.; Krajinski, F. Expression Pattern Suggests a Role of MiR399 in the Regulation of the Cellular Response to Local Pi Increase During Arbuscular Mycorrhizal Symbiosis. Mol. Plant Microbe. In. 2010, 23, 915–926. [Google Scholar] [CrossRef]
- Li, Z.X.; Zhang, X.R.; Liu, X.X.; Zhao, Y.J.; Wang, B.M.; Zhang, J.R. miRNA alterations are important mechanism in maize adaptations to low-phosphate environments. Plant Sci. 2016, 252, 103–117. [Google Scholar] [CrossRef]
- Jabnoune, M.; Secco, D.; Lecampion, C.; Robaglia, C.; Shu, Q.; Poirier, Y. A rice cis-natural antisense RNA acts as a translational enhancer for its cognate mRNA and contributes to phosphate homeostasis and plant fitness. Plant. Cell 2013, 25, 4166–4182. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, H.; Walker, C.; Schussler, A. ‘Glomus intraradices DAOM197198’, a model fungus in arbuscular mycorrhiza research, is not Glomus intraradices. New Phytol. 2009, 183, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 1661–1669. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample. | Raw Reads | Number of Clean Reads | Uniquely Mapped Reads Number | Uniquely Mapped Reads (%) | Reads Mapped to Multiple Loci (%) | Reads Unmapped: Too Short (%) |
|---|---|---|---|---|---|---|
| 40-CK-1 | 43,293,767 | 37,470,252 | 23,577,438 | 62.92 | 5.18 | 31.90 |
| 40-CK-2 | 53,402,529 | 46,776,062 | 32,169,682 | 68.77 | 4.52 | 26.71 |
| 40-CK-3 | 55,730,952 | 49,755,232 | 32,920,777 | 66.17 | 4.89 | 28.95 |
| 40-GI-1 | 51,811,515 | 44,618,959 | 26,549,282 | 59.50 | 4.52 | 35.98 |
| 40-GI-2 | 57,804,926 | 49,070,512 | 31,196,619 | 63.58 | 4.06 | 32.36 |
| 40-GI-3 | 53,450,977 | 46,286,135 | 30,037,248 | 64.89 | 4.10 | 31.01 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, G.; Cheng, C.; Zheng, Y.; Wang, X.; Xu, Y.; Wang, W.; Zhu, S.; Cheng, B. Identification of Long Non-Coding RNAs and the Regulatory Network Responsive to Arbuscular Mycorrhizal Fungi Colonization in Maize Roots. Int. J. Mol. Sci. 2019, 20, 4491. https://doi.org/10.3390/ijms20184491
Han G, Cheng C, Zheng Y, Wang X, Xu Y, Wang W, Zhu S, Cheng B. Identification of Long Non-Coding RNAs and the Regulatory Network Responsive to Arbuscular Mycorrhizal Fungi Colonization in Maize Roots. International Journal of Molecular Sciences. 2019; 20(18):4491. https://doi.org/10.3390/ijms20184491
Chicago/Turabian StyleHan, Guomin, Chen Cheng, Yanmei Zheng, Xuewen Wang, Yunjian Xu, Wei Wang, Suwen Zhu, and Beijiu Cheng. 2019. "Identification of Long Non-Coding RNAs and the Regulatory Network Responsive to Arbuscular Mycorrhizal Fungi Colonization in Maize Roots" International Journal of Molecular Sciences 20, no. 18: 4491. https://doi.org/10.3390/ijms20184491
APA StyleHan, G., Cheng, C., Zheng, Y., Wang, X., Xu, Y., Wang, W., Zhu, S., & Cheng, B. (2019). Identification of Long Non-Coding RNAs and the Regulatory Network Responsive to Arbuscular Mycorrhizal Fungi Colonization in Maize Roots. International Journal of Molecular Sciences, 20(18), 4491. https://doi.org/10.3390/ijms20184491