Potential of the NKG2D/NKG2DL Axis in NK Cell-Mediated Clearance of the HIV-1 Reservoir


{kind=link}
Abstract
1. Introduction
2. Mechanisms Maintaining HIV-1 Latency Can Be Counteracted by LRAs
3. LRAs Can Affect NKG2DLs Expression
4. HIV-1 Affects NK Cell Recognition by Modulating NKG2DLs
5. Latent HIV-1 and NKG2DLs Are Regulated via Common Mechanisms That Can Be Modulated by LRAs
6. Impact of LRAs on NK Cell Phenotype and Function
7. NK Cell Response in HIV-1-Infected Patient on ART
8. NK Cell Function in HIV-1 Cure Clinical Trials
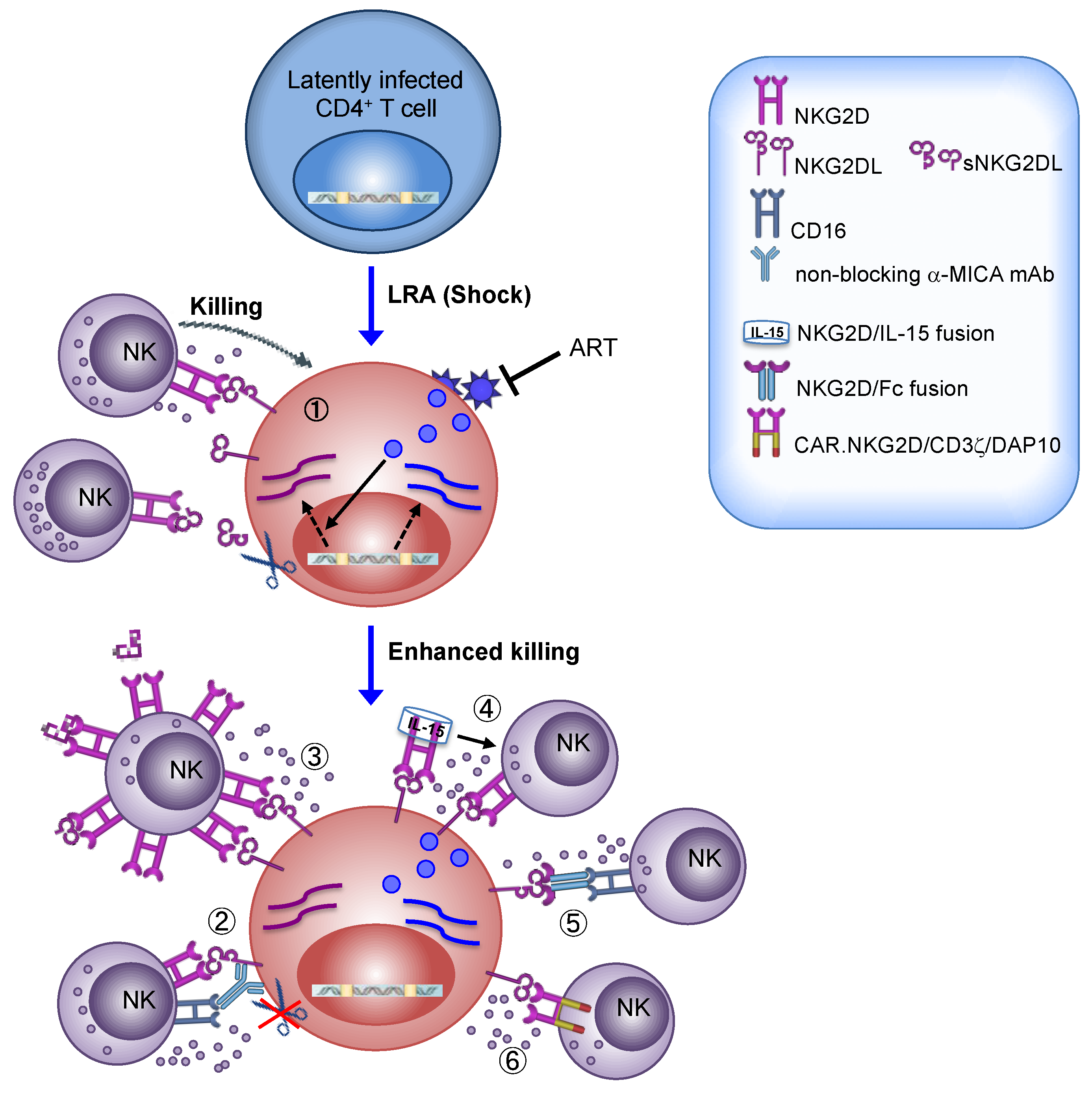
9. Exploiting NK Cells to Eradicate HIV-1
9.1. Questions to Be Addressed
9.2. NKG2D-Based Therapy: Lessons from Cancer Studies
10. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deeks, S.G. HIV: Shock and Kill. Nature 2012, 487, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Spivak, A.M.; Planelles, V. HIV-1 Eradication: Early Trials (and Tribulations). Trends Mol. Med. 2016, 22, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Thorlund, K.; Horwitz, M.S.; Fife, B.T.; Lester, R.; Cameron, D.W. Landscape Review of Current HIV ‘Kick and Kill’ Cure Research—Some Kicking, Not enough Killing. BMC Infect. Dis. 2017, 17, 595. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Deng, K.; Shroff, N.S.; Durand, C.M.; Rabi, S.A.; Yang, H.C.; Zhang, H.; Margolick, J.B.; Blankson, J.N.; Siliciano, R.F. Stimulation of HIV-1-Specific Cytolytic T Lymphocytes Facilitates Elimination of Latent Viral Reservoir After Virus Reactivation. Immunity 2012, 36, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Klatt, N.R.; Chomont, N.; Douek, D.C.; Deeks, S.G. Immune Activation and HIV Persistence: Implications for Curative Approaches to HIV Infection. Immunol. Rev. 2013, 254, 326–342. [Google Scholar] [CrossRef] [PubMed]
- Deng, K.; Pertea, M.; Rongvaux, A.; Wang, L.; Durand, C.M.; Ghiaur, G.; Lai, J.; McHugh, H.L.; Hao, H.; Zhang, H.; et al. Broad CTL Response is Required to Clear Latent HIV-1 due to Dominance of Escape Mutations. Nature 2015, 517, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Perreau, M.; Banga, R.; Pantaleo, G. Targeted Immune Interventions for an HIV-1 Cure. Trends Mol. Med. 2017, 23, 945–961. [Google Scholar] [CrossRef]
- Sengupta, S.; Siliciano, R.F. Targeting the Latent Reservoir for HIV-1. Immunity 2018, 48, 872–895. [Google Scholar] [CrossRef]
- Lanier, L.L. NK Cell Recognition. Annu. Rev. Immunol. 2005, 23, 225–274. [Google Scholar] [CrossRef]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of Natural Killer Cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Moretta, A.; Marcenaro, E.; Parolini, S.; Ferlazzo, G.; Moretta, L. NK Cells at the Interface between Innate and Adaptive Immunity. Cell Death Differ. 2008, 15, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Alter, G.; Altfeld, M. NK Cells in HIV-1 Infection: Evidence for their Role in the Control of HIV-1 Infection. J. Intern. Med. 2009, 265, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Alter, G.; Heckerman, D.; Schneidewind, A.; Fadda, L.; Kadie, C.M.; Carlson, J.M.; Oniangue-Ndza, C.; Martin, M.; Li, B.; Khakoo, S.I.; et al. HIV-1 Adaptation to NK-Cell-Mediated Immune Pressure. Nature 2011, 476, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Bradley, T.; Peppa, D.; Pedroza-Pacheco, I.; Li, D.; Cain, D.W.; Henao, R.; Venkat, V.; Hora, B.; Chen, Y.; Vandergrift, N.A.; et al. RAB11FIP5 Expression and Altered Natural Killer Cell Function Are Associated with Induction of HIV Broadly Neutralizing Antibody Responses. Cell 2018, 175, 387–399.e17. [Google Scholar] [CrossRef] [PubMed]
- Olesen, R.; Vigano, S.; Rasmussen, T.A.; Sogaard, O.S.; Ouyang, Z.; Buzon, M.; Bashirova, A.; Carrington, M.; Palmer, S.; Brinkmann, C.R.; et al. Innate Immune Activity Correlates with CD4 T Cell-Associated HIV-1 DNA Decline during Latency-Reversing Treatment with Panobinostat. J. Virol. 2015, 89, 10176–10189. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Tolstrup, M.; Sogaard, O.S.; Rasmussen, T.A.; Allard, B.; Soriano-Sarabia, N.; Archin, N.M.; Margolis, D.M. In-Vivo Administration of Histone Deacetylase Inhibitors does Not Impair Natural Killer Cell Function in HIV+ Individuals. AIDS 2019, 33, 605–613. [Google Scholar] [CrossRef]
- Marras, F.; Casabianca, A.; Bozzano, F.; Ascierto, M.L.; Orlandi, C.; Di Biagio, A.; Pontali, E.; Dentone, C.; Orofino, G.; Nicolini, L.; et al. Control of the HIV-1 DNA Reservoir is Associated in Vivo and in Vitro with NKp46/NKp30 (CD335 CD337) Inducibility and Interferon Gamma Production by Transcriptionally Unique NK Cells. J. Virol. 2017, 91, e00647-17. [Google Scholar] [CrossRef]
- Huot, N.; Jacquelin, B.; Garcia-Tellez, T.; Rascle, P.; Ploquin, M.J.; Madec, Y.; Reeves, R.K.; Derreudre-Bosquet, N.; Muller-Trutwin, M. Natural Killer Cells Migrate into and Control Simian Immunodeficiency Virus Replication in Lymph Node Follicles in African Green Monkeys. Nat. Med. 2017, 23, 1277–1286. [Google Scholar] [CrossRef]
- Cerboni, C.; Neri, F.; Casartelli, N.; Zingoni, A.; Cosman, D.; Rossi, P.; Santoni, A.; Doria, M. Human Immunodeficiency Virus 1 Nef Protein Downmodulates the Ligands of the Activating Receptor NKG2D and Inhibits Natural Killer Cell-Mediated Cytotoxicity. J. Gen. Virol. 2007, 88, 242–250. [Google Scholar] [CrossRef]
- Fogli, M.; Mavilio, D.; Brunetta, E.; Varchetta, S.; Ata, K.; Roby, G.; Kovacs, C.; Follmann, D.; Pende, D.; Ward, J.; et al. Lysis of Endogenously Infected CD4+ T Cell Blasts by rIL-2 Activated Autologous Natural Killer Cells from HIV-Infected Viremic Individuals. PLoS Pathog. 2008, 4, e1000101. [Google Scholar] [CrossRef]
- Ward, J.; Davis, Z.; DeHart, J.; Zimmerman, E.; Bosque, A.; Brunetta, E.; Mavilio, D.; Planelles, V.; Barker, E. HIV-1 Vpr Triggers Natural Killer Cell-Mediated Lysis of Infected Cells through Activation of the ATR-Mediated DNA Damage Response. PLoS Pathog. 2009, 5, e1000613. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.; Sindhu, S.; Pham, T.N.; Belzile, J.P.; Cohen, E.A. HIV-1 Vpr Up-Regulates Expression of Ligands for the Activating NKG2D Receptor and Promotes NK Cell-Mediated Killing. Blood 2010, 115, 1354–1363. [Google Scholar] [CrossRef] [PubMed]
- Matusali, G.; Tchidjou, H.K.; Pontrelli, G.; Bernardi, S.; D’Ettorre, G.; Vullo, V.; Buonomini, A.R.; Andreoni, M.; Santoni, A.; Cerboni, C.; et al. Soluble Ligands for the NKG2D Receptor are Released during HIV-1 Infection and Impair NKG2D Expression and Cytotoxicity of NK Cells. FASEB J. 2013, 27, 2440–2450. [Google Scholar] [CrossRef] [PubMed]
- Desimio, M.G.; Giuliani, E.; Doria, M. The Histone Deacetylase Inhibitor SAHA Simultaneously Reactivates HIV-1 from Latency and Up-Regulates NKG2D Ligands Sensitizing for Natural Killer Cell Cytotoxicity. Virology 2017, 510, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Desimio, M.G.; Giuliani, E.; Ferraro, A.S.; Adorno, G.; Doria, M. In Vitro Exposure to Prostratin but Not Bryostatin-1 Improves Natural Killer Cell Functions Including Killing of CD4(+) T Cells Harboring Reactivated Human Immunodeficiency Virus. Front. Immunol. 2018, 9, 1514. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of Ligands for the NKG2D Activating Receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK Cells and T Cells by NKG2D, a Receptor for Stress-Inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef]
- Groh, V.; Rhinehart, R.; Randolph-Habecker, J.; Topp, M.S.; Riddell, S.R.; Spies, T. Costimulation of CD8alphabeta T Cells by NKG2D Via Engagement by MIC Induced on Virus-Infected Cells. Nat. Immunol. 2001, 2, 255–260. [Google Scholar] [CrossRef]
- Maasho, K.; Opoku-Anane, J.; Marusina, A.I.; Coligan, J.E.; Borrego, F. NKG2D is a Costimulatory Receptor for Human Naive CD8+ T Cells. J. Immunol. 2005, 174, 4480–4484. [Google Scholar] [CrossRef]
- Bryceson, Y.T.; March, M.E.; Ljunggren, H.G.; Long, E.O. Synergy among Receptors on Resting NK Cells for the Activation of Natural Cytotoxicity and Cytokine Secretion. Blood 2006, 107, 159–166. [Google Scholar] [CrossRef]
- Barber, A.; Sentman, C.L. NKG2D Receptor Regulates Human Effector T-Cell Cytokine Production. Blood 2011, 117, 6571–6581. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pende, D.; Cantoni, C.; Rivera, P.; Vitale, M.; Castriconi, R.; Marcenaro, S.; Nanni, M.; Biassoni, R.; Bottino, C.; Moretta, A.; et al. Role of NKG2D in Tumor Cell Lysis Mediated by Human NK Cells: Cooperation with Natural Cytotoxicity Receptors and Capability of Recognizing Tumors of Nonepithelial Origin. Eur. J. Immunol. 2001, 31, 1076–1086. [Google Scholar] [CrossRef]
- Parsons, M.S.; Richard, J.; Lee, W.S.; Vanderven, H.; Grant, M.D.; Finzi, A.; Kent, S.J. NKG2D Acts as a Co-Receptor for Natural Killer Cell-Mediated Anti-HIV-1 Antibody-Dependent Cellular Cytotoxicity. AIDS Res. Hum. Retroviruses 2016, 32, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Krieg, S.; Ullrich, E. Novel Immune Modulators used in Hematology: Impact on NK Cells. Front. Immunol. 2013, 3, 388. [Google Scholar] [CrossRef] [PubMed]
- Chretien, A.S.; Le Roy, A.; Vey, N.; Prebet, T.; Blaise, D.; Fauriat, C.; Olive, D. Cancer-Induced Alterations of NK-Mediated Target Recognition: Current and Investigational Pharmacological Strategies Aiming at Restoring NK-Mediated Anti-Tumor Activity. Front. Immunol. 2014, 5, 122. [Google Scholar] [CrossRef] [PubMed]
- Cifaldi, L.; Locatelli, F.; Marasco, E.; Moretta, L.; Pistoia, V. Boosting Natural Killer Cell-Based Immunotherapy with Anticancer Drugs: A Perspective. Trends Mol. Med. 2017, 23, 1156–1175. [Google Scholar] [CrossRef]
- Zingoni, A.; Fionda, C.; Borrelli, C.; Cippitelli, M.; Santoni, A.; Soriani, A. Natural Killer Cell Response to Chemotherapy-Stressed Cancer Cells: Role in Tumor Immunosurveillance. Front. Immunol. 2017, 8, 1194. [Google Scholar] [CrossRef]
- Cary, D.C.; Fujinaga, K.; Peterlin, B.M. Molecular Mechanisms of HIV Latency. J. Clin. Investig. 2016, 126, 448–454. [Google Scholar] [CrossRef]
- Romani, B.; Allahbakhshi, E. Underlying Mechanisms of HIV-1 Latency. Virus Genes 2017, 53, 329–339. [Google Scholar] [CrossRef]
- Spivak, A.M.; Planelles, V. Novel Latency Reversal Agents for HIV-1 Cure. Annu. Rev. Med. 2018, 69, 421–436. [Google Scholar] [CrossRef]
- Zerbato, J.M.; Purves, H.V.; Lewin, S.R.; Rasmussen, T.A. Between a Shock and a Hard Place: Challenges and Developments in HIV Latency Reversal. Curr. Opin. Virol. 2019, 38, 1–9. [Google Scholar] [CrossRef]
- Abner, E.; Jordan, A. HIV “Shock and Kill” Therapy: In Need of Revision. Antiviral Res. 2019, 166, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, C.; Serrano-Villar, S.; Madrid-Elena, N.; Perez-Elias, M.J.; Martin, M.E.; Barbas, C.; Ruiperez, J.; Munoz, E.; Munoz-Fernandez, M.A.; Castor, T.; et al. Bryostatin-1 for Latent Virus Reactivation in HIV-Infected Patients on Antiretroviral Therapy. AIDS 2016, 30, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Vibholm, L.; Schleimann, M.H.; Hojen, J.F.; Benfield, T.; Offersen, R.; Rasmussen, K.; Olesen, R.; Dige, A.; Agnholt, J.; Grau, J.; et al. Short-Course Toll-Like Receptor 9 Agonist Treatment Impacts Innate Immunity and Plasma Viremia in Individuals with Human Immunodeficiency Virus Infection. Clin. Infect. Dis. 2017, 64, 1686–1695. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.H.; McMahon, J.H.; Chang, C.C.; Lee, S.A.; Hartogensis, W.; Bumpus, N.; Savic, R.; Roney, J.; Hoh, R.; Solomon, A.; et al. Short-Term Administration of Disulfiram for Reversal of Latent HIV Infection: A Phase 2 Dose-Escalation Study. Lancet HIV 2015, 2, e520–e529. [Google Scholar] [CrossRef]
- Schmiedel, D.; Mandelboim, O. NKG2D Ligands-Critical Targets for Cancer Immune Escape and Therapy. Front. Immunol. 2018, 9, 2040. [Google Scholar] [CrossRef] [PubMed]
- Zingoni, A.; Molfetta, R.; Fionda, C.; Soriani, A.; Paolini, R.; Cippitelli, M.; Cerboni, C.; Santoni, A. NKG2D and its Ligands: ‘One for all, all for One’. Front. Immunol. 2018, 9, 476. [Google Scholar] [CrossRef]
- Cohen, G.B.; Gandhi, R.T.; Davis, D.M.; Mandelboim, O.; Chen, B.K.; Strominger, J.L.; Baltimore, D. The Selective Downregulation of Class I Major Histocompatibility Complex Proteins by HIV-1 Protects HIV-Infected Cells from NK Cells. Immunity 1999, 10, 661–671. [Google Scholar] [CrossRef]
- Apps, R.; Del Prete, G.Q.; Chatterjee, P.; Lara, A.; Brumme, Z.L.; Brockman, M.A.; Neil, S.; Pickering, S.; Schneider, D.K.; Piechocka-Trocha, A.; et al. HIV-1 Vpu Mediates HLA-C Downregulation. Cell. Host Microbe 2016, 19, 686–695. [Google Scholar] [CrossRef]
- Korner, C.; Simoneau, C.R.; Schommers, P.; Granoff, M.; Ziegler, M.; Holzemer, A.; Lunemann, S.; Chukwukelu, J.; Corleis, B.; Naranbhai, V.; et al. HIV-1-Mediated Downmodulation of HLA-C Impacts Target Cell Recognition and Antiviral Activity of NK Cells. Cell. Host Microbe 2017, 22, 111–119.e4. [Google Scholar] [CrossRef]
- Ward, J.; Bonaparte, M.; Sacks, J.; Guterman, J.; Fogli, M.; Mavilio, D.; Barker, E. HIV Modulates the Expression of Ligands Important in Triggering Natural Killer Cell Cytotoxic Responses on Infected Primary T-Cell Blasts. Blood 2007, 110, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.; Pham, T.N.; Ishizaka, Y.; Cohen, E.A. Viral Protein R Upregulates Expression of ULBP2 on Uninfected Bystander Cells during HIV-1 Infection of Primary CD4+ T Lymphocytes. Virology 2013, 443, 248–256. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Le Rouzic, E.; Belaidouni, N.; Estrabaud, E.; Morel, M.; Rain, J.C.; Transy, C.; Margottin-Goguet, F. HIV1 Vpr Arrests the Cell Cycle by Recruiting DCAF1/VprBP, a Receptor of the Cul4-DDB1 Ubiquitin Ligase. Cell. Cycle 2007, 6, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Goh, W.C.; Rogel, M.E.; Kinsey, C.M.; Michael, S.F.; Fultz, P.N.; Nowak, M.A.; Hahn, B.H.; Emerman, M. HIV-1 Vpr Increases Viral Expression by Manipulation of the Cell Cycle: A Mechanism for Selection of Vpr in Vivo. Nat. Med. 1998, 4, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Vassena, L.; Giuliani, E.; Matusali, G.; Cohen, E.A.; Doria, M. The Human Immunodeficiency Virus Type 1 Vpr Protein Upregulates PVR Via Activation of the ATR-Mediated DNA Damage Response Pathway. J. Gen. Virol. 2013, 94, 2664–2669. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Swinbank, K.M.; Ahmed, P.S.; Taylor, D.L.; Jackson, S.P.; Smith, G.C.; O’Connor, M.J. Suppression of HIV-1 Infection by a Small Molecule Inhibitor of the ATM Kinase. Nat. Cell Biol. 2005, 7, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Norman, J.M.; Mashiba, M.; McNamara, L.A.; Onafuwa-Nuga, A.; Chiari-Fort, E.; Shen, W.; Collins, K.L. The Antiviral Factor APOBEC3G Enhances the Recognition of HIV-Infected Primary T Cells by Natural Killer Cells. Nat. Immunol. 2011, 12, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Roeth, J.F.; Collins, K.L. Human Immunodeficiency Virus Type 1 Nef: Adapting to Intracellular Trafficking Pathways. Microbiol. Mol. Biol. Rev. 2006, 70, 548–563. [Google Scholar] [CrossRef]
- Kirchhoff, F. Immune Evasion and Counteraction of Restriction Factors by HIV-1 and Other Primate Lentiviruses. Cell. Host Microbe 2010, 8, 55–67. [Google Scholar] [CrossRef]
- Galaski, J.; Ahmad, F.; Tibroni, N.; Pujol, F.M.; Muller, B.; Schmidt, R.E.; Fackler, O.T. Cell Surface Downregulation of NK Cell Ligands by Patient-Derived HIV-1 Vpu and Nef Alleles. J. Acquir. Immune Defic. Syndr. 2016, 72, 1–10. [Google Scholar] [CrossRef]
- Alsahafi, N.; Richard, J.; Prevost, J.; Coutu, M.; Brassard, N.; Parsons, M.S.; Kaufmann, D.E.; Brockman, M.; Finzi, A. Impaired Downregulation of NKG2D Ligands by Nef Proteins from Elite Controllers Sensitizes HIV-1-Infected Cells to Antibody-Dependent Cellular Cytotoxicity. J. Virol. 2017, 91, e00109-17. [Google Scholar] [CrossRef] [PubMed]
- Matusali, G.; Potesta, M.; Santoni, A.; Cerboni, C.; Doria, M. The Human Immunodeficiency Virus Type 1 Nef and Vpu Proteins Downregulate the Natural Killer Cell-Activating Ligand PVR. J. Virol. 2012, 86, 4496–4504. [Google Scholar] [CrossRef] [PubMed]
- Fausther-Bovendo, H.; Sol-Foulon, N.; Candotti, D.; Agut, H.; Schwartz, O.; Debre, P.; Vieillard, V. HIV Escape from Natural Killer Cytotoxicity: Nef Inhibits NKp44L Expression on CD4+ T Cells. AIDS 2009, 23, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.H.; Sowrirajan, B.; Davis, Z.B.; Ward, J.P.; Campbell, E.M.; Planelles, V.; Barker, E. Degranulation of Natural Killer Cells Following Interaction with HIV-1-Infected Cells is Hindered by Downmodulation of NTB-A by Vpu. Cell. Host Microbe 2010, 8, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Nolting, A.; Dugast, A.S.; Rihn, S.; Luteijn, R.; Carrington, M.F.; Kane, K.; Jost, S.; Toth, I.; Nagami, E.; Faetkenheuer, G.; et al. MHC Class I Chain-Related Protein A Shedding in Chronic HIV-1 Infection is Associated with Profound NK Cell Dysfunction. Virology 2010, 406, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Mastroianni, C.M.; Liuzzi, G.M. Matrix Metalloproteinase Dysregulation in HIV Infection: Implications for Therapeutic Strategies. Trends Mol. Med. 2007, 13, 449–459. [Google Scholar] [CrossRef]
- Baragano Raneros, A.; Suarez-Alvarez, B.; Lopez-Larrea, C. Secretory Pathways Generating Immunosuppressive NKG2D Ligands: New Targets for Therapeutic Intervention. Oncoimmunology 2014, 3, e28497. [Google Scholar] [CrossRef]
- Deng, W.; Gowen, B.G.; Zhang, L.; Wang, L.; Lau, S.; Iannello, A.; Xu, J.; Rovis, T.L.; Xiong, N.; Raulet, D.H. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science 2015, 348, 136–139. [Google Scholar] [CrossRef]
- Giuliani, E.; Vassena, L.; Cerboni, C.; Doria, M. Release of Soluble Ligands for the Activating NKG2D Receptor: One More Immune Evasion Strategy Evolved by HIV-1? Curr. Drug Targets 2016, 17, 54–64. [Google Scholar] [CrossRef]
- Berghuis, D.; Schilham, M.W.; Vos, H.I.; Santos, S.J.; Kloess, S.; Buddingh’, E.P.; Egeler, R.M.; Hogendoorn, P.C.; Lankester, A.C. Histone Deacetylase Inhibitors Enhance Expression of NKG2D Ligands in Ewing Sarcoma and Sensitize for Natural Killer Cell-Mediated Cytolysis. Clin. Sarcoma Res. 2012, 2, 8. [Google Scholar] [CrossRef]
- Andersen, J.L.; Le Rouzic, E.; Planelles, V. HIV-1 Vpr: Mechanisms of G2 Arrest and Apoptosis. Exp. Mol. Pathol. 2008, 85, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Xu, W.S. Histone Deacetylase Inhibitors: Potential in Cancer Therapy. J. Cell. Biochem. 2009, 107, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Espeseth, A.; Parker, D.; Cheema, M.; Hazuda, D.; Margolis, D.M. Expression of Latent HIV Induced by the Potent HDAC Inhibitor Suberoylanilide Hydroxamic Acid. AIDS Res. Hum. Retroviruses 2009, 25, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.C.; Lim, K.I.; Calafi, A.; Rossi, J.J.; Schaffer, D.V.; Arkin, A.P. Combinatorial Latency Reactivation for HIV-1 Subtypes and Variants. J. Virol. 2010, 84, 5958–5974. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, S.; Lopez-Soto, A.; Suarez-Alvarez, B.; Lopez-Vazquez, A.; Lopez-Larrea, C. NKG2D Ligands: Key Targets of the Immune Response. Trends Immunol. 2008, 29, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Contreras, X.; Schweneker, M.; Chen, C.S.; McCune, J.M.; Deeks, S.G.; Martin, J.; Peterlin, B.M. Suberoylanilide Hydroxamic Acid Reactivates HIV from Latently Infected Cells. J. Biol. Chem. 2009, 284, 6782–6789. [Google Scholar] [CrossRef] [PubMed]
- Skov, S.; Pedersen, M.T.; Andresen, L.; Straten, P.T.; Woetmann, A.; Odum, N. Cancer Cells Become Susceptible to Natural Killer Cell Killing After Exposure to Histone Deacetylase Inhibitors due to Glycogen Synthase Kinase-3-Dependent Expression of MHC Class I-Related Chain A and B. Cancer Res. 2005, 65, 11136–11145. [Google Scholar] [CrossRef] [PubMed]
- McKernan, L.N.; Momjian, D.; Kulkosky, J. Protein Kinase C: One Pathway Towards the Eradication of Latent HIV-1 Reservoirs. Adv. Virol. 2012, 2012, 805347. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Dandekar, S. Targeting NF-κB signaling with protein kinase C agonists as an emerging strategy for combating HIV latency. AIDS Res. Hum. Retroviruses 2015, 31, 4–12. [Google Scholar] [CrossRef]
- Jones, R.B.; O’Connor, R.; Mueller, S.; Foley, M.; Szeto, G.L.; Karel, D.; Lichterfeld, M.; Kovacs, C.; Ostrowski, M.A.; Trocha, A.; et al. Histone Deacetylase Inhibitors Impair the Elimination of HIV-Infected Cells by Cytotoxic T-Lymphocytes. PLoS Pathog. 2014, 10, e1004287. [Google Scholar] [CrossRef]
- Clutton, G.; Xu, Y.; Baldoni, P.L.; Mollan, K.R.; Kirchherr, J.; Newhard, W.; Cox, K.; Kuruc, J.D.; Kashuba, A.; Barnard, R.; et al. The Differential Short- and Long-Term Effects of HIV-1 Latency-Reversing Agents on T Cell Function. Sci. Rep. 2016, 6, 30749. [Google Scholar] [CrossRef] [PubMed]
- Walker-Sperling, V.E.; Pohlmeyer, C.W.; Tarwater, P.M.; Blankson, J.N. The Effect of Latency Reversal Agents on Primary CD8+ T Cells: Implications for Shock and Kill Strategies for Human Immunodeficiency Virus Eradication. EBioMedicine 2016, 8, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Ogbomo, H.; Michaelis, M.; Kreuter, J.; Doerr, H.W.; Cinatl, J. Histone Deacetylase Inhibitors Suppress Natural Killer Cell Cytolytic Activity. FEBS Lett. 2007, 581, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Spivak, A.M.; Soriano-Sarabia, N.; Checkley, M.A.; Barker, E.; Karn, J.; Planelles, V.; Margolis, D.M. HIV Latency-Reversing Agents have Diverse Effects on Natural Killer Cell Function. Front. Immunol. 2016, 7, 356. [Google Scholar] [CrossRef] [PubMed]
- Pace, M.; Williams, J.; Kurioka, A.; Gerry, A.B.; Jakobsen, B.; Klenerman, P.; Nwokolo, N.; Fox, J.; Fidler, S.; Frater, J.; et al. Histone Deacetylase Inhibitors Enhance CD4 T Cell Susceptibility to NK Cell Killing but Reduce NK Cell Function. PLoS Pathog. 2016, 12, e1005782. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Wang, L.; Yao, C.; Ni, Z.; Liu, F.; Gong, C.; Zhu, X.; Yan, X.; Watowich, S.S.; Lee, D.A.; et al. The Histone Deacetylase Inhibitor Valproic Acid Inhibits NKG2D Expression in Natural Killer Cells through Suppression of STAT3 and HDAC3. Sci. Rep. 2017, 7, 45266. [Google Scholar] [CrossRef]
- Giuliani, E.; Desimio, M.G.; Doria, M. Hexamethylene Bisacetamide Impairs NK Cell-Mediated Clearance of Acute T Lymphoblastic Leukemia Cells and HIV-1-Infected T Cells that Exit Viral Latency. Sci. Rep. 2019, 9, 4373. [Google Scholar] [CrossRef]
- Lai, W.; Huang, L.; Zhu, L.; Ferrari, G.; Chan, C.; Li, W.; Lee, K.H.; Chen, C.H. Gnidimacrin, a Potent Anti-HIV Diterpene, can Eliminate Latent HIV-1 Ex Vivo by Activation of Protein Kinase C Beta. J. Med. Chem. 2015, 58, 8638–8646. [Google Scholar] [CrossRef]
- Lundqvist, A.; Yokoyama, H.; Smith, A.; Berg, M.; Childs, R. Bortezomib Treatment and Regulatory T-Cell Depletion Enhance the Antitumor Effects of Adoptively Infused NK Cells. Blood 2009, 113, 6120–6127. [Google Scholar] [CrossRef]
- Mikulak, J.; Oriolo, F.; Zaghi, E.; Di Vito, C.; Mavilio, D. Natural Killer Cells in HIV-1 Infection and Therapy. AIDS 2017, 31, 2317–2330. [Google Scholar] [CrossRef]
- Florez-Alvarez, L.; Hernandez, J.C.; Zapata, W. NK Cells in HIV-1 Infection: From Basic Science to Vaccine Strategies. Front. Immunol. 2018, 9, 2290. [Google Scholar] [CrossRef] [PubMed]
- Mavilio, D.; Benjamin, J.; Daucher, M.; Lombardo, G.; Kottilil, S.; Planta, M.A.; Marcenaro, E.; Bottino, C.; Moretta, L.; Moretta, A.; et al. Natural Killer Cells in HIV-1 Infection: Dichotomous Effects of Viremia on Inhibitory and Activating Receptors and their Functional Correlates. Proc. Natl. Acad. Sci. USA 2003, 100, 15011–15016. [Google Scholar] [CrossRef] [PubMed]
- Bisio, F.; Bozzano, F.; Marras, F.; Di Biagio, A.; Moretta, L.; De Maria, A. Successfully Treated HIV-Infected Patients have Differential Expression of NK Cell Receptors (NKp46 and NKp30) According to AIDS Status at Presentation. Immunol. Lett. 2013, 152, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Lecuroux, C.; Saez-Cirion, A.; Noel, N.; Ben-Lamine, L.; Girault, I.; Caillat-Zucman, S.; Scott-Algara, D.; Venet, A.; Lambotte, O. NKG2D Expression on HIV-Specific CD8+ T Cells is Reduced in Viremic HIV-1-Infected Patients but Maintained in HIV Controllers. J. Acquir. Immune Defic. Syndr. 2013, 62, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Sung, J.M.; Garrido, C.; Soriano-Sarabia, N.; Margolis, D.M. Eradicating HIV-1 Infection: Seeking to Clear a Persistent Pathogen. Nat. Rev. Microbiol. 2014, 12, 750–764. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.H.; Wightman, F.; Solomon, A.; Ghneim, K.; Ahlers, J.; Cameron, M.J.; Smith, M.Z.; Spelman, T.; McMahon, J.; Velayudham, P.; et al. Activation of HIV Transcription with Short-Course Vorinostat in HIV-Infected Patients on Suppressive Antiretroviral Therapy. PLoS Pathog. 2014, 10, e1004473. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.A.; Tolstrup, M.; Brinkmann, C.R.; Olesen, R.; Erikstrup, C.; Solomon, A.; Winckelmann, A.; Palmer, S.; Dinarello, C.; Buzon, M.; et al. Panobinostat, a Histone Deacetylase Inhibitor, for Latent-Virus Reactivation in HIV-Infected Patients on Suppressive Antiretroviral Therapy: A Phase 1/2, Single Group, Clinical Trial. Lancet HIV 2014, 1, e13–e21. [Google Scholar] [CrossRef]
- Archin, N.M.; Kirchherr, J.L.; Sung, J.A.; Clutton, G.; Sholtis, K.; Xu, Y.; Allard, B.; Stuelke, E.; Kashuba, A.D.; Kuruc, J.D.; et al. Interval Dosing with the HDAC Inhibitor Vorinostat Effectively Reverses HIV Latency. J. Clin. Investig. 2017, 127, 3126–3135. [Google Scholar] [CrossRef]
- Seay, K.; Church, C.; Zheng, J.H.; Deneroff, K.; Ochsenbauer, C.; Kappes, J.C.; Liu, B.; Jeng, E.K.; Wong, H.C.; Goldstein, H. In Vivo Activation of Human NK Cells by Treatment with an Interleukin-15 Superagonist Potently Inhibits Acute in Vivo HIV-1 Infection in Humanized Mice. J. Virol. 2015, 89, 6264–6274. [Google Scholar] [CrossRef]
- Garrido, C.; Abad-Fernandez, M.; Tuyishime, M.; Pollara, J.J.; Ferrari, G.; Soriano-Sarabia, N.; Margolis, D.M. Interleukin-15-Stimulated Natural Killer Cells Clear HIV-1-Infected Cells Following Latency Reversal Ex Vivo. J. Virol. 2018, 92, e00235-18. [Google Scholar] [CrossRef]
- Tomescu, C.; Mavilio, D.; Montaner, L.J. Lysis of HIV-1-Infected Autologous CD4+ Primary T Cells by Interferon-Alpha-Activated NK Cells Requires NKp46 and NKG2D. AIDS 2015, 29, 1767–1773. [Google Scholar] [CrossRef] [PubMed]
- Hua, S.; Vigano, S.; Tse, S.; Zhengyu, O.; Harrington, S.; Negron, J.; Garcia-Broncano, P.; Marchetti, G.; Genebat, M.; Leal, M.; et al. Pegylated Interferon-Alpha-Induced Natural Killer Cell Activation is Associated with Human Immunodeficiency Virus-1 DNA Decline in Antiretroviral Therapy-Treated HIV-1/Hepatitis C Virus-Coinfected Patients. Clin. Infect. Dis. 2018, 66, 1910–1917. [Google Scholar] [CrossRef] [PubMed]
- Offersen, R.; Nissen, S.K.; Rasmussen, T.A.; Ostergaard, L.; Denton, P.W.; Sogaard, O.S.; Tolstrup, M. A Novel Toll-Like Receptor 9 Agonist, MGN1703, Enhances HIV-1 Transcription and NK Cell-Mediated Inhibition of HIV-1-Infected Autologous CD4+ T Cells. J. Virol. 2016, 90, 4441–4453. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.; Irrinki, A.; Kaur, J.; Cihlar, T.; Kukolj, G.; Sloan, D.D.; Murry, J.P. Toll-Like Receptor 7 Agonist GS-9620 Induces HIV Expression and HIV-Specific Immunity in Cells from HIV-Infected Individuals on Suppressive Antiretroviral Therapy. J. Virol. 2017, 91, e02166-16. [Google Scholar] [CrossRef] [PubMed]
- Dietsch, G.N.; Lu, H.; Yang, Y.; Morishima, C.; Chow, L.Q.; Disis, M.L.; Hershberg, R.M. Coordinated Activation of Toll-Like Receptor8 (TLR8) and NLRP3 by the TLR8 Agonist, VTX-2337, Ignites Tumoricidal Natural Killer Cell Activity. PLoS ONE 2016, 11, e0148764. [Google Scholar] [CrossRef]
- Peppa, D. Natural Killer Cells in Human Immunodeficiency Virus-1 Infection: Spotlight on the Impact of Human Cytomegalovirus. Front. Immunol. 2017, 8, 1322. [Google Scholar] [CrossRef]
- Peng, H.; Tian, Z. Natural Killer Cell Memory: Progress and Implications. Front. Immunol. 2017, 8, 1143. [Google Scholar] [CrossRef]
- Reeves, R.K.; Li, H.; Jost, S.; Blass, E.; Li, H.; Schafer, J.L.; Varner, V.; Manickam, C.; Eslamizar, L.; Altfeld, M.; et al. Antigen-Specific NK Cell Memory in Rhesus Macaques. Nat. Immunol. 2015, 16, 927–932. [Google Scholar] [CrossRef]
- Lisovsky, I.; Isitman, G.; Song, R.; DaFonseca, S.; Tremblay-McLean, A.; Lebouche, B.; Routy, J.P.; Bruneau, J.; Bernard, N.F. A Higher Frequency of NKG2A+ than of NKG2A- NK Cells Responds to Autologous HIV-Infected CD4 Cells Irrespective of Whether Or Not they Coexpress KIR3DL1. J. Virol. 2015, 89, 9909–9919. [Google Scholar] [CrossRef]
- Guma, M.; Angulo, A.; Vilches, C.; Gomez-Lozano, N.; Malats, N.; Lopez-Botet, M. Imprint of Human Cytomegalovirus Infection on the NK Cell Receptor Repertoire. Blood 2004, 104, 3664–3671. [Google Scholar] [CrossRef]
- Bruel, T.; Guivel-Benhassine, F.; Amraoui, S.; Malbec, M.; Richard, L.; Bourdic, K.; Donahue, D.A.; Lorin, V.; Casartelli, N.; Noel, N.; et al. Elimination of HIV-1-Infected Cells by Broadly Neutralizing Antibodies. Nat. Commun. 2016, 7, 10844. [Google Scholar] [CrossRef]
- Guillerey, C.; Huntington, N.D.; Smyth, M.J. Targeting Natural Killer Cells in Cancer Immunotherapy. Nat. Immunol. 2016, 17, 1025–1036. [Google Scholar] [CrossRef]
- Veluchamy, J.P.; Kok, N.; van der Vliet, H.J.; Verheul, H.M.W.; de Gruijl, T.D.; Spanholtz, J. The Rise of Allogeneic Natural Killer Cells as a Platform for Cancer Immunotherapy: Recent Innovations and Future Developments. Front. Immunol. 2017, 8, 631. [Google Scholar] [CrossRef]
- Matosevic, S. Viral and Nonviral Engineering of Natural Killer Cells as Emerging Adoptive Cancer Immunotherapies. J. Immunol. Res. 2018, 2018, 4054815. [Google Scholar] [CrossRef]
- Hu, W.; Wang, G.; Huang, D.; Sui, M.; Xu, Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. Front. Immunol. 2019, 10, 1205. [Google Scholar] [CrossRef]
- Spear, P.; Wu, M.R.; Sentman, M.L.; Sentman, C.L. NKG2D Ligands as Therapeutic Targets. Cancer. Immun. 2013, 13, 8. [Google Scholar]
- Lu, S.; Zhang, J.; Liu, D.; Li, G.; Staveley-O’Carroll, K.F.; Li, Z.; Wu, J.D. Nonblocking Monoclonal Antibody Targeting Soluble MIC Revamps Endogenous Innate and Adaptive Antitumor Responses and Eliminates Primary and Metastatic Tumors. Clin. Cancer Res. 2015, 21, 4819–4830. [Google Scholar] [CrossRef]
- Ferrari de Andrade, L.; Tay, R.E.; Pan, D.; Luoma, A.M.; Ito, Y.; Badrinath, S.; Tsoucas, D.; Franz, B.; May, K.F., Jr.; Harvey, C.J.; et al. Antibody-Mediated Inhibition of MICA and MICB Shedding Promotes NK Cell-Driven Tumor Immunity. Science 2018, 359, 1537–1542. [Google Scholar] [CrossRef]
- Kloess, S.; Huenecke, S.; Piechulek, D.; Esser, R.; Koch, J.; Brehm, C.; Soerensen, J.; Gardlowski, T.; Brinkmann, A.; Bader, P.; et al. IL-2-Activated Haploidentical NK Cells Restore NKG2D-Mediated NK-Cell Cytotoxicity in Neuroblastoma Patients by Scavenging of Plasma MICA. Eur. J. Immunol. 2010, 40, 3255–3267. [Google Scholar] [CrossRef]
- Xia, Y.; Chen, B.; Shao, X.; Xiao, W.; Qian, L.; Ding, Y.; Ji, M.; Gong, W. Treatment with a Fusion Protein of the Extracellular Domains of NKG2D to IL-15 Retards Colon Cancer Growth in Mice. J. Immunother. 2014, 37, 257–266. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, B.; Yang, T.; Xiao, W.; Qian, L.; Ding, Y.; Ji, M.; Ge, X.; Gong, W. Human Fused NKG2D-IL-15 Protein Controls Xenografted Human Gastric Cancer through the Recruitment and Activation of NK Cells. Cell. Mol. Immunol. 2017, 14, 293–307. [Google Scholar] [CrossRef]
- Raab, S.; Steinbacher, J.; Schmiedel, B.J.; Kousis, P.C.; Steinle, A.; Jung, G.; Grosse-Hovest, L.; Salih, H.R. Fc-Optimized NKG2D-Fc Constructs Induce NK Cell Antibody-Dependent Cellular Cytotoxicity Against Breast Cancer Cells Independently of HER2/neu Expression Status. J. Immunol. 2014, 193, 4261–4272. [Google Scholar] [CrossRef]
- Steinbacher, J.; Baltz-Ghahremanpour, K.; Schmiedel, B.J.; Steinle, A.; Jung, G.; Kubler, A.; Andre, M.C.; Grosse-Hovest, L.; Salih, H.R. An Fc-Optimized NKG2D-Immunoglobulin G Fusion Protein for Induction of Natural Killer Cell Reactivity against Leukemia. Int. J. Cancer 2015, 136, 1073–1084. [Google Scholar] [CrossRef]
- Chang, Y.H.; Connolly, J.; Shimasaki, N.; Mimura, K.; Kono, K.; Campana, D. A Chimeric Receptor with NKG2D Specificity Enhances Natural Killer Cell Activation and Killing of Tumor Cells. Cancer Res. 2013, 73, 1777–1786. [Google Scholar] [CrossRef]
- Kamiya, T.; Chang, Y.H.; Campana, D. Expanded and Activated Natural Killer Cells for Immunotherapy of Hepatocellular Carcinoma. Cancer. Immunol. Res. 2016, 4, 574–581. [Google Scholar] [CrossRef]
- Hu, X.T.; Zhu, B.L.; Zhao, L.G.; Wang, J.W.; Liu, L.; Lai, Y.J.; He, L.; Deng, X.J.; Chen, G.J. Histone Deacetylase Inhibitor Apicidin Increases Expression of the Alpha-Secretase ADAM10 through Transcription Factor USF1-Mediated Mechanisms. FASEB J. 2017, 31, 1482–1493. [Google Scholar] [CrossRef]
- Raneros, A.B.; Minguela, A.; Rodriguez, R.M.; Colado, E.; Bernal, T.; Anguita, E.; Mogorron, A.V.; Gil, A.C.; Vidal-Castineira, J.R.; Marquez-Kisinousky, L.; et al. Increasing TIMP3 Expression by Hypomethylating Agents Diminishes Soluble MICA, MICB and ULBP2 Shedding in Acute Myeloid Leukemia, Facilitating NK Cell-Mediated Immune Recognition. Oncotarget 2017, 8, 31959–31976. [Google Scholar] [CrossRef]
- Salih, H.R.; Holdenrieder, S.; Steinle, A. Soluble NKG2D Ligands: Prevalence, Release, and Functional Impact. Front. Biosci. 2008, 13, 3448–3456. [Google Scholar] [CrossRef]
- Bryceson, Y.T.; Ljunggren, H.G.; Long, E.O. Minimal Requirement for Induction of Natural Cytotoxicity and Intersection of Activation Signals by Inhibitory Receptors. Blood 2009, 114, 2657–2666. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desimio, M.G.; Covino, D.A.; Doria, M. Potential of the NKG2D/NKG2DL Axis in NK Cell-Mediated Clearance of the HIV-1 Reservoir. Int. J. Mol. Sci. 2019, 20, 4490. https://doi.org/10.3390/ijms20184490
Desimio MG, Covino DA, Doria M. Potential of the NKG2D/NKG2DL Axis in NK Cell-Mediated Clearance of the HIV-1 Reservoir. International Journal of Molecular Sciences. 2019; 20(18):4490. https://doi.org/10.3390/ijms20184490
Chicago/Turabian StyleDesimio, Maria G., Daniela A. Covino, and Margherita Doria. 2019. "Potential of the NKG2D/NKG2DL Axis in NK Cell-Mediated Clearance of the HIV-1 Reservoir" International Journal of Molecular Sciences 20, no. 18: 4490. https://doi.org/10.3390/ijms20184490
APA StyleDesimio, M. G., Covino, D. A., & Doria, M. (2019). Potential of the NKG2D/NKG2DL Axis in NK Cell-Mediated Clearance of the HIV-1 Reservoir. International Journal of Molecular Sciences, 20(18), 4490. https://doi.org/10.3390/ijms20184490