Mitochondrial Carriers for Aspartate, Glutamate and Other Amino Acids: A Review


 and
and 
Abstract
| Contents | |
| 1. Introduction | 2 |
| 2. Mitochondrial Carriers for Acidic Amino Acids | 7 |
| 2.1. The Human and Yeast Aspartate-glutamate Carriers | 7 |
| 2.2. Arabidopsis AtUCP1 and AtUCP2 Transport Aspartate and Glutamate | 9 |
| 2.3. The Human Glutamate Carriers | 10 |
| 2.4. Arabidopsis BOU and Yeast Ymc2p Transport Glutamate | 11 |
| 2.5. Human UCP2 Transports Aspartate | 12 |
| 3. Mitochondrial Carriers for Basic Amino Acids | 13 |
| 3.1. The Human and Yeast Ornithine Carriers | 13 |
| 3.2. Human SLC25A29 Transports Basic Amino Acids with a Preference for Lysine and Arginine | 15 |
| 3.3. Arabidopsis BAC1 and BAC2 | 15 |
| 4. Mitochondrial Carriers for Neutral Amino Acids | 16 |
| 4.1. The Human and Yeast Glycine Carriers | 16 |
| 5. Future Perspectives | 17 |
1. Introduction
2. Mitochondrial Carriers for Acidic Amino Acids
2.1. The Human and Yeast Aspartate-glutamate Carriers
2.2. Arabidopsis AtUCP1 and AtUCP2 Transport Aspartate and Glutamate
2.3. The Human Glutamate Carriers
2.4. Arabidopsis BOU and Yeast Ymc2p Transport Glutamate
2.5. Human UCP2 Transports Aspartate
3. Mitochondrial Carriers for Basic Amino Acids
3.1. The Human and Yeast Ornithine Carriers
3.2. Human SLC25A29 Transports Basic Amino Acids with a Preference for Lysine and Arginine
3.3. Arabidopsis BAC1 and BAC2
4. Mitochondrial Carriers for Neutral Amino Acids
4.1. The Human and Yeast Glycine Carriers
5. Future Perspectives
Note added during revision
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAC | ADP/ATP carrier, adenine nucleotide carrier or translocase |
| ADMA | asymmetric dimethylarginine |
| AGC | aspartate glutamate carrier |
| BAC | basic amino acid carrier |
| GC | glutamate carrier |
| EPRA | expression-purification-reconstitution-assay |
| MC | mitochondrial carrier |
| OGC | 2-oxoglutarate carrier |
| ORC | ornithine carrier |
| TCA | tricarboxylic acid |
| UCP | uncoupling protein |
References
- Krämer, R.; Palmieri, F. Metabolite carriers in mitochondria. In Molecular Mechanisms in Bioenergetics; Ernster, L., Ed.; New Comprehensive Biochemistry; Elsevier: Amsterdam, the Netherlands, 1992; Volume 23, pp. 359–384. [Google Scholar]
- Palmieri, F. The mitochondrial transporter family SLC25: Identification, properties and physiopathology. Mol. Asp. Med. 2013, 34, 465–484. [Google Scholar] [CrossRef] [PubMed]
- Saraste, M.; Walker, J.E. Internal sequence repeats and the path of polypeptide in mitochondrial ADP/ATP translocase. FEBS Lett. 1982, 144, 250–254. [Google Scholar] [CrossRef]
- Palmieri, F. Mitochondrial carrier proteins. FEBS Lett. 1994, 346, 48–54. [Google Scholar] [CrossRef]
- Palmieri, F.; Pierri, C.L. Mitochondrial metabolite transport. Essays Biochem. 2010, 47, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F.; Pierri, C.L.; De Grassi, A.; Nunes-Nesi, A.; Fernie, A.R. Evolution, structure and function of mitochondrial carriers: A review with new insights. Plant J. 2011, 66, 161–181. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F.; Pierri, C.L. Structure and function of mitochondrial carriers - role of the transmembrane helix P and G residues in the gating and transport mechanism. FEBS Lett. 2010, 584, 1931–1939. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F.; Monné, M. Discoveries, metabolic roles and diseases of mitochondrial carriers: A review. Biochim. Biophys. Acta 2016, 1863, 2362–2378. [Google Scholar] [CrossRef]
- Fiermonte, G.; Walker, J.E.; Palmieri, F. Abundant bacterial expression and reconstitution of an intrinsic membrane-transport protein from bovine mitochondria. Biochem. J. 1993, 294, 293–299. [Google Scholar] [CrossRef]
- Palmieri, F.; Klingenberg, M. Direct methods for measuring metabolite transport and distribution in mitochondria. Methods Enzymol. 1979, 56, 279–301. [Google Scholar]
- Palmieri, F.; Indiveri, C.; Bisaccia, F.; Iacobazzi, V. Mitochondrial metabolite carrier proteins: Purification, reconstitution, and transport studies. Methods Enzymol. 1995, 260, 349–369. [Google Scholar]
- Monné, M.; Palmieri, F. Antiporters of the mitochondrial carrier family. Curr. Top. Membr. 2014, 73, 289–320. [Google Scholar] [PubMed]
- Pebay-Peyroula, E.; Dahout-Gonzalez, C.; Kahn, R.; Trézéguet, V.; Lauquin, G.J.-M.; Brandolin, G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 2003, 426, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Ruprecht, J.J.; Hellawell, A.M.; Harding, M.; Crichton, P.G.; McCoy, A.J.; Kunji, E.R.S. Structures of yeast mitochondrial ADP/ATP carriers support a domain-based alternating-access transport mechanism. Proc. Natl. Acad. Sci. USA 2014, 111, E426–E434. [Google Scholar] [CrossRef] [PubMed]
- Ruprecht, J.J.; King, M.S.; Zögg, T.; Aleksandrova, A.A.; Pardon, E.; Crichton, P.G.; Steyaert, J.; Kunji, E.R.S. The Molecular Mechanism of Transport by the Mitochondrial ADP/ATP Carrier. Cell 2019, 176, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Klingenberg, M. The ADP, ATP shuttle of the mitochondrion. Trends Biochem. Sci. 1979, 4, 249–252. [Google Scholar] [CrossRef]
- Klingenberg, M. The ADP and ATP transport in mitochondria and its carrier. Biochim. Biophys. Acta 2008, 1778, 1978–2021. [Google Scholar] [CrossRef] [PubMed]
- Klingenberg, M. Ligand-protein interaction in biomembrane carriers. The induced transition fit of transport catalysis. Biochemistry 2005, 44, 8563–8570. [Google Scholar] [CrossRef]
- Robinson, A.J.; Kunji, E.R.S. Mitochondrial carriers in the cytoplasmic state have a common substrate binding site. Proc. Natl. Acad. Sci. USA 2006, 103, 2617–2622. [Google Scholar] [CrossRef]
- Marobbio, C.M.T.; Giannuzzi, G.; Paradies, E.; Pierri, C.L.; Palmieri, F. α-Isopropylmalate, a leucine biosynthesis intermediate in yeast, is transported by the mitochondrial oxalacetate carrier. J. Biol. Chem. 2008, 283, 28445–28553. [Google Scholar] [CrossRef]
- Indiveri, C.; Tonazzi, A.; Palmieri, F. The reconstituted carnitine carrier from rat liver mitochondria: Evidence for a transport mechanism different from that of the other mitochondrial translocators. Biochim. Biophys. Acta 1994, 1189, 65–73. [Google Scholar] [CrossRef]
- Palmieri, L.; Pardo, B.; Lasorsa, F.M.; del Arco, A.; Kobayashi, K.; Iijima, M.; Runswick, M.J.; Walker, J.E.; Saheki, T.; Satrústegui, J.; et al. Citrin and aralar1 are Ca(2+)-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 2001, 20, 5060–5069. [Google Scholar] [CrossRef] [PubMed]
- Lasorsa, F.M.; Pinton, P.; Palmieri, L.; Fiermonte, G.; Rizzuto, R.; Palmieri, F. Recombinant expression of the Ca(2+)-sensitive aspartate/glutamate carrier increases mitochondrial ATP production in agonist-stimulated Chinese hamster ovary cells. J. Biol. Chem. 2003, 278, 38686–38692. [Google Scholar] [CrossRef] [PubMed]
- Thangaratnarajah, C.; Ruprecht, J.J.; Kunji, E.R.S. Calcium-induced conformational changes of the regulatory domain of human mitochondrial aspartate/glutamate carriers. Nat. Commun. 2014, 5, 5491. [Google Scholar] [CrossRef] [PubMed]
- del Arco, A.; Satrústegui, J. Molecular cloning of Aralar, a new member of the mitochondrial carrier superfamily that binds calcium and is present in human muscle and brain. J. Biol. Chem. 1998, 273, 23327–23334. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Sinasac, D.S.; Iijima, M.; Boright, A.P.; Begum, L.; Lee, J.R.; Yasuda, T.; Ikeda, S.; Hirano, R.; Terazono, H.; et al. The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein. Nat. Genet. 1999, 22, 159–163. [Google Scholar] [CrossRef] [PubMed]
- del Arco, A.; Agudo, M.; Satrústegui, J. Characterization of a second member of the subfamily of calcium-binding mitochondrial carriers expressed in human non-excitable tissues. Biochem. J. 2000, 345, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Iijima, M.; Jalil, A.; Begum, L.; Yasuda, T.; Yamaguchi, N.; Xian Li, M.; Kawada, N.; Endou, H.; Kobayashi, K.; Saheki, T. Pathogenesis of adult-onset type II citrullinemia caused by deficiency of citrin, a mitochondrial solute carrier protein: Tissue and subcellular localization of citrin. Adv. Enzyme Regul. 2001, 41, 325–342. [Google Scholar] [CrossRef]
- Indiveri, C.; Krämer, R.; Palmieri, F. Reconstitution of the malate/aspartate shuttle from mitochondria. J. Biol. Chem. 1987, 262, 15979–15983. [Google Scholar] [PubMed]
- Palmieri, F. The mitochondrial transporter family (SLC25): Physiological and pathological implications. Pflüg. Arch. 2004, 447, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Wibom, R.; Lasorsa, F.M.; Töhönen, V.; Barbaro, M.; Sterky, F.H.; Kucinski, T.; Naess, K.; Jonsson, M.; Pierri, C.L.; Palmieri, F.; et al. AGC1 deficiency associated with global cerebral hypomyelination. N. Engl. J. Med. 2009, 361, 489–495. [Google Scholar] [CrossRef]
- Falk, M.J.; Li, D.; Gai, X.; McCormick, E.; Place, E.; Lasorsa, F.M.; Otieno, F.G.; Hou, C.; Kim, C.E.; Abdel-Magid, N.; et al. AGC1 Deficiency Causes Infantile Epilepsy, Abnormal Myelination, and Reduced N-Acetylaspartate. JIMD Rep. 2014, 14, 77–85. [Google Scholar] [PubMed]
- Profilo, E.; Peña-Altamira, L.E.; Corricelli, M.; Castegna, A.; Danese, A.; Agrimi, G.; Petralla, S.; Giannuzzi, G.; Porcelli, V.; Sbano, L.; et al. Down-regulation of the mitochondrial aspartate-glutamate carrier isoform 1 AGC1 inhibits proliferation and N-acetylaspartate synthesis in Neuro2A cells. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1422–1435. [Google Scholar] [CrossRef] [PubMed]
- Jalil, M.A.; Begum, L.; Contreras, L.; Pardo, B.; Iijima, M.; Li, M.X.; Ramos, M.; Marmol, P.; Horiuchi, M.; Shimotsu, K.; et al. Reduced N-acetylaspartate levels in mice lacking aralar, a brain- and muscle-type mitochondrial aspartate-glutamate carrier. J. Biol. Chem. 2005, 280, 31333–31339. [Google Scholar] [CrossRef] [PubMed]
- Petralla, S.; Peña-Altamira, L.E.; Poeta, E.; Massenzio, F.; Virgili, M.; Barile, S.N.; Sbano, L.; Profilo, E.; Corricelli, M.; Danese, A.; et al. Deficiency of mitochondrial aspartate/glutamate carrier 1 leads to oligodendrocyte precursor cell proliferation defects both in vitro and in vivo. Int. J. Mol. Sci. 2019, 20, 4486. [Google Scholar] [CrossRef]
- Palmieri, F. Mitochondrial transporters of the SLC25 family and associated diseases: A review. J. Inherit. Metab. Dis. 2014, 37, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Saheki, T.; Kobayashi, K. Mitochondrial aspartate glutamate carrier (citrin) deficiency as the cause of adult-onset type II citrullinemia (CTLN2) and idiopathic neonatal hepatitis (NICCD). J. Hum. Genet. 2002, 47, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Fiermonte, G.; Parisi, G.; Martinelli, D.; De Leonardis, F.; Torre, G.; Pierri, C.L.; Saccari, A.; Lasorsa, F.M.; Vozza, A.; Palmieri, F.; et al. A new Caucasian case of neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD): A clinical, molecular, and functional study. Mol. Genet. Metab. 2011, 104, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Fiermonte, G.; Soon, D.; Chaudhuri, A.; Paradies, E.; Lee, P.J.; Krywawych, S.; Palmieri, F.; Lachmann, R.H. An adult with type 2 citrullinemia presenting in Europe. N. Engl. J. Med. 2008, 358, 1408–1409. [Google Scholar] [CrossRef]
- Woo, H.I.; Park, H.-D.; Lee, Y.-W. Molecular genetics of citrullinemia types I and II. Clin. Chim. Acta 2014, 431, 1–8. [Google Scholar] [CrossRef]
- Begum, L.; Jalil, M.A.; Kobayashi, K.; Iijima, M.; Li, M.X.; Yasuda, T.; Horiuchi, M.; del Arco, A.; Satrústegui, J.; Saheki, T. Expression of three mitochondrial solute carriers, citrin, aralar1 and ornithine transporter, in relation to urea cycle in mice. Biochim. Biophys. Acta 2002, 1574, 283–292. [Google Scholar] [CrossRef]
- Convertini, P.; Todisco, S.; De Santis, F.; Pappalardo, I.; Iacobazzi, D.; Castiglione Morelli, M.A.; Fondufe-Mittendorf, Y.N.; Martelli, G.; Palmieri, F.; Infantino, V. Transcriptional Regulation Factors of the Human Mitochondrial Aspartate/Glutamate Carrier Gene, Isoform 2 (SLC25A13): USF1 as Basal Factor and FOXA2 as Activator in Liver Cells. Int. J. Mol. Sci. 2019, 20, 1888. [Google Scholar] [CrossRef]
- Menga, A.; Iacobazzi, V.; Infantino, V.; Avantaggiati, M.L.; Palmieri, F. The mitochondrial aspartate/glutamate carrier isoform 1 gene expression is regulated by CREB in neuronal cells. Int. J. Biochem. Cell Biol. 2015, 60, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Infantino, V.; Dituri, F.; Convertini, P.; Santarsiero, A.; Palmieri, F.; Todisco, S.; Mancarella, S.; Giannelli, G.; Iacobazzi, V. Epigenetic upregulation and functional role of the mitochondrial aspartate/glutamate carrier isoform 1 in hepatocellular carcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Casimir, M.; Rubi, B.; Frigerio, F.; Chaffard, G.; Maechler, P. Silencing of the mitochondrial NADH shuttle component aspartate-glutamate carrier AGC1/Aralar1 in INS-1E cells and rat islets. Biochem. J. 2009, 424, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Cavero, S.; Vozza, A.; Del Arco, A.; Palmieri, L.; Villa, A.; Blanco, E.; Runswick, M.J.; Walker, J.E.; Cerdán, S.; Palmieri, F.; et al. Identification and metabolic role of the mitochondrial aspartate-glutamate transporter in Saccharomyces cerevisiae. Mol. Microbiol. 2003, 50, 1257–1269. [Google Scholar] [CrossRef] [PubMed]
- Scarcia, P.; Palmieri, L.; Agrimi, G.; Palmieri, F.; Rottensteiner, H. Three mitochondrial transporters of Saccharomyces cerevisiae are essential for ammonium fixation and lysine biosynthesis in synthetic minimal medium. Mol. Genet. Metab. 2017, 122, 54–60. [Google Scholar] [CrossRef]
- Scarcia, P.; Agrimi, G.; Germinario, L.; Ibrahim, A.; Rottensteiner, H.; Palmieri, F.; Palmieri, L. In Saccharomyces cerevisiae grown in synthetic minimal medium supplemented with non-fermentable carbon sources glutamate is synthesized within mitochondria. Rendiconti Lincei 2018, 29, 483–490. [Google Scholar] [CrossRef]
- Monné, M.; Daddabbo, L.; Gagneul, D.; Obata, T.; Hielscher, B.; Palmieri, L.; Miniero, D.V.; Fernie, A.R.; Weber, A.P.M.; Palmieri, F. Uncoupling proteins 1 and 2 (UCP1 and UCP2) from Arabidopsis thaliana are mitochondrial transporters of aspartate, glutamate, and dicarboxylates. J. Biol. Chem. 2018, 293, 4213–4227. [Google Scholar] [CrossRef]
- Sweetlove, L.J.; Lytovchenko, A.; Morgan, M.; Nunes-Nesi, A.; Taylor, N.L.; Baxter, C.J.; Eickmeier, I.; Fernie, A.R. Mitochondrial uncoupling protein is required for efficient photosynthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 19587–19592. [Google Scholar] [CrossRef]
- Fiermonte, G.; Palmieri, L.; Todisco, S.; Agrimi, G.; Palmieri, F.; Walker, J.E. Identification of the mitochondrial glutamate transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution of two human isoforms. J. Biol. Chem. 2002, 277, 19289–19294. [Google Scholar] [CrossRef]
- Lunetti, P.; Cappello, A.R.; Marsano, R.M.; Pierri, C.L.; Carrisi, C.; Martello, E.; Caggese, C.; Dolce, V.; Capobianco, L. Mitochondrial glutamate carriers from Drosophila melanogaster: Biochemical, evolutionary and modeling studies. Biochim. Biophys. Acta 2013, 1827, 1245–1255. [Google Scholar] [CrossRef]
- Brosnan, J.T. Glutamate, at the interface between amino acid and carbohydrate metabolism. J. Nutr. 2000, 130, 988S–990S. [Google Scholar] [CrossRef] [PubMed]
- Casimir, M.; Lasorsa, F.M.; Rubi, B.; Caille, D.; Palmieri, F.; Meda, P.; Maechler, P. Mitochondrial glutamate carrier GC1 as a newly identified player in the control of glucose-stimulated insulin secretion. J. Biol. Chem. 2009, 284, 25004–25014. [Google Scholar] [CrossRef] [PubMed]
- Molinari, F.; Raas-Rothschild, A.; Rio, M.; Fiermonte, G.; Encha-Razavi, F.; Palmieri, L.; Palmieri, F.; Ben-Neriah, Z.; Kadhom, N.; Vekemans, M.; et al. Impaired mitochondrial glutamate transport in autosomal recessive neonatal myoclonic epilepsy. Am. J. Hum. Genet. 2005, 76, 334–339. [Google Scholar] [CrossRef]
- Molinari, F.; Kaminska, A.; Fiermonte, G.; Boddaert, N.; Raas-Rothschild, A.; Plouin, P.; Palmieri, L.; Brunelle, F.; Palmieri, F.; Dulac, O.; et al. Mutations in the mitochondrial glutamate carrier SLC25A22 in neonatal epileptic encephalopathy with suppression bursts. Clin. Genet. 2009, 76, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Poduri, A.; Heinzen, E.; Chitsazzadeh, V.; Lasorsa, F.; LaCoursiere, C.; Martin, E.; Yusakaitis, C.; Hill, R.; Elhosary, P.; Atabay, K.; et al. SLC25A22 is a novel gene for migrating partial seizures in infancy. Ann. Neurol. 2013, 76, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Lemattre, C.; Imbert-Bouteille, M.; Gatinois, V.; Benit, P.; Sanchez, E.; Guignard, T.; Tran Mau-Them, F.; Haquet, E.; Rivier, F.; Carme, E.; et al. Report on three additional patients and genotype-phenotype correlation in SLC25A22-related disorders group. Eur. J. Hum. Genet. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Goubert, E.; Mircheva, Y.; Lasorsa, F.M.; Melon, C.; Profilo, E.; Sutera, J.; Becq, H.; Palmieri, F.; Palmieri, L.; Aniksztejn, L.; et al. Inhibition of the Mitochondrial Glutamate Carrier SLC25A22 in Astrocytes Leads to Intracellular Glutamate Accumulation. Front. Cell. Neurosci. 2017, 11, 149. [Google Scholar] [CrossRef]
- Porcelli, V.; Vozza, A.; Calcagnile, V.; Gorgoglione, R.; Arrigoni, R.; Fontanesi, F.; Marobbio, C.M.T.; Castegna, A.; Palmieri, F.; Palmieri, L. Molecular identification and functional characterization of a novel glutamate transporter in yeast and plant mitochondria. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 1249–1258. [Google Scholar] [CrossRef]
- Trotter, P.J.; Adamson, A.L.; Ghrist, A.C.; Rowe, L.; Scott, L.R.; Sherman, M.P.; Stites, N.C.; Sun, Y.; Tawiah-Boateng, M.A.; Tibbetts, A.S.; et al. Mitochondrial transporters involved in oleic acid utilization and glutamate metabolism in yeast. Arch. Biochem. Biophys. 2005, 442, 21–32. [Google Scholar] [CrossRef]
- Hildebrandt, T.M.; Nunes Nesi, A.; Araújo, W.L.; Braun, H.-P. Amino Acid Catabolism in Plants. Mol. Plant 2015, 8, 1563–1579. [Google Scholar] [CrossRef]
- Eisenhut, M.; Planchais, S.; Cabassa, C.; Guivarc’h, A.; Justin, A.-M.; Taconnat, L.; Renou, J.-P.; Linka, M.; Gagneul, D.; Timm, S.; et al. Arabidopsis A BOUT DE SOUFFLE is a putative mitochondrial transporter involved in photorespiratory metabolism and is required for meristem growth at ambient CO₂ levels. Plant J. Cell Mol. Biol. 2013, 73, 836–849. [Google Scholar] [CrossRef] [PubMed]
- Klingenberg, M.; Winkler, E. The reconstituted isolated uncoupling protein is a membrane potential driven H+ translocator. EMBO J. 1985, 4, 3087–3092. [Google Scholar] [CrossRef]
- Nicholls, D.G. The physiological regulation of uncoupling proteins. Biochim. Biophys. Acta 2006, 1757, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, L.; Picault, N.; Arrigoni, R.; Besin, E.; Palmieri, F.; Hodges, M. Molecular identification of three Arabidopsis thaliana mitochondrial dicarboxylate carrier isoforms: Organ distribution, bacterial expression, reconstitution into liposomes and functional characterization. Biochem. J. 2008, 410, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Vozza, A.; Parisi, G.; De Leonardis, F.; Lasorsa, F.M.; Castegna, A.; Amorese, D.; Marmo, R.; Calcagnile, V.M.; Palmieri, L.; Ricquier, D.; et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, 960–965. [Google Scholar] [CrossRef] [PubMed]
- Gorgoglione, R.; Porcelli, V.; Santoro, A.; Daddabbo, L.; Vozza, A.; Monné, M.; Di Noia, M.A.; Palmieri, L.; Fiermonte, G.; Palmieri, F. The human uncoupling proteins 5 and 6 (UCP5/SLC25A14 and UCP6/SLC25A30) transport sulfur oxyanions, phosphate and dicarboxylates. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 724–733. [Google Scholar] [CrossRef]
- Pecqueur, C.; Bui, T.; Gelly, C.; Hauchard, J.; Barbot, C.; Bouillaud, F.; Ricquier, D.; Miroux, B.; Thompson, C.B. Uncoupling protein-2 controls proliferation by promoting fatty acid oxidation and limiting glycolysis-derived pyruvate utilization. FASEB J. 2007, 22, 9–18. [Google Scholar] [CrossRef]
- Fleury, C.; Neverova, M.; Collins, S.; Raimbault, S.; Champigny, O.; Levi-Meyrueis, C.; Bouillaud, F.; Seldin, M.F.; Surwit, R.S.; Ricquier, D.; et al. Uncoupling protein-2: A novel gene linked to obesity and hyperinsulinemia. Nat. Genet. 1997, 15, 269–272. [Google Scholar] [CrossRef]
- González-Barroso, M.M.; Giurgea, I.; Bouillaud, F.; Anedda, A.; Bellanné-Chantelot, C.; Hubert, L.; de Keyzer, Y.; de Lonlay, P.; Ricquier, D. Mutations in UCP2 in congenital hyperinsulinism reveal a role for regulation of insulin secretion. PLoS One 2008, 3, e3850. [Google Scholar] [CrossRef]
- Ferrara, C.T.; Boodhansingh, K.E.; Paradies, E.; Fiermonte, G.; Steinkrauss, L.J.; Topor, L.S.; Quintos, J.B.; Ganguly, A.; De Leon, D.D.; Palmieri, F.; et al. Novel Hypoglycemia Phenotype in Congenital Hyperinsulinism Due to Dominant Mutations of Uncoupling Protein 2. J. Clin. Endocrinol. Metab. 2017, 102, 942–949. [Google Scholar] [CrossRef]
- Esterbauer, H.; Schneitler, C.; Oberkofler, H.; Ebenbichler, C.; Paulweber, B.; Sandhofer, F.; Ladurner, G.; Hell, E.; Strosberg, A.D.; Patsch, J.R.; et al. A common polymorphism in the promoter of UCP2 is associated with decreased risk of obesity in middle-aged humans. Nat Genet. 2001, 28, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Dik, E.; Naamati, A.; Asraf, H.; Lehming, N.; Pines, O. Human Fumarate Hydratase Is Dual Localized by an Alternative Transcription Initiation Mechanism. Traffic Cph. Den. 2016, 17, 720–732. [Google Scholar] [CrossRef]
- Palmieri, L.; De Marco, V.; Iacobazzi, V.; Palmieri, F.; Runswick, M.J.; Walker, J.E. Identification of the yeast ARG-11 gene as a mitochondrial ornithine carrier involved in arginine biosynthesis. FEBS Lett. 1997, 410, 447–451. [Google Scholar] [CrossRef]
- Crabeel, M.; Soetens, O.; De Rijcke, M.; Pratiwi, R.; Pankiewicz, R. The ARG11 gene of Saccharomyces cerevisiae encodes a mitochondrial integral membrane protein required for arginine biosynthesis. J. Biol. Chem. 1996, 271, 25011–25018. [Google Scholar] [CrossRef] [PubMed]
- Fiermonte, G.; Dolce, V.; David, L.; Santorelli, F.M.; Dionisi-Vici, C.; Palmieri, F.; Walker, J.E. The mitochondrial ornithine transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution of two human isoforms. J. Biol. Chem. 2003, 278, 32778–32783. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, V.; Longo, A.; Palmieri, L.; Closs, E.I.; Palmieri, F. Asymmetric dimethylarginine is transported by the mitochondrial carrier SLC25A2. Amino Acids 2016, 48, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Monné, M.; Miniero, D.V.; Daddabbo, L.; Robinson, A.J.; Kunji, E.R.S.; Palmieri, F. Substrate specificity of the two mitochondrial ornithine carriers can be swapped by single mutation in substrate binding site. J. Biol. Chem. 2012, 287, 7925–7934. [Google Scholar] [CrossRef] [PubMed]
- Indiveri, C.; Tonazzi, A.; Palmieri, F. Identification and purification of the ornithine/citrulline carrier from rat liver mitochondria. Eur. J. Biochem. 1992, 207, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Indiveri, C.; Palmieri, L.; Palmieri, F. Kinetic characterization of the reconstituted ornithine carrier from rat liver mitochondria. Biochim. Biophys. Acta 1994, 1188, 293–301. [Google Scholar] [CrossRef]
- Indiveri, C.; Tonazzi, A.; Stipani, I.; Palmieri, F. The purified and reconstituted ornithine/citrulline carrier from rat liver mitochondria: Electrical nature and coupling of the exchange reaction with H+ translocation. Biochem. J. 1997, 327, 349–355. [Google Scholar] [CrossRef]
- Morris, S.M.; Kepka-Lenhart, D. Hormonal induction of hepatic mitochondrial ornithine/citrulline transporter mRNA. Biochem. Biophys. Res. Commun. 2002, 294, 749–752. [Google Scholar] [CrossRef]
- Wu, G.; Morris, S.M. Arginine metabolism: Nitric oxide and beyond. Biochem. J. 1998, 336, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Monné, M.; Miniero, D.V.; Daddabbo, L.; Palmieri, L.; Porcelli, V.; Palmieri, F. Mitochondrial transporters for ornithine and related amino acids: A review. Amino Acids 2015, 47, 1763–1777. [Google Scholar] [CrossRef] [PubMed]
- Kishita, Y.; Pajak, A.; Bolar, N.A.; Marobbio, C.M.T.; Maffezzini, C.; Miniero, D.V.; Monné, M.; Kohda, M.; Stranneheim, H.; Murayama, K.; et al. Intra-mitochondrial Methylation Deficiency Due to Mutations in SLC25A26. Am. J. Med. Genet. 2015, 97, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Marobbio, C.M.T.; Agrimi, G.; Lasorsa, F.M.; Palmieri, F. Identification and functional reconstitution of yeast mitochondrial carrier for S-adenosylmethionine. EMBO J. 2003, 22, 5975–5982. [Google Scholar] [CrossRef] [PubMed]
- Agrimi, G.; Di Noia, M.A.; Marobbio, C.M.T.; Fiermonte, G.; Lasorsa, F.M.; Palmieri, F. Identification of the human mitochondrial S-adenosylmethionine transporter: Bacterial expression, reconstitution, functional characterization and tissue distribution. Biochem. J. 2004, 379, 183–190. [Google Scholar] [CrossRef]
- Palmieri, L.; Arrigoni, R.; Blanco, E.; Carrari, F.; Zanor, M.I.; Studart-Guimaraes, C.; Fernie, A.R.; Palmieri, F. Molecular identification of an Arabidopsis S-adenosylmethionine transporter. Analysis of organ distribution, bacterial expression, reconstitution into liposomes, and functional characterization. Plant Physiol. 2006, 142, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Tessa, A.; Fiermonte, G.; Dionisi-Vici, C.; Paradies, E.; Baumgartner, M.R.; Chien, Y.-H.; Loguercio, C.; de Baulny, H.O.; Nassogne, M.-C.; Schiff, M.; et al. Identification of novel mutations in the SLC25A15 gene in hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome: A clinical, molecular, and functional study. Hum. Mutat. 2009, 30, 741–748. [Google Scholar] [CrossRef]
- Ersoy Tunali, N.; Marobbio, C.M.T.; Tiryakioğlu, N.O.; Punzi, G.; Saygılı, S.K.; Onal, H.; Palmieri, F. A novel mutation in the SLC25A15 gene in a Turkish patient with HHH syndrome: Functional analysis of the mutant protein. Mol Genet Metab 2014, 112, 25–29. [Google Scholar] [CrossRef]
- Marobbio, C.M.T.; Punzi, G.; Pierri, C.L.; Palmieri, L.; Calvello, R.; Panaro, M.A.; Palmieri, F. Pathogenic potential of SLC25A15 mutations assessed by transport assays and complementation of Saccharomyces cerevisiae ORT1 null mutant. Mol. Genet. Metab. 2015, 115, 27–32. [Google Scholar] [CrossRef]
- Martinelli, D.; Diodato, D.; Ponzi, E.; Monné, M.; Boenzi, S.; Bertini, E.; Fiermonte, G.; Dionisi-Vici, C. The hyperornithinemia-hyperammonemia-homocitrullinuria syndrome. Orphanet J. Rare Dis. 2015, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Sekoguchi, E.; Sato, N.; Yasui, A.; Fukada, S.; Nimura, Y.; Aburatani, H.; Ikeda, K.; Matsuura, A. A novel mitochondrial carnitine-acylcarnitine translocase induced by partial hepatectomy and fasting. J. Biol. Chem. 2003, 278, 38796–38802. [Google Scholar] [CrossRef] [PubMed]
- Camacho, J.A.; Rioseco-Camacho, N. The human and mouse SLC25A29 mitochondrial transporters rescue the deficient ornithine metabolism in fibroblasts of patients with the hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome. Pediatr. Res. 2009, 66, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, V.; Fiermonte, G.; Longo, A.; Palmieri, F. The human gene SLC25A29, of solute carrier family 25, encodes a mitochondrial transporter of basic amino acids. J. Biol. Chem. 2014, 289, 13374–13384. [Google Scholar] [CrossRef] [PubMed]
- Papes, F.; Kemper, E.L.; Cord-Neto, G.; Langone, F.; Arruda, P. Lysine degradation through the saccharopine pathway in mammals: Involvement of both bifunctional and monofunctional lysine-degrading enzymes in mouse. Biochem. J. 1999, 344, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, X.; Wang, M.; Chang, Y.; Zhang, F.; Ban, Z.; Tang, R.; Gan, Q.; Wu, S.; Guo, Y.; et al. The lysine catabolite saccharopine impairs development by disrupting mitochondrial homeostasis. J. Cell Biol. 2019, 218, 580–597. [Google Scholar] [CrossRef] [PubMed]
- Litvinova, L.; Atochin, D.N.; Fattakhov, N.; Vasilenko, M.; Zatolokin, P.; Kirienkova, E. Nitric oxide and mitochondria in metabolic syndrome. Front. Physiol. 2015, 6, 20. [Google Scholar] [CrossRef]
- Ghafourifar, P.; Cadenas, E. Mitochondrial nitric oxide synthase. Trends Pharmacol. Sci. 2005, 26, 190–195. [Google Scholar] [CrossRef]
- Hoyos, M.E.; Palmieri, L.; Wertin, T.; Arrigoni, R.; Polacco, J.C.; Palmieri, F. Identification of a mitochondrial transporter for basic amino acids in Arabidopsis thaliana by functional reconstitution into liposomes and complementation in yeast. Plant J. 2003, 33, 1027–1035. [Google Scholar] [CrossRef]
- Catoni, E.; Desimone, M.; Hilpert, M.; Wipf, D.; Kunze, R.; Schneider, A.; Flügge, U.-I.; Schumacher, K.; Frommer, W.B. Expression pattern of a nuclear encoded mitochondrial arginine-ornithine translocator gene from Arabidopsis. BMC Plant Biol. 2003, 3, 1. [Google Scholar] [CrossRef]
- Palmieri, L.; Todd, C.D.; Arrigoni, R.; Hoyos, M.E.; Santoro, A.; Polacco, J.C.; Palmieri, F. Arabidopsis mitochondria have two basic amino acid transporters with partially overlapping specificities and differential expression in seedling development. Biochim. Biophys. Acta 2006, 1757, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Planchais, S.; Cabassa, C.; Toka, I.; Justin, A.-M.; Renou, J.-P.; Savouré, A.; Carol, P. Basic amino acid carrier 2 gene expression modulates arginine and urea content and stress recovery in Arabidopsis leaves. Front. Plant Sci. 2014, 5, 330. [Google Scholar] [CrossRef] [PubMed]
- Toka, I.; Planchais, S.; Cabassa, C.; Justin, A.-M.; De Vos, D.; Richard, L.; Savouré, A.; Carol, P. Mutations in the hyperosmotic stress-responsive mitochondrial BASIC AMINO ACID CARRIER2 enhance proline accumulation in Arabidopsis. Plant Physiol. 2010, 152, 1851–1862. [Google Scholar] [CrossRef] [PubMed]
- Guernsey, D.L.; Jiang, H.; Campagna, D.R.; Evans, S.C.; Ferguson, M.; Kellogg, M.D.; Lachance, M.; Matsuoka, M.; Nightingale, M.; Rideout, A.; et al. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat. Genet. 2009, 41, 651–653. [Google Scholar] [CrossRef] [PubMed]
- Lunetti, P.; Damiano, F.; De Benedetto, G.; Siculella, L.; Pennetta, A.; Muto, L.; Paradies, E.; Marobbio, C.M.T.; Dolce, V.; Capobianco, L. Characterization of Human and Yeast Mitochondrial Glycine Carriers with Implications for Heme Biosynthesis and Anemia. J. Biol. Chem. 2016, 291, 19746–19759. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Murray, J.P.; Prykhozhij, S.V.; Dufay, J.N.; Steele, S.L.; Gaston, D.; Nasrallah, G.K.; Coombs, A.J.; Liwski, R.S.; Fernandez, C.V.; Berman, J.N.; et al. Glycine and Folate Ameliorate Models of Congenital Sideroblastic Anemia. PLoS Genet. 2016, 12, e1005783. [Google Scholar] [CrossRef] [PubMed]
- Kory, N.; Wyant, G.A.; Prakash, G.; Uit de Bos, J.; Bottanelli, F.; Pacold, M.E.; Chan, S.H.; Lewis, C.A.; Wang, T.; Keys, H.R.; et al. SFXN1 is a mitochondrial serine transporter required for one-carbon metabolism. Science 2018, 362, eaat9528. [Google Scholar] [CrossRef]
- Chacinska, A.; Koehler, C.M.; Milenkovic, D.; Lithgow, T.; Pfanner, N. Importing mitochondrial proteins: Machineries and mechanisms. Cell 2009, 138, 628–644. [Google Scholar] [CrossRef]




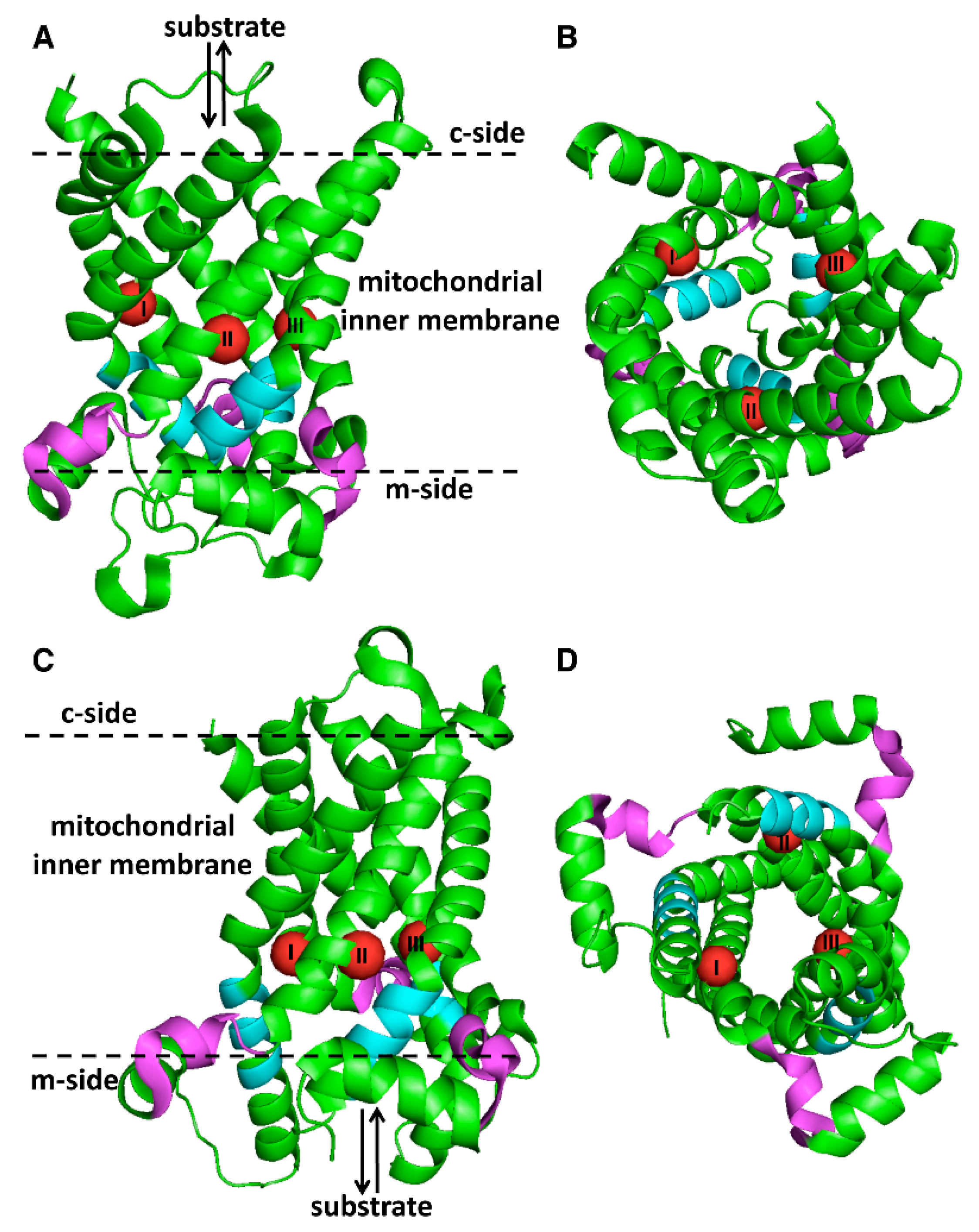{kind=link}
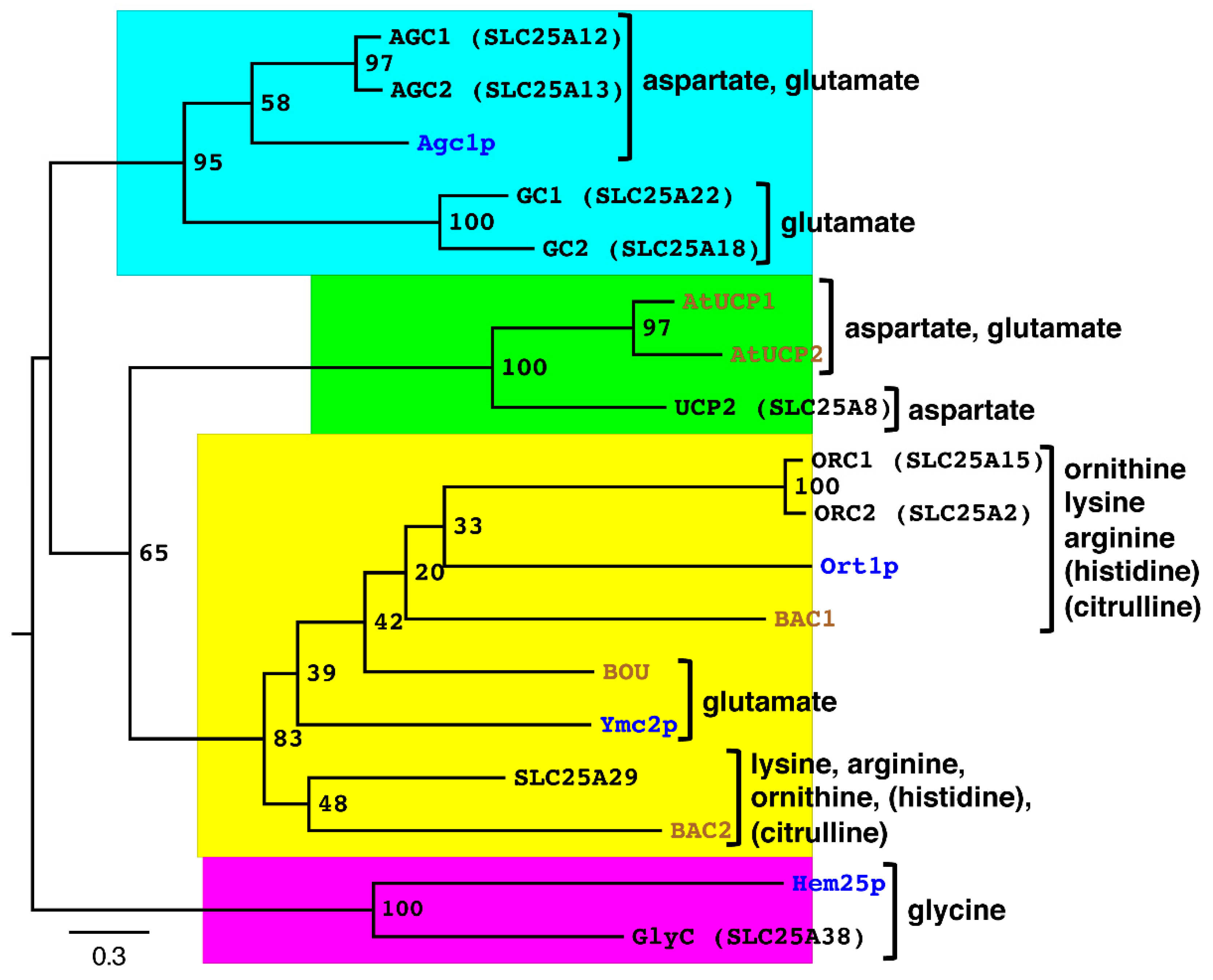{kind=link}
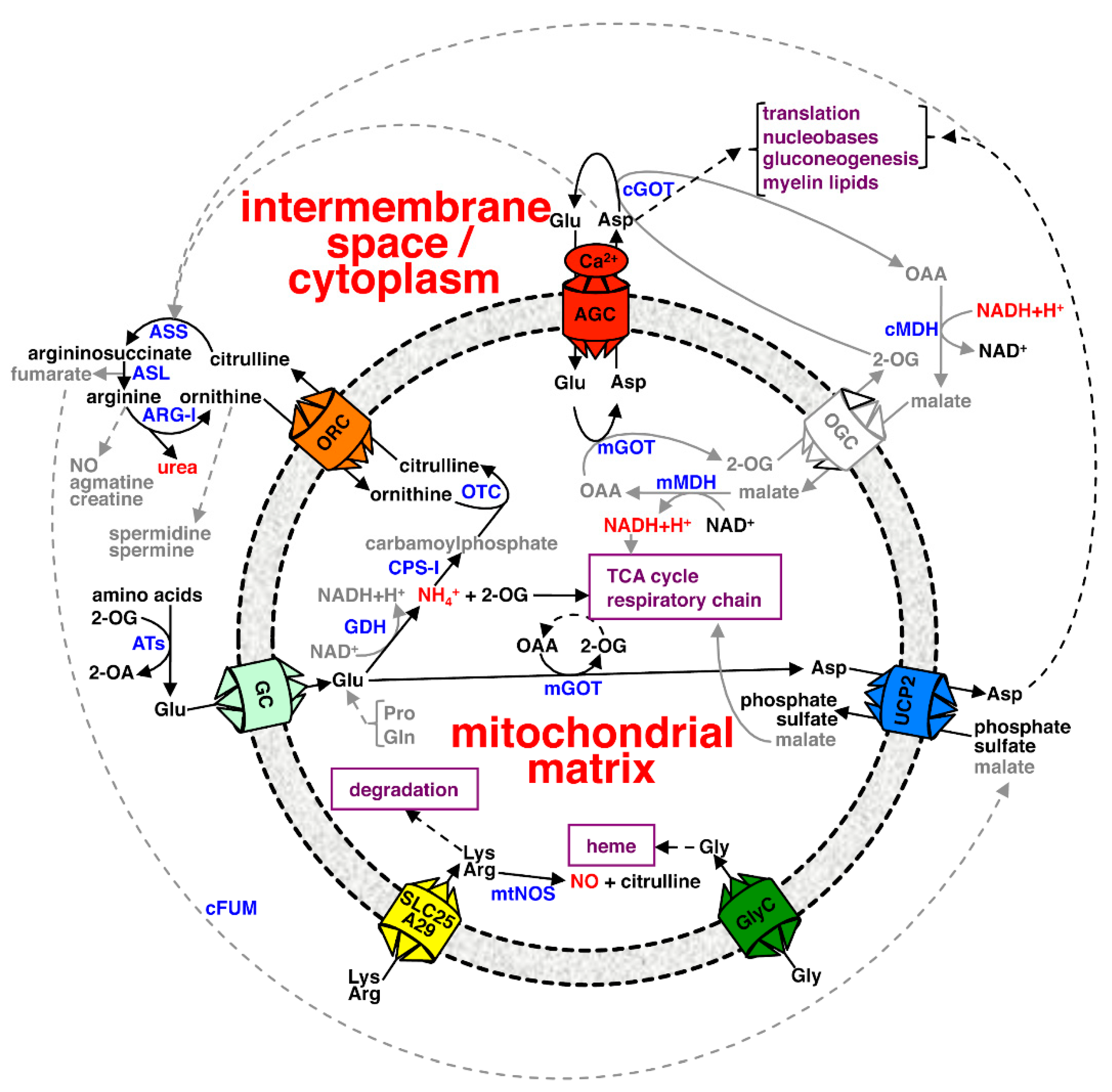{kind=link}
| AGC1 | AGC2 | Agc1p | GC1 | GC2 | AtUCP1 | AtUCP2 | UCP2 | Ymc2p | BOU | ORC1 | ORC2 | Ort1p | BAC1 | A29 | BAC2 | Hem25p | GlyC | |
| AGC1 | 88 | 57 | 46 | 46 | 28 | 28 | 27 | 24 | 32 | 27 | 27 | 27 | 25 | 30 | 33 | 21 | 25 | |
| AGC2 | 88 | 58 | 46 | 46 | 29 | 28 | 25 | 25 | 31 | 28 | 28 | 27 | 27 | 29 | 33 | 21 | 26 | |
| Agc1p | 57 | 58 | 42 | 39 | 24 | 26 | 26 | 28 | 32 | 27 | 28 | 28 | 28 | 33 | 30 | 27 | 29 | |
| GC1 | 46 | 46 | 42 | 68 | 25 | 24 | 25 | 21 | 29 | 26 | 27 | 26 | 24 | 29 | 29 | 18 | 26 | |
| GC2 | 46 | 46 | 39 | 68 | 25 | 25 | 25 | 24 | 27 | 25 | 25 | 24 | 26 | 28 | 31 | 23 | 26 | |
| AtUCP1 | 28 | 29 | 24 | 25 | 25 | 74 | 51 | 26 | 32 | 24 | 22 | 24 | 24 | 30 | 27 | 19 | 23 | |
| AtUCP2 | 28 | 28 | 26 | 24 | 25 | 74 | 45 | 28 | 30 | 23 | 21 | 24 | 27 | 31 | 25 | 20 | 23 | |
| UCP2 | 27 | 25 | 26 | 25 | 25 | 51 | 45 | 25 | 29 | 25 | 25 | 25 | 24 | 27 | 27 | 20 | 25 | |
| Ymc2p | 24 | 25 | 28 | 21 | 24 | 26 | 28 | 25 | 41 | 31 | 28 | 30 | 31 | 39 | 30 | 23 | 26 | |
| BOU | 32 | 31 | 32 | 29 | 27 | 32 | 30 | 29 | 41 | 40 | 38 | 33 | 36 | 38 | 36 | 19 | 26 | |
| ORC1 | 27 | 28 | 27 | 26 | 25 | 24 | 23 | 25 | 31 | 40 | 88 | 33 | 33 | 34 | 31 | 24 | 25 | |
| ORC2 | 27 | 28 | 28 | 27 | 25 | 22 | 21 | 25 | 28 | 38 | 88 | 31 | 34 | 34 | 28 | 21 | 25 | |
| Ort1p | 27 | 27 | 28 | 26 | 24 | 24 | 24 | 25 | 30 | 33 | 33 | 31 | 31 | 31 | 28 | 21 | 21 | |
| BAC1 | 25 | 27 | 28 | 24 | 26 | 24 | 27 | 24 | 31 | 36 | 33 | 34 | 31 | 34 | 29 | 20 | 26 | |
| SLC25A29 | 30 | 29 | 33 | 29 | 28 | 30 | 31 | 27 | 39 | 38 | 34 | 34 | 31 | 34 | 40 | 19 | 27 | |
| BAC2 | 33 | 33 | 30 | 29 | 31 | 27 | 25 | 27 | 30 | 36 | 31 | 28 | 28 | 29 | 40 | 26 | 26 | |
| Hem25p | 21 | 21 | 27 | 18 | 23 | 19 | 20 | 20 | 23 | 19 | 24 | 21 | 21 | 20 | 19 | 26 | 35 | |
| GlyC | 25 | 26 | 29 | 26 | 26 | 23 | 23 | 25 | 26 | 26 | 25 | 25 | 21 | 26 | 27 | 26 | 35 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monné, M.; Vozza, A.; Lasorsa, F.M.; Porcelli, V.; Palmieri, F. Mitochondrial Carriers for Aspartate, Glutamate and Other Amino Acids: A Review. Int. J. Mol. Sci. 2019, 20, 4456. https://doi.org/10.3390/ijms20184456
Monné M, Vozza A, Lasorsa FM, Porcelli V, Palmieri F. Mitochondrial Carriers for Aspartate, Glutamate and Other Amino Acids: A Review. International Journal of Molecular Sciences. 2019; 20(18):4456. https://doi.org/10.3390/ijms20184456
Chicago/Turabian StyleMonné, Magnus, Angelo Vozza, Francesco Massimo Lasorsa, Vito Porcelli, and Ferdinando Palmieri. 2019. "Mitochondrial Carriers for Aspartate, Glutamate and Other Amino Acids: A Review" International Journal of Molecular Sciences 20, no. 18: 4456. https://doi.org/10.3390/ijms20184456
APA StyleMonné, M., Vozza, A., Lasorsa, F. M., Porcelli, V., & Palmieri, F. (2019). Mitochondrial Carriers for Aspartate, Glutamate and Other Amino Acids: A Review. International Journal of Molecular Sciences, 20(18), 4456. https://doi.org/10.3390/ijms20184456