Inflammaging and Oxidative Stress in Human Diseases: From Molecular Mechanisms to Novel Treatments

 ,
,
Abstract
1. Introduction
2. Inflammaging in Cardiovascular Disease (CVD)
2.1. Cardiovascular Diseases in Aging Population
2.2. General Background and Risk Factors
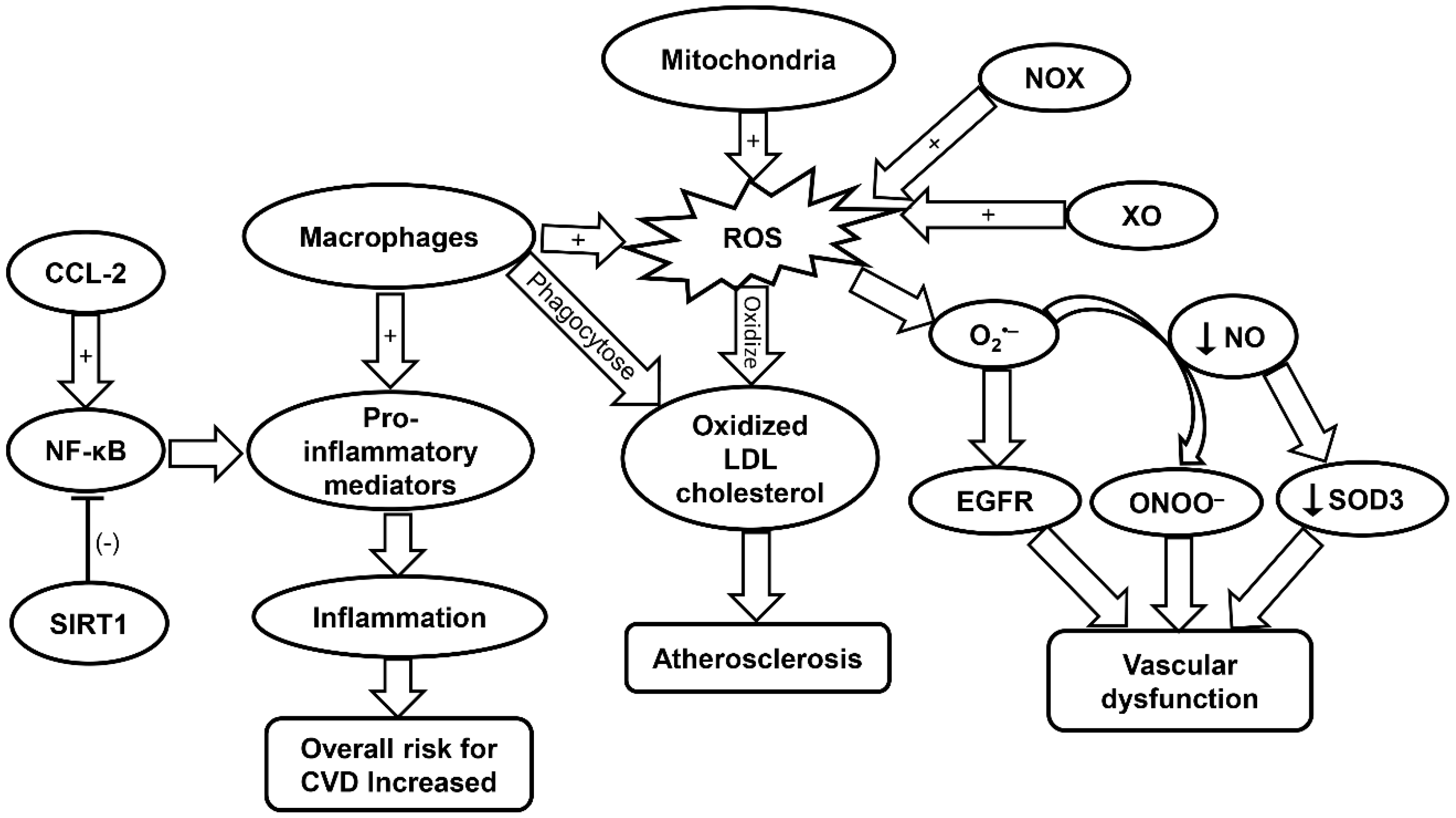
2.3. Oxidative Stress and Inflammaging in Cardiovascular Aging
2.4. Roles of Inflammaging in CVD
2.5. Senescence Cells and CVD
2.6. Immunosenescence and CVD
3. Inflammaging in Cancer
3.1. Cancer in Aging Population
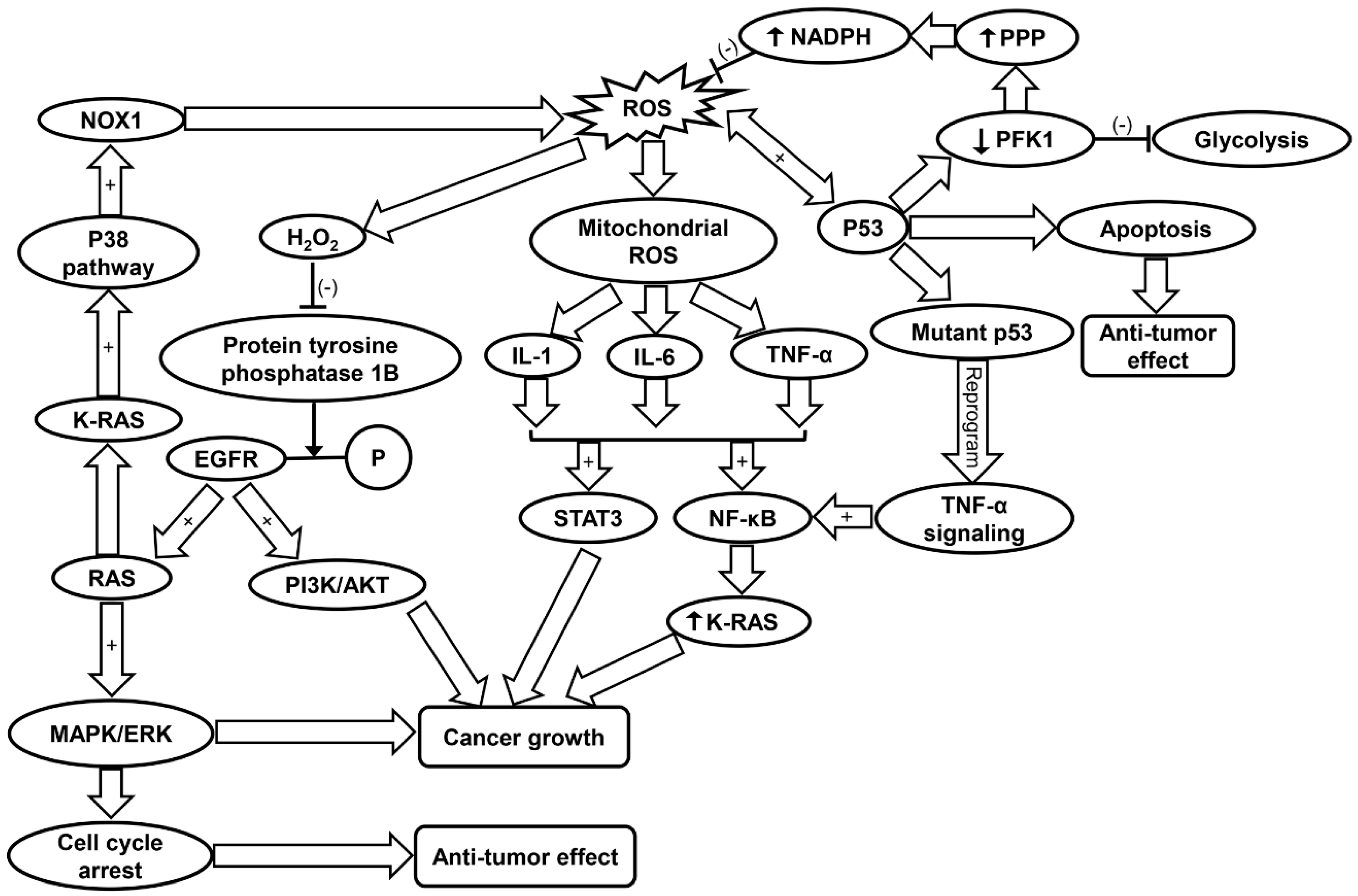
3.2. Oxidative Stress in Cancer
3.3. Inflammaging in Cancer
4. Inflammaging in Neurodegenerative Diseases
4.1. Inflammaging in Neurodegeneration
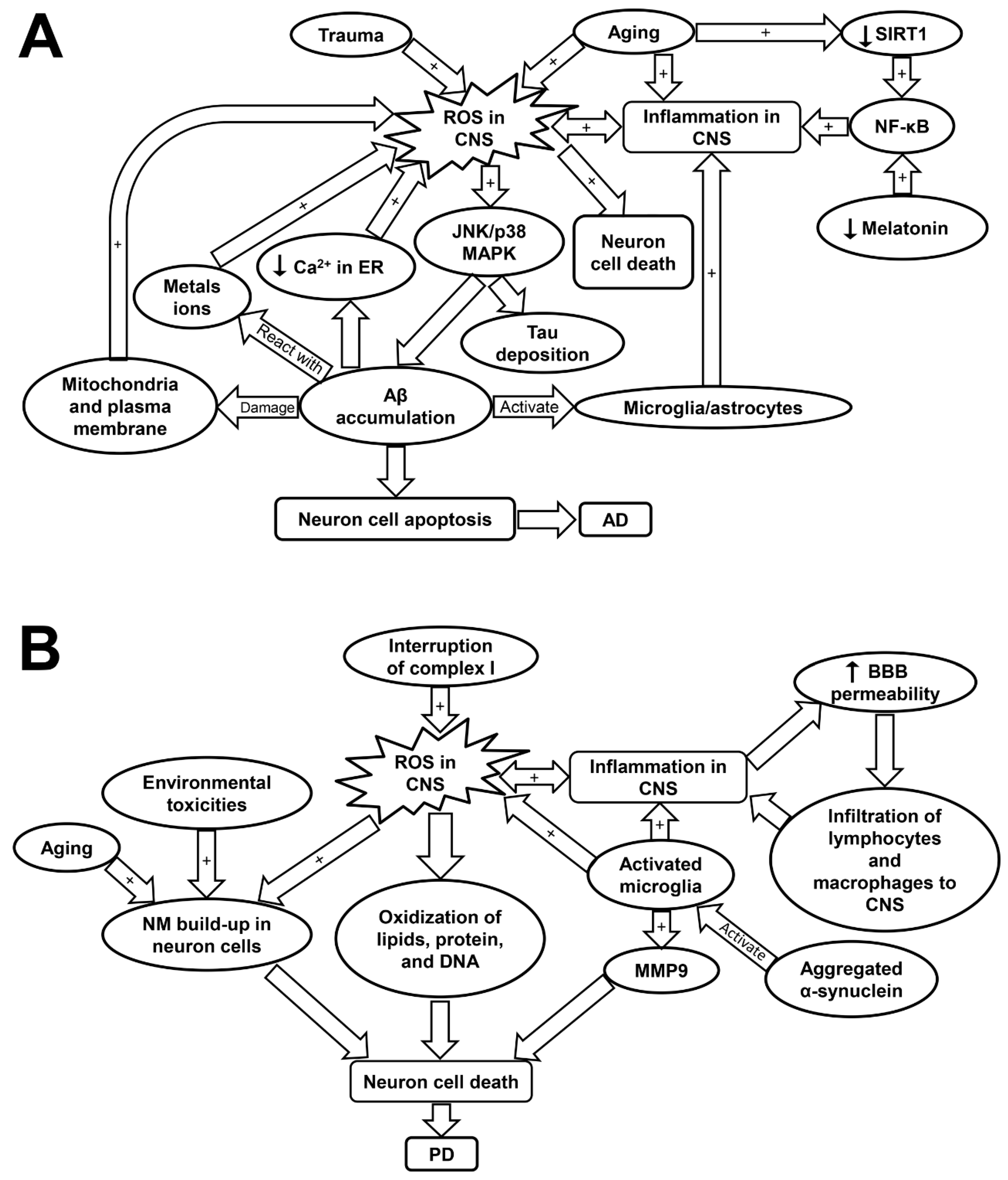
4.2. Inflammaging and Oxidative Stress in Alzheimer’s Disease (AD)
4.3. Inflammaging and Oxidative Stress in Parkinson’s Disease (PD)
5. Inflammaging in Chronic Obstructive Pulmonary Disease (COPD)
5.1. Introduction of COPD
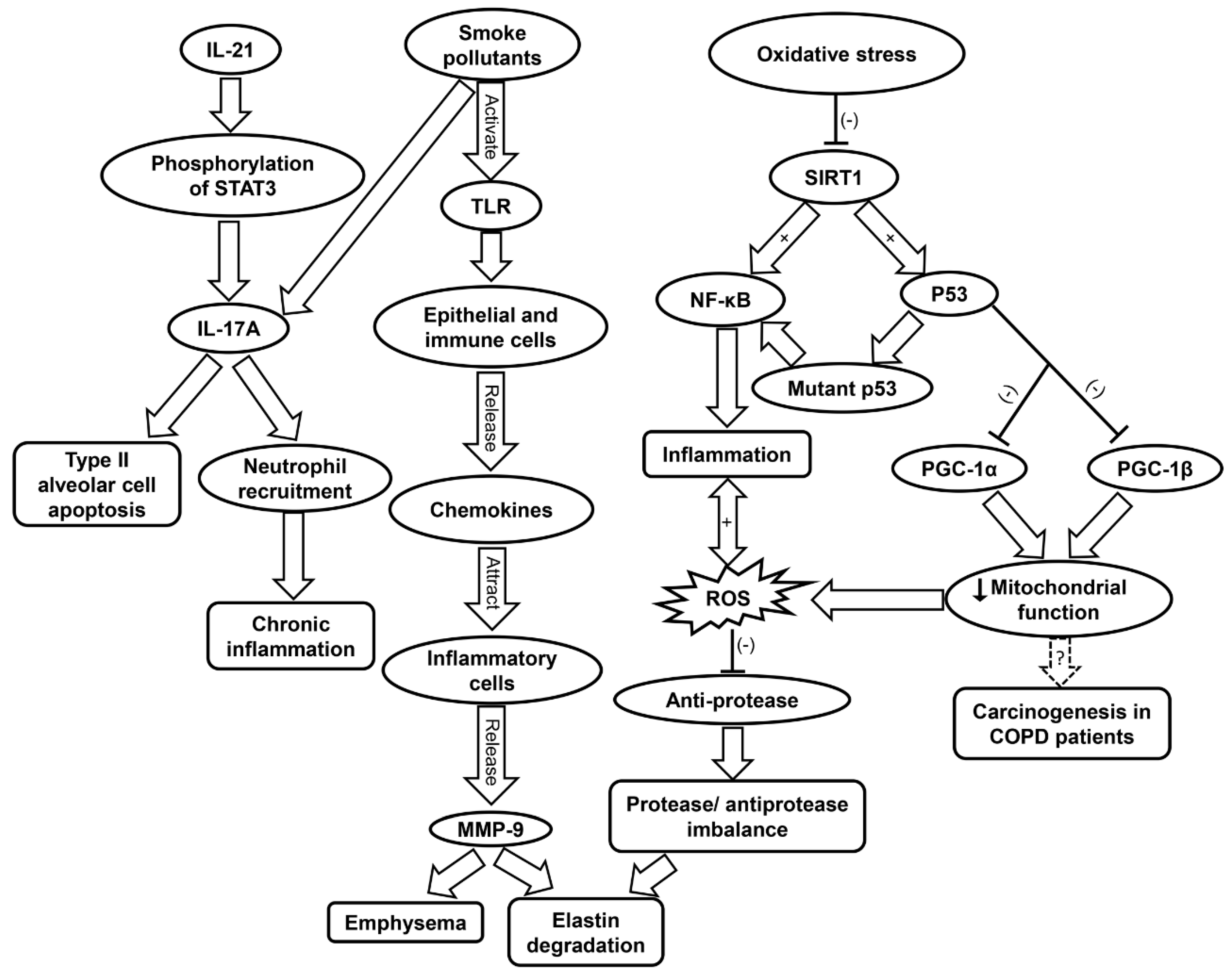
5.2. Inflammatory Cells Involved in COPD
5.3. Inflammaging and Oxidative Stress in COPD
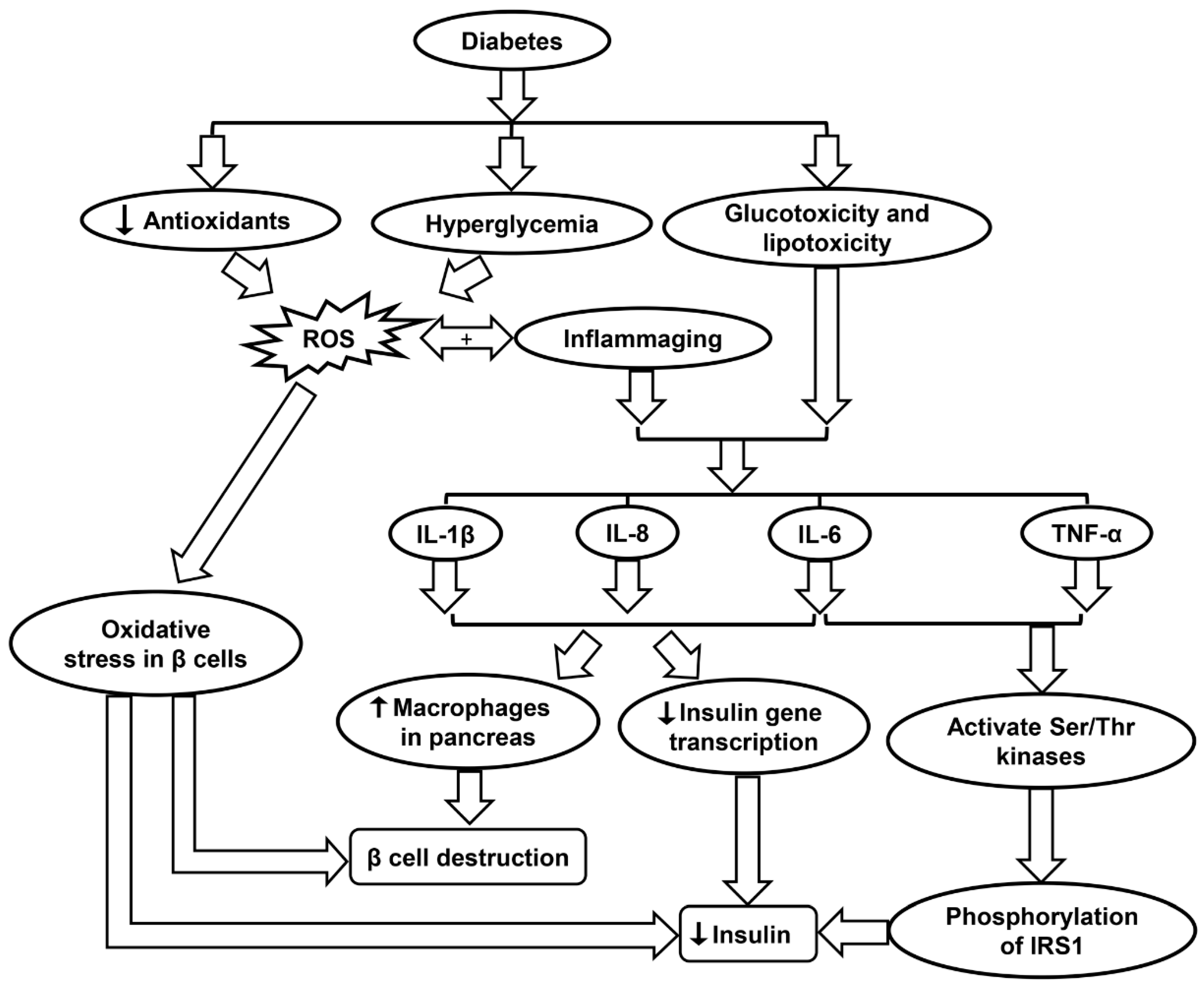
6. Inflammaging in Diabetes
6.1. Type I vs. Type II Diabetes
6.2. Role of Oxidative Stress in Diabetes
6.3. Inflammaging and Diabetes
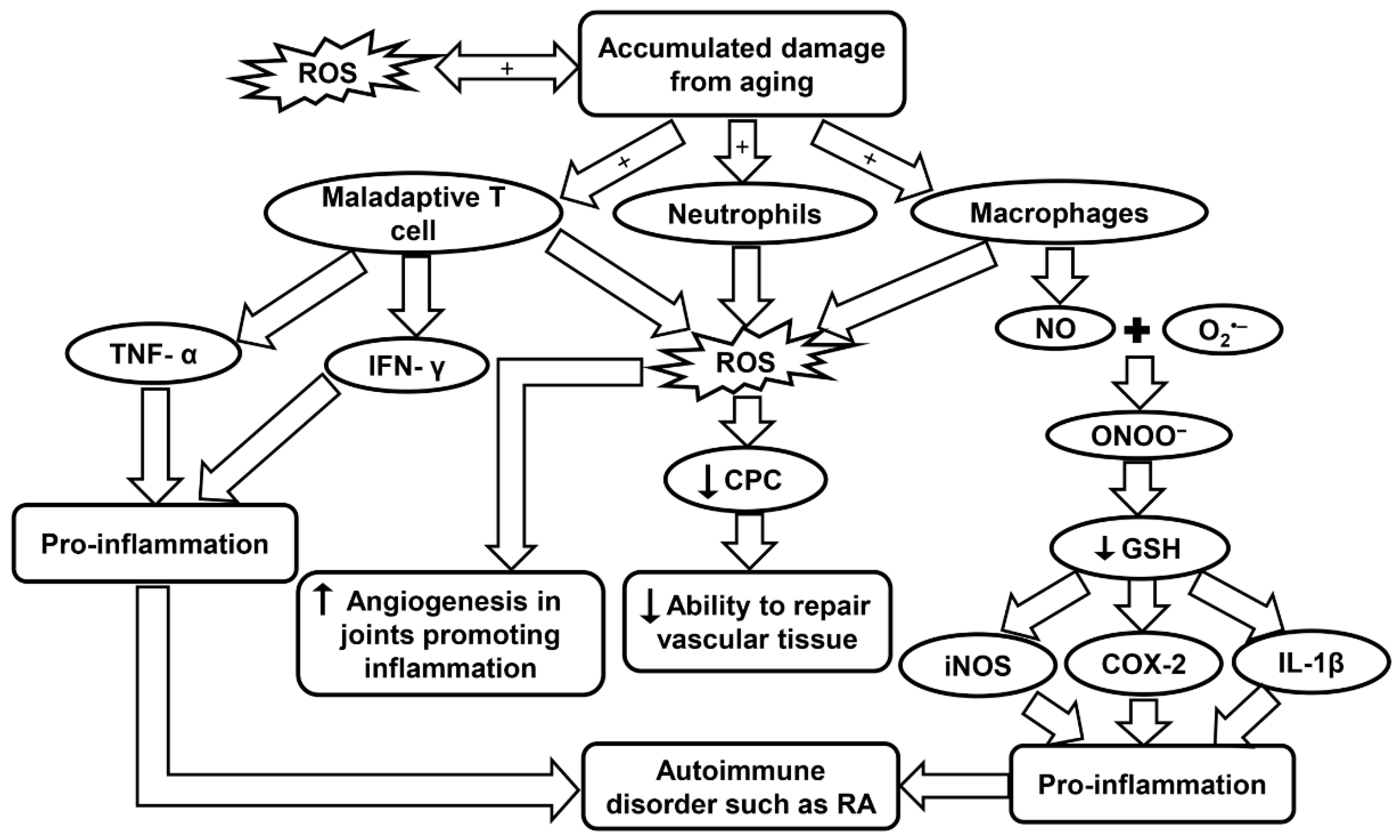
7. Inflammaging in Rheumatoid Arthritis (RA)
7.1. RA in Aging Population
7.2. Causes of RA
7.3. Inflammaging and the Innate Immune System in RA
7.4. Inflammaging and Oxidative Stress in RA
8. Potential Treatment for Inflammaging-Related Diseases
8.1. Drugs
8.2. Stem Cell Interventions
8.3. Diet
8.4. Plant Supplements
8.5. Gut Microbiome
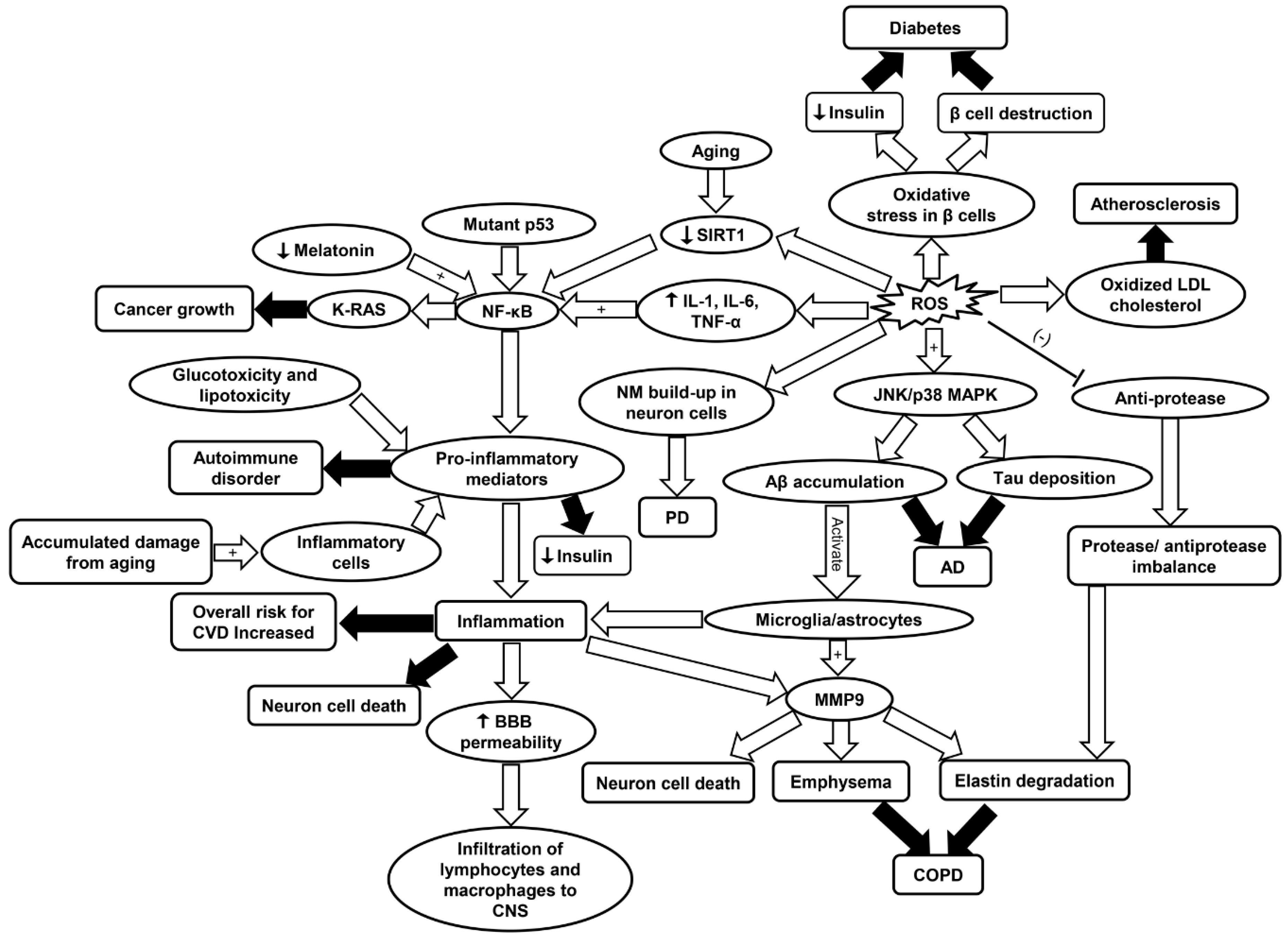
9. Summary and Future Perspective
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACPA | Anti-citrullinated protein antibody |
| AD | Alzheimer’s disease |
| BDB | 3-bromo-4, 5-dihydroxybenzaldehyde |
| CCL11 | CC Chemokine ligand 11 |
| COPD | Chronic obstructive pulmonary disease |
| CCL-2 | Chemokine (C-C motif) ligand 2 |
| CPCs | Circulating progenitor cells |
| CRP | C-reactive protein |
| CSF | Cerebrospinal fluid |
| CVD | Cardiovascular disease |
| EF | Epimedium total flavonoids |
| EGFR | Epidermal growth factor receptor |
| EPCs | Endothelial progenitor cells |
| ER | Endoplasmic reticulum |
| G-MDSCs | Granylocytic-myeloid derived suppressor cells |
| GPX | Glutathione peroxidase |
| GSH | Glutathione |
| Hb | Hemoglobin |
| HDAC2 | Histone deacetylase 2 |
| HSCs | Hematopoietic stem cell |
| Ica | Icariin |
| IL-1 | Interleukin-1 |
| iNOS | Inducible nitric oxide synthase |
| IP-10 | Interferon–gamma induced protein 10 |
| JNK | c-Jun N-terminal kinase |
| KCNB1 | Voltage-gated potassium (K+) channel sub-family B member 1 |
| KEAP1 | Kelch-like ECH-associated protein 1 |
| LDL | Low density lipoprotein |
| LO | Lipoxygenase |
| LRRK2 | Leucine-rich repeat kinase 2 (LRRK2) |
| LTL | Leukocyte telomere length |
| LW | Liuwei Dihuang |
| LW-AFC | Active fraction combination from Liuwei Dihuang decoction |
| MAPK | Mitogen-activated protein kinase |
| MMP | Matrix metalloprotease |
| MPO | Myeloperoxidase |
| MS | Multiple sclerosis |
| mTOR | Mammalian target of rapamycin |
| mtDNA | Mitochondrial DNA |
| mtROS | Mitochondrial ROS |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B |
| NLRP3 | Pryin domain containing-3 protein |
| NM | Neuromelanin |
| NO | Nitric oxide |
| NOX | NADPH oxidase |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| PD | Parkinson’s disease |
| PFK | Phosphofructokinase |
| PINK1 | PTEN-induced putative kinase 1 (PINK1) |
| PI3K | Phosphatidyl inositol 3-kinase |
| PGC-1α | Peroxisome proliferator-activated receptor-γ coactivator 1alpha |
| PPP | Pentose phosphate pathway |
| RA | Rheumatoid arthritis |
| ROS | Reactive oxygen species |
| SASP | Senescence-associated secretory phenotype |
| SMR | Standardized mortality ratio |
| SNpc | Substantia nigra pars compacta |
| STAT3 | Signal transducers and activators of transcription 3 |
| TGF-β | Transforming growth factor-β |
| Th1 | T helper cell 1 |
| TIGAR | TP53-induced glycolysis and apoptosis regulator |
| TLR | Toll-like receptor |
| TNF-α | Tumor necrosis factor alpha |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| VSMCs | Vascular smooth-muscle cells |
| XO | Xanthine oxidase |
| 8-iso-PGF2α | 8-iso-prostaglandin F2α |
| 8-oxodG | 8-oxo-7,8-dihydro-2’-deoxyguanine |
References
- Xu, J.; Murphy, S.L.; Kochanek, K.D.; Arias, E. Mortality in the United States, 2015. NCHS Data Brief 2016, 1–8. Available online: http://www.ncbi.nlm.nih.gov/pubmed/27930283 (accessed on 5 January 2019).
- Jin, K. Modern Biological Theories of Aging. Aging Dis. 2010, 1, 72–74. Available online: http://www.ncbi.nlm.nih.gov/pubmed/21132086 (accessed on 9 June 2019).
- Sergiev, P.V.; Dontsova, O.A.; Berezkin, G.V. Theories of aging: An ever-evolving field. Acta Nat. 2015, 7, 9–18. Available online: http://www.ncbi.nlm.nih.gov/pubmed/25926998 (accessed on 11 November 2018). [CrossRef]
- Campisi, J.; Robert, L. Cell Senescence: Role in Aging and Age-Related Diseases. Aging Facts Theor. 2014, 39, 45–61. [Google Scholar] [CrossRef]
- Salvioli, S.; Monti, D.; Lanzarini, C.; Conte, M.; Pirazzini, C.; Bacalini, M.G.; Garagnani, P.; Giuliani, C.; Fontanesi, E.; Ostan, R.; et al. Immune system, cell senescence, aging and longevity--inflamm-aging reappraised. Curr. Pharm. Des. 2013, 19, 1675–1679. Available online: http://www.ncbi.nlm.nih.gov/pubmed/23589904 (accessed on 6 May 2019).
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Cheeseman, K.H.; Slater, T.F. An Introduction to Free-Radical Biochemistry. Br. Med. Bull. 1993, 49, 481–493. [Google Scholar] [CrossRef]
- Zuo, L.; Christofi, F.L.; Wright, V.P.; Bao, S.Y.; Clanton, T.L. Lipoxygenase-dependent superoxide release in skeletal muscle. J. Appl. Physiol. 2004, 97, 661–668. [Google Scholar] [CrossRef]
- Marosi, K.; Bori, Z.; Hart, N.; Sarga, L.; Koltai, E.; Radak, Z.; Nyakas, C. Long-Term Exercise Treatment Reduces Oxidative Stress in the Hippocampus of Aging Rats. Neuroscience 2012, 226, 21–28. [Google Scholar] [CrossRef]
- Wang, H.J.; Pan, Y.X.; Wang, W.Z.; Zucker, I.H.; Wang, W. NADPH oxidase-derived reactive oxygen species in skeletal muscle modulates the exercise pressor reflex. J. Appl. Physiol. 2009, 107, 450–459. [Google Scholar] [CrossRef]
- Baeeri, M.; Bahadar, H.; Rahimifard, M.; Navaei-Nigjeh, M.; Khorasani, R.; Rezvanfar, M.A.; Gholami, M.; Abdollahi, M. Alpha-Lipoic acid prevents senescence, cell cycle arrest, and inflammatory cues in fibroblasts by inhibiting oxidative stress. Pharm. Res. 2019, 141, 214–223. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef]
- Itsara, L.S.; Kennedy, S.R.; Fox, E.J.; Yu, S.; Hewitt, J.J.; Sanchez-Contreras, M.; Cardozo-Pelaez, F.; Pallanck, L.J. Oxidative stress is not a major contributor to somatic mitochondrial DNA mutations. PLoS Genet. 2014, 10, e1003974. [Google Scholar] [CrossRef]
- Liu, Z.; Ren, Z.; Zhang, J.; Chuang, C.C.; Kandaswamy, E.; Zhou, T.; Zuo, L. Role of ROS and Nutritional Antioxidants in Human Diseases. Front. Physiol. 2018, 9, 477. [Google Scholar] [CrossRef]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox Regulation of NLRP3 Inflammasomes: ROS as Trigger or Effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef]
- Lowe, K.; Kubra, K.T.; He, Z.Y.; Carey, K. Vitamin D Supplementation to Treat Statin-Associated Muscle Symptoms: A Review. Sr. Care Pharm. 2019, 34, 253–257. [Google Scholar] [CrossRef]
- Lakatta, E.G. So! What’s aging? Is cardiovascular aging a disease? J. Mol. Cell. Cardiol. 2015, 83, 1–13. [Google Scholar] [CrossRef]
- Lloyd-Jones, D.M.; Leip, E.P.; Larson, M.G.; D’Agostino, R.B.; Beiser, A.; Wilson, P.W.; Wolf, P.A.; Levy, D. Prediction of lifetime risk for cardiovascular disease by risk factor burden at 50 years of age. Circulation 2006, 113, 791–798. [Google Scholar] [CrossRef]
- North, B.J.; Sinclair, D.A. The Intersection Between Aging and Cardiovascular Disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Yazdanyar, A.; Newman, A.B. The Burden of Cardiovascular Disease in the Elderly: Morbidity, Mortality, and Costs. Clin. Geriatr. Med. 2009, 25, 563–577. [Google Scholar] [CrossRef]
- Xu, X.L.; Wang, B.; Ren, C.H.; Hu, J.N.; Greenberg, D.A.; Chen, T.X.; Xie, L.P.; Jin, K.L. Age-related Impairment of Vascular Structure and Functions. Aging Dis. 2017, 8, 590–610. [Google Scholar] [CrossRef]
- Olivieri, F.; Recchioni, R.; Marcheselli, F.; Abbatecola, A.M.; Santini, G.; Borghetti, G.; Antonicelli, R.; Procopio, A.D. Cellular Senescence in Cardiovascular Diseases: Potential Age-Related Mechanisms and Implications for Treatment. Curr. Pharm. Des. 2013, 19, 1710–1719. [Google Scholar] [PubMed]
- Pirillo, A.; Norata, G.D.; Catapano, A.L. LOX-1, OxLDL, and Atherosclerosis. Mediat. Inflamm. 2013. [Google Scholar] [CrossRef] [PubMed]
- Cervantes Gracia, K.; Llanas-Cornejo, D.; Husi, H. CVD and Oxidative Stress. J. Clin. Med. 2017, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Hazen, S.L. Myeloperoxidase and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1102–1111. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef]
- Yu, H.T.; Park, S.; Shin, E.C.; Lee, W.W. T cell senescence and cardiovascular diseases. Clin. Exp. Med. 2016, 16, 257–263. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Wu, J.; Xia, S.; Kalionis, B.; Wan, W.; Sun, T. The role of oxidative stress and inflammation in cardiovascular aging. Biomed Res. Int. 2014, 2014, 615312. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, A.; Pantell, M.S.; Puterman, E.; Dhabhar, F.S.; Blackburn, E.H.; Yaffe, K.; Cawthon, R.M.; Opresko, P.L.; Hsueh, W.C.; Satterfield, S.; et al. Cumulative inflammatory load is associated with short leukocyte telomere length in the Health, Aging and Body Composition Study. PLoS ONE 2011, 6, e19687. [Google Scholar] [CrossRef]
- Cai, W.; He, J.C.; Zhu, L.; Chen, X.; Zheng, F.; Striker, G.E.; Vlassara, H. Oral glycotoxins determine the effects of calorie restriction on oxidant stress, age-related diseases, and lifespan. Am. J. Pathol. 2008, 173, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Mocchegiani, E.; Costarelli, L.; Giacconi, R.; Cipriano, C.; Muti, E.; Tesei, S.; Malavolta, M. Nutrient-gene interaction in ageing and successful ageing. A single nutrient (zinc) and some target genes related to inflammatory/immune response. Mech. Ageing Dev. 2006, 127, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Shayganni, E.; Bahmani, M.; Asgary, S.; Rafieian-Kopaei, M. Inflammaging and cardiovascular disease: Management by medicinal plants. Phytomedicine 2016, 23, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Scalbert, A.; Morand, C.; Remesy, C.; Jimenez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [PubMed]
- Park, M.S.; Youn, J.C. A New Era of Targeting Pathogenic Immune Mechanisms in Cardiovascular Disease. Korean Circ. J. 2018, 48, 944–946. [Google Scholar] [CrossRef]
- Accardi, G.; Aiello, A.; Gambino, C.M.; Virruso, C.; Caruso, C.; Candore, G. Mediterranean nutraceutical foods: Strategy to improve vascular ageing. Mech. Ageing Dev. 2016, 159, 63–70. [Google Scholar] [CrossRef]
- Vaiserman, A.M.; Koliada, A.K.; Marotta, F. Gut microbiota: A player in aging and a target for anti-aging intervention. Ageing Res. Rev. 2017, 35, 36–45. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Sato, A.; Okada, M.; Shibuya, K.; Watanabe, E.; Seino, S.; Narita, Y.; Shibui, S.; Kayama, T.; Kitanaka, C. Pivotal role for ROS activation of p38 MAPK in the control of differentiation and tumor-initiating capacity of glioma-initiating cells. Stem Cell Res. 2014, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Leelahavanichkul, K.; Amornphimoltham, P.; Molinolo, A.A.; Basile, J.R.; Koontongkaew, S.; Gutkind, J.S. A role for p38 MAPK in head and neck cancer cell growth and tumor-induced angiogenesis and lymphangiogenesis. Mol. Oncol. 2014, 8, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Nikulenkov, F.; Zawacka-Pankau, J.; Li, H.; Gabdoulline, R.; Xu, J.; Eriksson, S.; Hedstrom, E.; Issaeva, N.; Kel, A.; et al. ROS-dependent activation of JNK converts p53 into an efficient inhibitor of oncogenes leading to robust apoptosis. Cell Death Differ. 2014, 21, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Cooks, T.; Harris, C.C.; Oren, M. Caught in the cross fire: p53 in inflammation. Carcinogenesis 2014, 35, 1680–1690. [Google Scholar] [CrossRef] [PubMed]
- Ostan, R.; Lanzarini, C.; Pini, E.; Scurti, M.; Vianello, D.; Bertarelli, C.; Fabbri, C.; Izzi, M.; Palmas, G.; Biondi, F.; et al. Inflammaging and Cancer: A Challenge for the Mediterranean Diet. Nutrients 2015, 7, 2589–2621. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Vitale, G.; Capri, M.; Salvioli, S. Inflammaging and ‘Garb-aging’. Trends Endocrinol. Metab. 2017, 28, 199–212. [Google Scholar] [CrossRef]
- Kim, M.K.; Song, Y.S. Stress Response, Inflammaging, and Cancer. Inflamm. Adv. Age Nutr. Res. Clin. Interv. 2014, 49–53. [Google Scholar] [CrossRef]
- De Simone, V.; Franze, E.; Ronchetti, G.; Colantoni, A.; Fantini, M.C.; Di Fusco, D.; Sica, G.S.; Sileri, P.; MacDonald, T.T.; Pallone, F.; et al. Th17-type cytokines, IL-6 and TNF-alpha synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene 2015, 34, 3493–3503. [Google Scholar] [CrossRef] [PubMed]
- Civenni, G.; Shinde, D.; Zoma, M.; Albino, D.; Costales, P.; Moris, F.; Carbone, G.; Catapano, C. The multi-kinase inhibitor EC-70124 delivers a double-hit to prostate cancer stem cells interfering with both STAT3 and NF-kB signaling. Eur. Urol. 2017, 16, e1294. [Google Scholar] [CrossRef]
- Di Minin, G.; Bellazzo, A.; Dal Ferro, M.; Chiaruttini, G.; Nuzzo, S.; Bicciato, S.; Piazza, S.; Rami, D.; Bulla, R.; Sommaggio, R.; et al. Mutant p53 reprograms TNF signaling in cancer cells through interaction with the tumor suppressor DAB2IP. Mol. Cell 2014, 56, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Ji, B.; Daniluk, J.; Liu, Y.; Deng, D.F.; Huang, H.J.; Wang, H.M.; Logsdon, C.D. An NF-kappa B pathway mediated positive feedback loop amplifies Ras activity to pathological levels in mice. Cancer Res. 2012, 72. [Google Scholar] [CrossRef]
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Mishalian, I.; Granot, Z.; Fridlender, Z.G. The diversity of circulating neutrophils in cancer. Immunobiology 2017, 222, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kensler, T.W.; Cho, C.G.; Posner, G.H.; Talalay, P. Anticarcinogenic activities of sulforaphane and structurally related synthetic norbornyl isothiocyanates. Proc. Natl. Acad. Sci. USA 1994, 91, 3147–3150. Available online: http://www.ncbi.nlm.nih.gov/pubmed/8159717 (accessed on 11 May 2018). [CrossRef]
- Fahey, J.W.; Talalay, P. Antioxidant functions of sulforaphane: A potent inducer of Phase II detoxication enzymes. Food Chem. Toxicol. 1999, 37, 973–979. Available online: http://www.ncbi.nlm.nih.gov/pubmed/10541453 (accessed on 11 May 2018). [CrossRef]
- Yu, W.; Zhang, H.; Shin, M.R.; Sesti, F. Oxidation of KCNB1 potassium channels in the murine brain during aging is associated with cognitive impairment. Biochem. Biophys. Res. Commun. 2019. [Google Scholar] [CrossRef]
- Buttner, R.; Schulz, A.; Reuter, M.; Akula, A.K.; Mindos, T.; Carlstedt, A.; Riecken, L.B.; Baader, S.L.; Bauer, R.; Morrison, H. Inflammaging impairs peripheral nerve maintenance and regeneration. Aging Cell 2018, 17, e12833. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxid. Med. Cell. Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Gutierrez, E.; Munoz-Arenas, G.; Trevino, S.; Espinosa, B.; Chavez, R.; Rojas, K.; Flores, G.; Diaz, A.; Guevara, J. Alzheimer’s disease and metabolic syndrome: A link from oxidative stress and inflammation to neurodegeneration. Synapse 2017, 71. [Google Scholar] [CrossRef] [PubMed]
- Mudo, G.; Frinchi, M.; Nuzzo, D.; Scaduto, P.; Plescia, F.; Massenti, M.F.; Di Carlo, M.; Cannizzaro, C.; Cassata, G.; Cicero, L.; et al. Anti-inflammatory and cognitive effects of interferon-beta1a (IFNbeta1a) in a rat model of Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 44. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, X.; Cheng, X.; Cheng, J.; Liu, F.; Xu, Y.; Zeng, J.; Qiao, S.; Zhou, W.; Zhang, Y. LW-AFC, A New Formula Derived from Liuwei Dihuang Decoction, Ameliorates Cognitive Deterioration and Modulates Neuroendocrine-Immune System in SAMP8 Mouse. Curr. Alzheimer Res. 2017, 14, 221–238. Available online: http://www.ncbi.nlm.nih.gov/pubmed/27335033 (accessed on 4 April 2019). [CrossRef] [PubMed]
- Deb, S.; Phukan, B.C.; Mazumder, M.K.; Dutta, A.; Paul, R.; Bhattacharya, P.; Sandhir, R.; Borah, A. Garcinol, a multifaceted sword for the treatment of Parkinson’s disease. Neurochem. Int. 2019, 128, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Scorza, F.A.; Fiorini, A.C.; Scorza, C.A.; Finsterer, J. Cardiac abnormalities in Parkinson’s disease and Parkinsonism. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2018, 53, 1–5. [Google Scholar] [CrossRef]
- Valdez, L.B.; Zaobornyj, T.; Bandez, M.J.; Lopez-Cepero, J.M.; Boveris, A.; Navarro, A. Complex I syndrome in striatum and frontal cortex in a rat model of Parkinson disease. Free Radic. Biol. Med. 2019, 135, 274–282. [Google Scholar] [CrossRef]
- Storelli, E.; Cassina, N.; Rasini, E.; Marino, F.; Cosentino, M. Do Th17 Lymphocytes and IL-17 Contribute to Parkinson’s Disease? A Systematic Review of Available Evidence. Front. Neurol. 2019, 10. [Google Scholar] [CrossRef]
- Gonzalez, H.; Pacheco, R. T-cell-mediated regulation of neuroinflammation involved in neurodegenerative diseases. J. Neuroinflamm. 2014, 11, 201. [Google Scholar] [CrossRef]
- Di Benedetto, S.; Muller, L.; Wenger, E.; Duzel, S.; Pawelec, G. Contribution of neuroinflammation and immunity to brain aging and the mitigating effects of physical and cognitive interventions. Neurosci. Biobehav. Rev. 2017, 75, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, M.Z.; Andrabi, S.S.; Salman, M.; Tabassum, H.; Shaquiquzzaman, M.; Parveen, S.; Parvez, S. Melatonin Improves Behavioral and Biochemical Outcomes in a Rotenone-Induced Rat Model of Parkinson’s Disease. J. Environ. Pathol. Toxicol. Oncol. 2018, 37, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Ansari Dezfouli, M.; Zahmatkesh, M.; Farahmandfar, M.; Khodagholi, F. Melatonin protective effect against amyloid beta-induced neurotoxicity mediated by mitochondrial biogenesis; involvement of hippocampal Sirtuin-1 signaling pathway. Physiol. Behav. 2019, 204, 65–75. [Google Scholar] [CrossRef] [PubMed]
- John-Schuster, G.; Gunter, S.; Hager, K.; Conlon, T.M.; Eickelberg, O.; Yildirim, A.O. Inflammaging increases susceptibility to cigarette smoke-induced COPD. Oncotarget 2016, 7, 30068–30083. [Google Scholar] [CrossRef] [PubMed]
- Bathri, R.; Bose, P.; Gujar, V.S.; Kumar, L. The Role of ROS in COPD Progression and Therapeutic Strategies. React. Oxyg. Species 2017, 4, 237–250. [Google Scholar] [CrossRef]
- McGuinness, A.J.; Sapey, E. Oxidative Stress in COPD: Sources, Markers, and Potential Mechanisms. J. Clin. Med. 2017, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.W.; Sundar, I.K.; Yao, H.W.; Rahman, I. SIRT1 and Inflammaging in Chronic Obstructive Pulmonary Disease. Inflamm. Adv. Age Nutr. Res. Clin. Interv. 2014, 183–191. [Google Scholar] [CrossRef]
- Lerner, C.A.; Sundar, I.K.; Rahman, I. Mitochondrial redox system, dynamics, and dysfunction in lung inflammaging and COPD. Int. J. Biochem. Cell Biol. 2016, 81, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, S.; Ishizaki, T.; Kadowaki, M.; Akai, M.; Shiozaki, K.; Iguchi, M.; Oikawa, T.; Nakagawa, K.; Osanai, K.; Toga, H.; et al. p53 Signaling Pathway Polymorphisms Associated with Emphysematous Changes in Patients With COPD. Chest 2017, 152, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Ng Kee Kwong, F.; Nicholson, A.G.; Harrison, C.L.; Hansbro, P.M.; Adcock, I.M.; Chung, K.F. Is mitochondrial dysfunction a driving mechanism linking COPD to nonsmall cell lung carcinoma? Eur. Respir. Rev. 2017, 26. [Google Scholar] [CrossRef]
- Wiegman, C.H.; Michaeloudes, C.; Haji, G.; Narang, P.; Clarke, C.J.; Russell, K.E.; Bao, W.; Pavlidis, S.; Barnes, P.J.; Kanerva, J.; et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2015, 136, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; Kinnula, V.L.; Gorbunova, V.; Yao, H. SIRT1 as a therapeutic target in inflammaging of the pulmonary disease. Prev. Med. 2012, 54, S20–S28. [Google Scholar] [CrossRef] [PubMed]
- Varraso, R.; Chiuve, S.E.; Fung, T.T.; Barr, R.G.; Hu, F.B.; Willett, W.C.; Camargo, C.A. Alternate Healthy Eating Index 2010 and risk of chronic obstructive pulmonary disease among US women and men: Prospective study. BMJ 2015, 350, h286. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Rahman, I. Role of histone deacetylase 2 in epigenetics and cellular senescence: Implications in lung inflammaging and COPD. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L557–L566. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Oxidative stress and stress-activated signaling pathways: A unifying hypothesis of type 2 diabetes. Endocr. Rev. 2002, 23, 599–622. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, U.; Park, K.G. A systematic review of oxidative stress and safety of antioxidants in diabetes: Focus on islets and their defense. Diabetes Metab. J. 2013, 37, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 1996, 271, 665–668. [Google Scholar] [CrossRef]
- Goto, M. Inflammaging (inflammation plus aging): A driving force for human aging based on an evolutionarily antagonistic pleiotropy theory? Biosci. Trends 2008, 2, 218–230. [Google Scholar]
- Moriscot, C.; Richard, M.J.; Favrot, M.C.; Benhamou, P.Y. Protection of insulin-secreting INS-1 cells against oxidative stress through adenoviral-mediated glutathione peroxidase overexpression. Diabetes Metab. 2003, 29, 145–151. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Goronzy, J.J.; Weyand, C.M. DNA damage, metabolism and aging in pro-inflammatory T cells: Rheumatoid arthritis as a model system. Exp. Gerontol. 2017, 105, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Shao, L.; Colmegna, I.; Goronzy, J.J.; Weyand, C.M. Telomerase insufficiency in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2009, 106, 4360–4365. [Google Scholar] [CrossRef] [PubMed]
- Weyand, C.M.; Yang, Z.; Goronzy, J.J. T-cell aging in rheumatoid arthritis. Curr. Opin. Rheumatol. 2014, 26, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Bala, A.; Ghosh, P.; Mukhopadhyay, D.; Mitra, A.; Sarkar, A.; Bauri, A.K.; Ghosh, A.; Chattopadhyay, S.; Chatterjee, M. Attenuation of oxidative stress by Allylpyrocatechol in synovial cellular infiltrate of patients with Rheumatoid Arthritis. Free Radic. Res. 2011, 45, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Ghosh, P.; Datta, S.; Ghosh, A.; Chattopadhyay, S.; Chatterjee, M. Oxidative stress as a potential biomarker for determining disease activity in patients with Rheumatoid Arthritis. Free Radic. Res. 2012, 46, 1482–1489. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.T.; Wei, J.; Tang, Y.W.; Wang, B.; Zhang, Y.; Shi, L.; Guo, J.P.; Hu, F.L.; Li, X. Leptin-induced migration and angiogenesis in rheumatoid arthritis is mediated by reactive oxygen species. FEBS Open Bio 2017, 7, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Goto, M. A comparative study of anti-inflammatory and antidyslipidemic effects of fenofibrate and statins on rheumatoid arthritis. Mod. Rheumatol. 2010, 20, 238–243. [Google Scholar] [CrossRef]
- Dhimolea, E. Canakinumab. MAbs 2010, 2, 3–13. [Google Scholar] [CrossRef]
- Li, J.; Stouffs, M.; Serrander, L.; Banfi, B.; Bettiol, E.; Charnay, Y.; Steger, K.; Krause, K.H.; Jaconi, M.E. The NADPH oxidase NOX4 drives cardiac differentiation: Role in regulating cardiac transcription factors and MAP kinase activation. Mol. Biol. Cell 2006, 17, 3978–3988. [Google Scholar] [CrossRef]
- Shekher, A.; Singh, M. Role of eicosanoid inhibition in ischemia reperfusion injury: Intact and isolated rat heart studies. Methods Find. Exp. Clin. Pharm. 1997, 19, 223–229. [Google Scholar]
- El Assar, M.; Angulo, J.; Vallejo, S.; Peiro, C.; Sanchez-Ferrer, C.F.; Rodriguez-Manas, L. Mechanisms involved in the aging-induced vascular dysfunction. Front. Physiol. 2012, 3, 132. [Google Scholar] [CrossRef] [PubMed]
- Wickremasinghe, D.; Peiris, H.; Chandrasena, L.G.; Senaratne, V.; Perera, R. Case control feasibility study assessing the association between severity of coronary artery disease with Glutathione Peroxidase-1 (GPX-1) and GPX-1 polymorphism (Pro198Leu). BMC Cardiovasc. Disord. 2016, 16, 111. [Google Scholar] [CrossRef] [PubMed]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione Peroxidase-1 in Health and Disease: From Molecular Mechanisms to Therapeutic Opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.C.; Mahoney, C.E.; Anderson, L.; Ottaviano, F.; Croce, K.; Leopold, J.A.; Zhang, Y.Y.; Tang, S.S.; Handy, D.E.; Loscalzo, J. Glutathione Peroxidase-3 Deficiency Promotes Platelet-Dependent Thrombosis In Vivo. Circulation 2011, 123, 1963–1973. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.W.E.; Ramji, D.P. Cytokines: Roles in atherosclerosis disease progression and potential therapeutic targets. Future Med. Chem. 2016, 8, 1317–1330. [Google Scholar] [CrossRef] [PubMed]
- Rippo, M.R.; Olivieri, F.; Monsurro, V.; Prattichizzo, F.; Albertini, M.C.; Procopio, A.D. MitomiRs in human inflamm-aging: A hypothesis involving miR-181a, miR-34a and miR-146a. Exp. Gerontol. 2014, 56, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Landi, F.; Bernabei, R.; Marzetti, E. Fueling Inflamm-Aging through Mitochondrial Dysfunction: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2017, 18, 933. [Google Scholar] [CrossRef]
- Paneni, F.; Diaz Canestro, C.; Libby, P.; Luscher, T.F.; Camici, G.G. The Aging Cardiovascular System: Understanding It at the Cellular and Clinical Levels. J. Am. Coll. Cardiol. 2017, 69, 1952–1967. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Fuster, J.J.; Andres, V. Telomere biology and cardiovascular disease. Circ. Res. 2006, 99, 1167–1180. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Huuskonen, J.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Activation of innate immunity system during aging: NF-kappa B signaling is the molecular culprit of inflamm-aging. Ageing Res. Rev. 2008, 7, 83–105. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Singh, V.; Pal Singh, M.; Shrivastava, P.; Singh, N.; Gambhir, I.S.; Singh, S.M. Effect of immunosenescence on the induction of cardiovascular disease pathogenesis: Role of peripheral blood mononuclear cells. Immunopharmacol. Immunotoxicol. 2008, 30, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.R.; Zibadi, S.; Vazquez, R.; Larson, D. Nutritional regulation of immunosenescence for heart health. J. Nutr. Biochem. 2005, 16, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Xydonas, S.; Parissis, J.; Lioni, L.; Kapsimali, V.; Psarra, E.; Farmakis, D.; Kremastinos, D.; Lekakis, J.; Sideris, A.; Tsirogianni, A.; et al. Immunosenescence in patients with chronic systolic heart failure. J. Cardiovasc. Med. (Hagerstown) 2016, 17, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Moro-Garcia, M.A.; Echeverria, A.; Galan-Artimez, M.C.; Suarez-Garcia, F.M.; Solano-Jaurrieta, J.J.; Avanzas-Fernandez, P.; Diaz-Molina, B.; Lambert, J.L.; Lopez-Larrea, C.; Moris de la Tassa, C.; et al. Immunosenescence and inflammation characterize chronic heart failure patients with more advanced disease. Int. J. Cardiol. 2014, 174, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Darby, S.C.; Ewertz, M.; McGale, P.; Bennet, A.M.; Blom-Goldman, U.; Bronnum, D.; Correa, C.; Cutter, D.; Gagliardi, G.; Gigante, B.; et al. Risk of ischemic heart disease in women after radiotherapy for breast cancer. N. Engl. J. Med. 2013, 368, 987–998. [Google Scholar] [CrossRef]
- Li, D.; de Glas, N.A.; Hurria, A. Cancer and Aging: General Principles, Biology, and Geriatric Assessment. Clin. Geriatr. Med. 2016, 32, 1–15. [Google Scholar] [CrossRef]
- Sosa, V.; Moline, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; LLeonart, M.E. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef]
- Wang, Q.; Huang, L.; Yue, J. Oxidative stress activates the TRPM2-Ca(2+)-CaMKII-ROS signaling loop to induce cell death in cancer cells. Biochim. Biophys. Acta 2017, 1864, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Noh, J.; Kwon, B.; Han, E.; Park, M.; Yang, W.; Cho, W.; Yoo, W.; Khang, G.; Lee, D. Amplification of oxidative stress by a dual stimuli-responsive hybrid drug enhances cancer cell death. Nat. Commun. 2015, 6, 6907. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT signaling pathway and cancer: An updated review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Stuhlmiller, T.J.; Angus, S.P.; Zawistowski, J.S.; Graves, L.M. Molecular pathways: Adaptive kinome reprogramming in response to targeted inhibition of the BRAF-MEK-ERK pathway in cancer. Clin. Cancer Res. 2014, 20, 2516–2522. [Google Scholar] [CrossRef] [PubMed]
- Park, M.T.; Kim, M.J.; Suh, Y.; Kim, R.K.; Kim, H.; Lim, E.J.; Yoo, K.C.; Lee, G.H.; Kim, Y.H.; Hwang, S.G.; et al. Novel signaling axis for ROS generation during K-Ras-induced cellular transformation. Cell Death Differ. 2014, 21, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Martini, M.; Ciraolo, E.; Gulluni, F.; Hirsch, E. Targeting PI3K in Cancer: Any Good News? Front. Oncol. 2013, 3, 108. [Google Scholar] [CrossRef] [PubMed]
- Deschenes-Simard, X.; Gaumont-Leclerc, M.F.; Bourdeau, V.; Lessard, F.; Moiseeva, O.; Forest, V.; Igelmann, S.; Mallette, F.A.; Saba-El-Leil, M.K.; Meloche, S.; et al. Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation. Genes Dev. 2013, 27, 900–915. [Google Scholar] [CrossRef]
- Deschenes-Simard, X.; Kottakis, F.; Meloche, S.; Ferbeyre, G. ERKs in cancer: Friends or foes? Cancer Res. 2014, 74, 412–419. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef] [PubMed]
- McGrath-Morrow, S.; Lauer, T.; Yee, M.; Neptune, E.; Podowski, M.; Thimmulappa, R.K.; O’Reilly, M.; Biswal, S. Nrf2 increases survival and attenuates alveolar growth inhibition in neonatal mice exposed to hyperoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L565–L573. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Luo, X.; Nie, P.; Wu, B.; Xu, W.; Shi, X.; Chang, H.; Li, B.; Yu, X.; Zou, Z. CQ synergistically sensitizes human colorectal cancer cells to SN-38/CPT-11 through lysosomal and mitochondrial apoptotic pathway via p53-ROS cross-talk. Free Radic Biol. Med. 2017, 104, 280–297. [Google Scholar] [CrossRef] [PubMed]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Berg, M.O. Antioxidants markedly accelerate tumor growth and reduce survival in mice with KRAS- and BRAF-induced lung cancer by disrupting the ROS-p53 axis. Cancer Res. 2014, 74. [Google Scholar] [CrossRef]
- Kawanishi, S.; Ohnishi, S.; Ma, N.; Hiraku, Y.; Murata, M. Crosstalk between DNA Damage and Inflammation in the Multiple Steps of Carcinogenesis. Int. J. Mol. Sci. 2017, 18, 1808. [Google Scholar] [CrossRef] [PubMed]
- Brancato, B.; Munnia, A.; Cellai, F.; Ceni, E.; Mello, T.; Bianchi, S.; Catarzi, S.; Risso, G.G.; Galli, A.; Peluso, M.E. 8-Oxo-7,8-dihydro-2’-deoxyguanosine and other lesions along the coding strand of the exon 5 of the tumour suppressor gene P53 in a breast cancer case-control study. DNA Res. 2016, 23, 395–402. [Google Scholar] [CrossRef]
- DeBalsi, K.L.; Hoff, K.E.; Copeland, W.C. Role of the mitochondrial DNA replication machinery in mitochondrial DNA mutagenesis, aging and age-related diseases. Ageing Res. Rev. 2017, 33, 89–104. [Google Scholar] [CrossRef]
- Halsne, R.; Esbensen, Y.; Wang, W.; Scheffler, K.; Suganthan, R.; Bjoras, M.; Eide, L. Lack of the DNA glycosylases MYH and OGG1 in the cancer prone double mutant mouse does not increase mitochondrial DNA mutagenesis. DNA Repair (Amst) 2012, 11, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.; Skola, D.; Cassetta, L.; Bain, C.; Louwe, P.; Lynch, R.; Jenkins, S.; Glass, C.; Pollard, J. Transcriptional alterations to blood monocytes in breast cancer reveal down regulation of anti-cancer pathways. Cancer Genet. 2018, 226–227, 47–48. [Google Scholar] [CrossRef]
- Kamp, D.W.; Shacter, E.; Weitzman, S.A. Chronic inflammation and cancer: The role of the mitochondria. Oncology (Williston Park) 2011, 25, 400–410, 413. Available online: http://www.ncbi.nlm.nih.gov/pubmed/21710835 (accessed on 9 March 2018). [PubMed]
- Candido, J.; Hagemann, T. Cancer-related inflammation. J. Clin. Immunol. 2013, 33, S79–S84. [Google Scholar] [CrossRef] [PubMed]
- Simons, A.; Stanam, A.; Koch, A.; Espinosa-Cotton, M.; Gibson-Corley, K. Targeting IL-1 signaling to improve tumor response to erlotinib in HNSCC. Clin. Cancer Res. 2017, 23. [Google Scholar] [CrossRef]
- Petrilli, V.; Bodnar, M.; Guey, B.; Hacot, S.; Lantuejoul, S. A novel role for the NLRP3 inflammasome in lung cancer. Cancer Res. 2015, 75. [Google Scholar] [CrossRef]
- Dupaul-Chicoine, J.; Arabzadeh, A.; Dagenais, M.; Douglas, T.; Champagne, C.; Morizot, A.; Rodrigue-Gervais, I.G.; Breton, V.; Colpitts, S.L.; Beauchemin, N.; et al. The Nlrp3 Inflammasome Suppresses Colorectal Cancer Metastatic Growth in the Liver by Promoting Natural Killer Cell Tumoricidal Activity. Immunity 2015, 43, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Mu, K.; Li, T.; Zhang, Y.; Yang, Z.; Jia, X.; Zhao, W.; Huai, W.; Guo, P.; Han, L. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab. Investig. 2014, 94, 52–62. [Google Scholar] [CrossRef]
- Bruchard, M.; Mignot, G.; Derangere, V.; Chalmin, F.; Chevriaux, A.; Vegran, F.; Boireau, W.; Simon, B.; Ryffel, B.; Connat, J.L.; et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat. Med. 2013, 19, 57–64. [Google Scholar] [CrossRef]
- Bae, J.Y.; Lee, S.W.; Shin, Y.H.; Lee, J.H.; Jahng, J.W.; Park, K. P2X7 receptor and NLRP3 inflammasome activation in head and neck cancer. Oncotarget 2017, 8, 48972–48982. [Google Scholar] [CrossRef]
- Hu, W.T.; Howell, J.C.; Ozturk, T.; Gangishetti, U.; Kollhoff, A.L.; Hatcher-Martin, J.M.; Anderson, A.M.; Tyor, W.R. CSF Cytokines in Aging, Multiple Sclerosis, and Dementia. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Nicaise, A.M.; Wagstaff, L.J.; Willis, C.M.; Paisie, C.; Chandok, H.; Robson, P.; Fossati, V.; Williams, A.; Crocker, S.J. Cellular senescence in progenitor cells contributes to diminished remyelination potential in progressive multiple sclerosis. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef] [PubMed]
- Cass, S.P. Alzheimer’s Disease and Exercise: A Literature Review. Curr. Sports Med. Rep. 2017, 16, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Perfeito, R.; Cunha-Oliveira, T.; Rego, A.C. Reprint of: Revisiting oxidative stress and mitochondrial dysfunction in the pathogenesis of Parkinson disease-resemblance to the effect of amphetamine drugs of abuse. Free Radic. Biol. Med. 2013, 62, 186–201. [Google Scholar] [CrossRef] [PubMed]
- Perfeito, R.; Cunha-Oliveira, T.; Rego, A.C. Revisiting oxidative stress and mitochondrial dysfunction in the pathogenesis of Parkinson disease-resemblance to the effect of amphetamine drugs of abuse. Free Radic. Biol. Med. 2012, 53, 1791–1806. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, M.; Giacomazza, D.; San Biagio, P.L. Alzheimer’s disease: Biological aspects, therapeutic perspectives and diagnostic tools. J. Phys. Condens. Matter 2012, 24. [Google Scholar] [CrossRef]
- Wilquet, V.; De Strooper, B. Amyloid-beta precursor protein processing in neurodegeneration. Curr. Opin. Neurobiol. 2004, 14, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Zhou, T.; Pannell, B.K.; Ziegler, A.C.; Best, T.M. Biological and physiological role of reactive oxygen species—the good, the bad and the ugly. Acta Physiol. 2015, 214, 329–348. [Google Scholar] [CrossRef]
- Mendelsohn, A.R.; Larrick, J.W. Sleep facilitates clearance of metabolites from the brain: Glymphatic function in aging and neurodegenerative diseases. Rejuvenation Res. 2013, 16, 518–523. [Google Scholar] [CrossRef]
- Hardeland, R. Aging, Melatonin, and the Pro- and Anti-Inflammatory Networks. Int. J. Mol. Sci. 2019, 20, 1223. [Google Scholar] [CrossRef]
- Pappolla, M.A.; Matsubara, E.; Vidal, R.; Pacheco-Quinto, J.; Poeggeler, B.; Zagorski, M.; Sambamurti, K. Melatonin Treatment Enhances A beta Lymphatic Clearance in a Transgenic Mouse Model of Amyloidosis. Curr. Alzheimer Res. 2018, 15, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Wang, W.Z.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X.W. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Et Biophys. Acta-Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Richartz-Salzburger, E.; Batra, A.; Stransky, E.; Laske, C.; Kohler, N.; Bartels, M.; Buchkremer, G.; Schott, K. Altered lymphocyte distribution in Alzheimer’s disease. J. Psychiatr. Res. 2007, 41, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Eikelenboom, P.; Veerhuis, R.; van Exel, E.; Hoozemans, J.J.M.; Rozemuller, A.J.M.; van Gool, W.A. The Early Involvement of the Innate Immunity in the Pathogenesis of Late-onset Alzheimer’s Disease: Neuropathological, Epidemiological and Genetic Evidence. Curr. Alzheimer Res. 2011, 8, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappa B signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Gomes, B.A.Q.; Silva, J.P.B.; Romeiro, C.F.R.; Dos Santos, S.M.; Rodrigues, C.A.; Goncalves, P.R.; Sakai, J.T.; Mendes, P.F.S.; Varela, E.L.P.; Monteiro, M.C. Neuroprotective Mechanisms of Resveratrol in Alzheimer’s Disease: Role of SIRT1. Oxid. Med. Cell. Longev. 2018, 2018, 8152373. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1255–1270. [Google Scholar] [CrossRef]
- Kluss, J.H.; Mamais, A.; Cookson, M.R. LRRK2 links genetic and sporadic Parkinson’s disease. Biochem. Soc. Trans. 2019, 47, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Carballo-Carbajal, I.; Laguna, A.; Romero-Gimenez, J.; Cuadros, T.; Bove, J.; Martinez-Vicente, M.; Parent, A.; Gonzalez-Sepulveda, M.; Penuelas, N.; Torra, A.; et al. Brain tyrosinase overexpression implicates age-dependent neuromelanin production in Parkinson’s disease pathogenesis. Nat. Commun. 2019, 10, 973. [Google Scholar] [CrossRef] [PubMed]
- Pinho, B.R.; Reis, S.D.; Hartley, R.C.; Murphy, M.P.; Oliveira, J.M.A. Mitochondrial superoxide generation induces a parkinsonian phenotype in zebrafish and huntingtin aggregation in human cells. Free Radic. Biol. Med. 2019, 130, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Raza, C.; Anjum, R.; Shakeel, N.U.A. Parkinson’s disease: Mechanisms, translational models and management strategies. Life Sci. 2019, 226, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Liou, C.J.; Yang, C.M.; Lee, T.H.; Liu, P.S.; Hsieh, H.L. Neuroprotective Effects of Dehydroepiandrosterone Sulfate Through Inhibiting Expression of Matrix Metalloproteinase-9 from Bradykinin-Challenged Astroglia. Mol. Neurobiol. 2019, 56, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Appel, S.H. CD4(+) T cells mediate cytotoxicity in neurodegenerative diseases. J. Clin. Investig. 2009, 119, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Brochard, V.; Combadiere, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.M.; et al. Infiltration of CD4(+) lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Investig. 2009, 119, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Mejias, N.H.; Martinez, C.C.; Stephens, M.E.; Vaccari, J.P.D. Contribution of the inflammasome to inflammaging. J. Inflamm. Lond. 2018, 15, 23. [Google Scholar] [CrossRef] [PubMed]
- MacNee, W. Is Chronic Obstructive Pulmonary Disease an Accelerated Aging Disease? Ann. Am. Thorac. Soc. 2016, 13, S429–S437. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Zhang, H.; Davies, K.J.A.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 2015, 88, 314–336. [Google Scholar] [CrossRef]
- Brandsma, C.A.; de Vries, M.; Costa, R.; Woldhuis, R.R.; Konigshoff, M.; Timens, W. Lung ageing and COPD: Is there a role for ageing in abnormal tissue repair? Eur. Respir. Rev. 2017, 26. [Google Scholar] [CrossRef]
- Fragoso, C.A. Epidemiology of Chronic Obstructive Pulmonary Disease (COPD) in Aging Populations. COPD 2016, 13, 125–129. [Google Scholar] [CrossRef]
- Papaioannou, A.I.; Rossios, C.; Kostikas, K.; Ito, K. Can we delay the accelerated lung aging in COPD? Anti-aging molecules and interventions. Curr. Drug Targets 2013, 14, 149–157. Available online: http://www.ncbi.nlm.nih.gov/pubmed/23256715 (accessed on 30 April 2019). [CrossRef] [PubMed]
- Barnes, P.J. Cellular and molecular mechanisms of asthma and COPD. Clin. Sci. (Lond.) 2017, 131, 1541–1558. [Google Scholar] [CrossRef] [PubMed]
- Aghasafari, P.; George, U.; Pidaparti, R. A review of inflammatory mechanism in airway diseases. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. [Et Al.] 2019, 68, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Richmond, B.W.; Du, R.H.; Han, W.; Benjamin, J.T.; van der Meer, R.; Gleaves, L.; Guo, M.; McKissack, A.; Zhang, Y.; Cheng, D.S.; et al. Bacterial-derived Neutrophilic Inflammation Drives Lung Remodeling in a Mouse Model of Chronic Obstructive Pulmonary Disease. Am. J. Respir. Cell Mol. Biol. 2018, 58, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Bazzan, E.; Turato, G.; Tine, M.; Radu, C.M.; Balestro, E.; Rigobello, C.; Biondini, D.; Schiavon, M.; Lunardi, F.; Baraldo, S.; et al. Dual polarization of human alveolar macrophages progressively increases with smoking and COPD severity. Respir. Res. 2017, 18, 40. [Google Scholar] [CrossRef]
- Cordoba-Lanus, E.; Cazorla-Rivero, S.; Espinoza-Jimenez, A.; de-Torres, J.P.; Pajares, M.J.; Aguirre-Jaime, A.; Celli, B.; Casanova, C. Telomere shortening and accelerated aging in COPD: Findings from the BODE cohort. Respir. Res. 2017, 18, 59. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Lee, E.C.; Ra, S.W.; Fishbane, N.; Tam, S.; Criner, G.J.; Woodruff, P.G.; Lazarus, S.C.; Albert, R.; Connett, J.E.; et al. Relationship of Absolute Telomere Length with Quality of Life, Exacerbations, and Mortality in COPD. Chest 2018, 154, 266–273. [Google Scholar] [CrossRef]
- Ding, Y.; Zhou, X.; Wu, C.; Li, Q.; Sun, J.; Niu, H.; Lin, D.; Sun, D.; Xie, P.; Wu, D.; et al. Telomere length, ZNF208 genetic variants and risk of chronic obstructive pulmonary disease in the Hainan Li population. J. Gene Med. 2018, 20, e3061. [Google Scholar] [CrossRef]
- Birch, J.; Anderson, R.K.; Correia-Melo, C.; Jurk, D.; Hewitt, G.; Marques, F.M.; Green, N.J.; Moisey, E.; Birrell, M.A.; Belvisi, M.G.; et al. DNA damage response at telomeres contributes to lung aging and chronic obstructive pulmonary disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L1124–L1137. [Google Scholar] [CrossRef]
- Everaerts, S.; Lammertyn, E.J.; Martens, D.S.; De Sadeleer, L.J.; Maes, K.; van Batenburg, A.A.; Goldschmeding, R.; van Moorsel, C.H.M.; Dupont, L.J.; Wuyts, W.A.; et al. The aging lung: Tissue telomere shortening in health and disease. Respir. Res. 2018, 19, 95. [Google Scholar] [CrossRef]
- Bartling, B.; Hofmann, H.S. Reduced proliferation capacity of lung cells in chronic obstructive pulmonary disease. Z. Gerontol. Geriatr. 2018, 52, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Pomiès, P.; Blaquière, M.; Gouzi, F.; Maury, J.; Mercier, J.; Hayot, M. Reactive oxygen species regulate autophagy in COPD myotubes in vitro. Eur. Respir. J. 2015, 46, PA4604. [Google Scholar] [CrossRef]
- Le Rouzic, O.; Pichavant, M.; Frealle, E.; Guillon, A.; Si-Tahar, M.; Gosset, P. Th17 cytokines: Novel potential therapeutic targets for COPD pathogenesis and exacerbations. Eur. Respir. J. 2017, 50, 1602434. [Google Scholar] [CrossRef] [PubMed]
- Halwani, R.; Sultana, A.; Vazquez-Tello, A.; Jamhawi, A.; Al-Masri, A.A.; Al-Muhsen, S. Th-17 regulatory cytokines IL-21, IL-23, and IL-6 enhance neutrophil production of IL-17 cytokines during asthma. J. Asthma 2017, 54, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Al-Alwan, L.; Audusseau, S.; Chouiali, F.; Carlevaro-Fita, J.; Iwakura, Y.; Baglole, C.J.; Eidelman, D.H.; Hamid, Q. Genetic deletion of IL-17A reduces cigarette smoke-induced inflammation and alveolar type II cell apoptosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L132–L143. [Google Scholar] [CrossRef] [PubMed]
- Menendez, D.; Shatz, M.; Azzam, K.; Garantziotis, S.; Fessler, M.B.; Resnick, M.A. The Toll-like receptor gene family is integrated into human DNA damage and p53 networks. PLoS Genet. 2011, 7, e1001360. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef]
- American Diabetes, A. Diagnosis and classification of diabetes mellitus. Diabetes Care 2014, 37, S81–S90. [Google Scholar] [CrossRef]
- Prattichizzo, F.; De Nigris, V.; La Sala, L.; Procopio, A.D.; Olivieri, F.; Ceriello, A. “Inflammaging” as a Druggable Target: A Senescence-Associated Secretory Phenotype-Centered View of Type 2 Diabetes. Oxid. Med. Cell. Longev. 2016. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging role of NF-kappaB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell. Signal. 2012, 24, 835–845. [Google Scholar] [CrossRef]
- Monnier, L.; Mas, E.; Ginet, C.; Michel, F.; Villon, L.; Cristol, J.P.; Colette, C. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic Hyperglycemia in patients with type 2 diabetes. JAMA J. Am. Med. Assoc. 2006, 295, 1681–1687. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Esposito, K.; Piconi, L.; Ihnat, M.A.; Thorpe, J.E.; Testa, R.; Boemi, M.; Giugliano, D. Oscillating glucose is more deleterious to endothelial function and oxidative stress than mean glucose in normal and type 2 diabetic patients. Diabetes 2008, 57, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.M.; Coughlan, M.T.; Cooper, M.E. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes 2008, 57, 1446–1454. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Neogi, T.; Silman, A.J. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative (vol 69, pg 1580, 2010). Ann. Rheum. Dis. 2010, 69, 1892. [Google Scholar] [CrossRef] [PubMed]
- Helmick, C.G.; Felson, D.T.; Lawrence, R.C.; Gabriel, S.; Hirsch, R.; Kwoh, C.K.; Liang, M.H.; Kremers, H.M.; Mayes, M.D.; Merkel, P.A.; et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Arthritis Rheum. 2008, 58, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, S.E.; Crowson, C.S.; Kremers, H.M.; Doran, M.F.; Turesson, C.; O’Fallon, W.M.; Matteson, E.L. Survival in rheumatoid arthritis—A population-based analysis of trends over 40 years. Arthritis Rheum. 2003, 48, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Scarpaci, S.; Frasca, D.; Barattini, P.; Guidi, L.; Doria, G. DNA damage recognition and repair capacities in human naive and memory T cells from peripheral blood of young and elderly subjects. Mech. Ageing Dev. 2003, 124, 517–524. Available online: http://www.ncbi.nlm.nih.gov/pubmed/12714261 (accessed on 11 January 2019). [CrossRef]
- Valmori, D.; Merlo, A.; Souleimanian, N.E.; Hesdorffer, C.S.; Ayyoub, M. A peripheral circulating compartment of natural naive CD4(+) Tregs. J. Clin. Investig. 2005, 115, 1953–1962. [Google Scholar] [CrossRef]
- Mateen, S.; Moin, S.; Khan, A.Q.; Zafar, A.; Fatima, N. Increased Reactive Oxygen Species Formation and Oxidative Stress in Rheumatoid Arthritis. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Vasanthi, P.; Nalini, G.; Rajasekhar, G. Status of oxidative stress in rheumatoid arthritis. Int. J. Rheum. Dis. 2009, 12, 29–33. [Google Scholar] [CrossRef]
- Jaswal, S.; Mehta, H.C.; Sood, A.K.; Kaur, J. Antioxidant status in rheumatoid arthritis and role of antioxidant therapy. Clin. Chim. Acta 2003, 338, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Lo Gullo, A.; Mandraffino, G.; Sardo, M.A.; D’Ascola, A.; Mamone, F.; Loddo, S.; Alibrandi, A.; Imbalzano, E.; Mandraffino, R.; Mormina, E.; et al. Circulating progenitor cells in rheumatoid arthritis: Association with inflammation and oxidative stress. Scand. J. Rheumatol. 2014, 43, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Elshabrawy, H.A.; Chen, Z.L.; Volin, M.V.; Ravella, S.; Virupannavar, S.; Shahrara, S. The pathogenic role of angiogenesis in rheumatoid arthritis. Angiogenesis 2015, 18, 433–448. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2017, 8, 1960. [Google Scholar] [CrossRef] [PubMed]
- Lal, H.; Cunningham, A.L.; Godeaux, O.; Chlibek, R.; Diez-Domingo, J.; Hwang, S.J.; Levin, M.J.; McElhaney, J.E.; Poder, A.; Puig-Barbera, J.; et al. Efficacy of an adjuvanted herpes zoster subunit vaccine in older adults. N. Engl. J. Med. 2015, 372, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Zhang, X.; Zheng, S.; Khanabdali, R.; Kalionis, B.; Wu, J.; Wan, W.; Tai, X. An Update on Inflamm-Aging: Mechanisms, Prevention, and Treatment. J. Immunol. Res. 2016, 2016, 8426874. [Google Scholar] [CrossRef]
- Pae, M.; Meydani, S.N.; Wu, D. The role of nutrition in enhancing immunity in aging. Aging Dis. 2012, 3, 91–129. Available online: http://www.ncbi.nlm.nih.gov/pubmed/22500273 (accessed on 10 March 2019).
- Alcain, F.J.; Villalba, J.M. Sirtuin activators. Expert Opin. Pat. 2009, 19, 403–414. [Google Scholar] [CrossRef]
- Conti, V.; Corbi, G.; Manzo, V.; Pelaia, G.; Filippelli, A.; Vatrella, A. Sirtuin 1 and aging theory for chronic obstructive pulmonary disease. Anal. Cell. Pathol. (Amst.) 2015, 2015, 897327. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Hardeland, R. Inflammaging, Metabolic Syndrome and Melatonin: A Call for Treatment Studies. Neuroendocrinology 2017, 104, 382–397. [Google Scholar] [CrossRef]
- Wang, J.; Ye, F.; Cheng, X.; Zhang, X.; Liu, F.; Liu, G.; Ni, M.; Qiao, S.; Zhou, W.; Zhang, Y. The Effects of LW-AFC on Intestinal Microbiome in Senescence-Accelerated Mouse Prone 8 Strain, a Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2016, 53, 907–919. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cheng, X.; Zhang, X.; Cheng, J.; Xu, Y.; Zeng, J.; Zhou, W.; Zhang, Y. The anti-aging effects of LW-AFC via correcting immune dysfunctions in senescence accelerated mouse resistant 1 (SAMR1) strain. Oncotarget 2016, 7, 26949–26965. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Witkowski, J.M.; Olivieri, F.; Larbi, A. The integration of inflammaging in age-related diseases. Semin. Immunol. 2018, 40, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.O.; Govind, S.; Aspinall, R. Reversing T cell immunosenescence: Why, who, and how. Age (Dordr.) 2013, 35, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Kovtonyuk, L.V.; Fritsch, K.; Feng, X.; Manz, M.G.; Takizawa, H. Inflamm-Aging of Hematopoiesis, Hematopoietic Stem Cells, and the Bone Marrow Microenvironment. Front. Immunol. 2016, 7, 502. [Google Scholar] [CrossRef] [PubMed]
- Wahlestedt, M.; Norddahl, G.L.; Sten, G.; Ugale, A.; Frisk, M.A.; Mattsson, R.; Deierborg, T.; Sigvardsson, M.; Bryder, D. An epigenetic component of hematopoietic stem cell aging amenable to reprogramming into a young state. Blood 2013, 121, 4257–4264. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, Y.; Liu, Y.; Zheng, P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci. Signal. 2009, 2, ra75. [Google Scholar] [CrossRef]
- Guidi, N.; Geiger, H. Rejuvenation of aged hematopoietic stem cells. Semin. Hematol. 2017, 54, 51–55. [Google Scholar] [CrossRef]
- Rojas, M.; Mora, A.L.; Kapetanaki, M.; Weathington, N.; Gladwin, M.; Eickelberg, O. Aging and Lung Disease. Clinical Impact and Cellular and Molecular Pathways. Ann. Am. Thorac. Soc. 2015, 12, S222–S2227. [Google Scholar] [CrossRef]
- Szarc vel Szic, K.; Declerck, K.; Vidakovic, M.; Vanden Berghe, W. From inflammaging to healthy aging by dietary lifestyle choices: Is epigenetics the key to personalized nutrition? Clin. Epig. 2015, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.; Rao, S. Mechanisms Underlying T Cell Immunosenescence: Aging and Cytomegalovirus Infection. Front. Microbiol. 2016, 7, 2111. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.Y.; Lee, C.; Longo, V.D. Nutrition and fasting mimicking diets in the prevention and treatment of autoimmune diseases and immunosenescence. Mol. Cell. Endocrinol. 2017, 455, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.P.; Son, K.H.; Chang, H.W.; Kang, S.S. Anti-inflammatory plant flavonoids and cellular action mechanisms. J. Pharm. Sci. 2004, 96, 229–245. Available online: http://www.ncbi.nlm.nih.gov/pubmed/15539763 (accessed on 25 November 2018). [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Causative Factors | Roles of Inflammaging and ROS | Potential Treatments | |
|---|---|---|---|---|
| CVD |
| |||
| Cancer | Oxidative stress [40] | |||
| Neurodegenerative Diseases | Aging | |||
| AD | ||||
| PD |
|
| ||
| COPD |
| |||
| Diabetes |
| |||
| RA |
|
| ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuo, L.; Prather, E.R.; Stetskiv, M.; Garrison, D.E.; Meade, J.R.; Peace, T.I.; Zhou, T. Inflammaging and Oxidative Stress in Human Diseases: From Molecular Mechanisms to Novel Treatments. Int. J. Mol. Sci. 2019, 20, 4472. https://doi.org/10.3390/ijms20184472
Zuo L, Prather ER, Stetskiv M, Garrison DE, Meade JR, Peace TI, Zhou T. Inflammaging and Oxidative Stress in Human Diseases: From Molecular Mechanisms to Novel Treatments. International Journal of Molecular Sciences. 2019; 20(18):4472. https://doi.org/10.3390/ijms20184472
Chicago/Turabian StyleZuo, Li, Evan R. Prather, Mykola Stetskiv, Davis E. Garrison, James R. Meade, Timotheus I. Peace, and Tingyang Zhou. 2019. "Inflammaging and Oxidative Stress in Human Diseases: From Molecular Mechanisms to Novel Treatments" International Journal of Molecular Sciences 20, no. 18: 4472. https://doi.org/10.3390/ijms20184472
APA StyleZuo, L., Prather, E. R., Stetskiv, M., Garrison, D. E., Meade, J. R., Peace, T. I., & Zhou, T. (2019). Inflammaging and Oxidative Stress in Human Diseases: From Molecular Mechanisms to Novel Treatments. International Journal of Molecular Sciences, 20(18), 4472. https://doi.org/10.3390/ijms20184472