Role of a RhoA/ROCK-Dependent Pathway on Renal Connexin43 Regulation in the Angiotensin II-Induced Renal Damage



Abstract
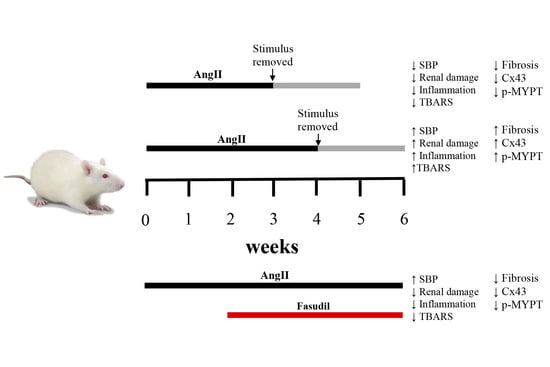
1. Introduction
2. Results
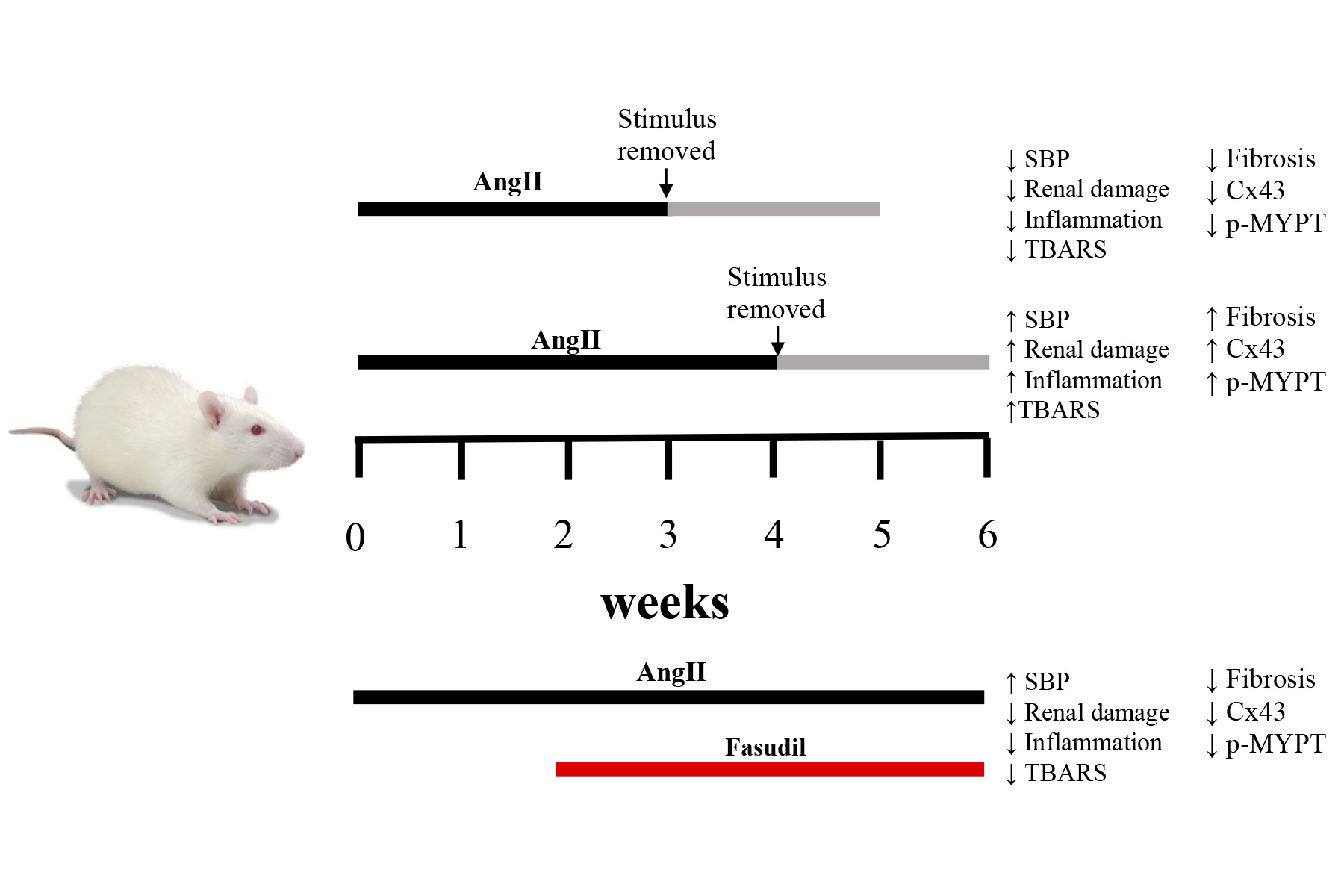
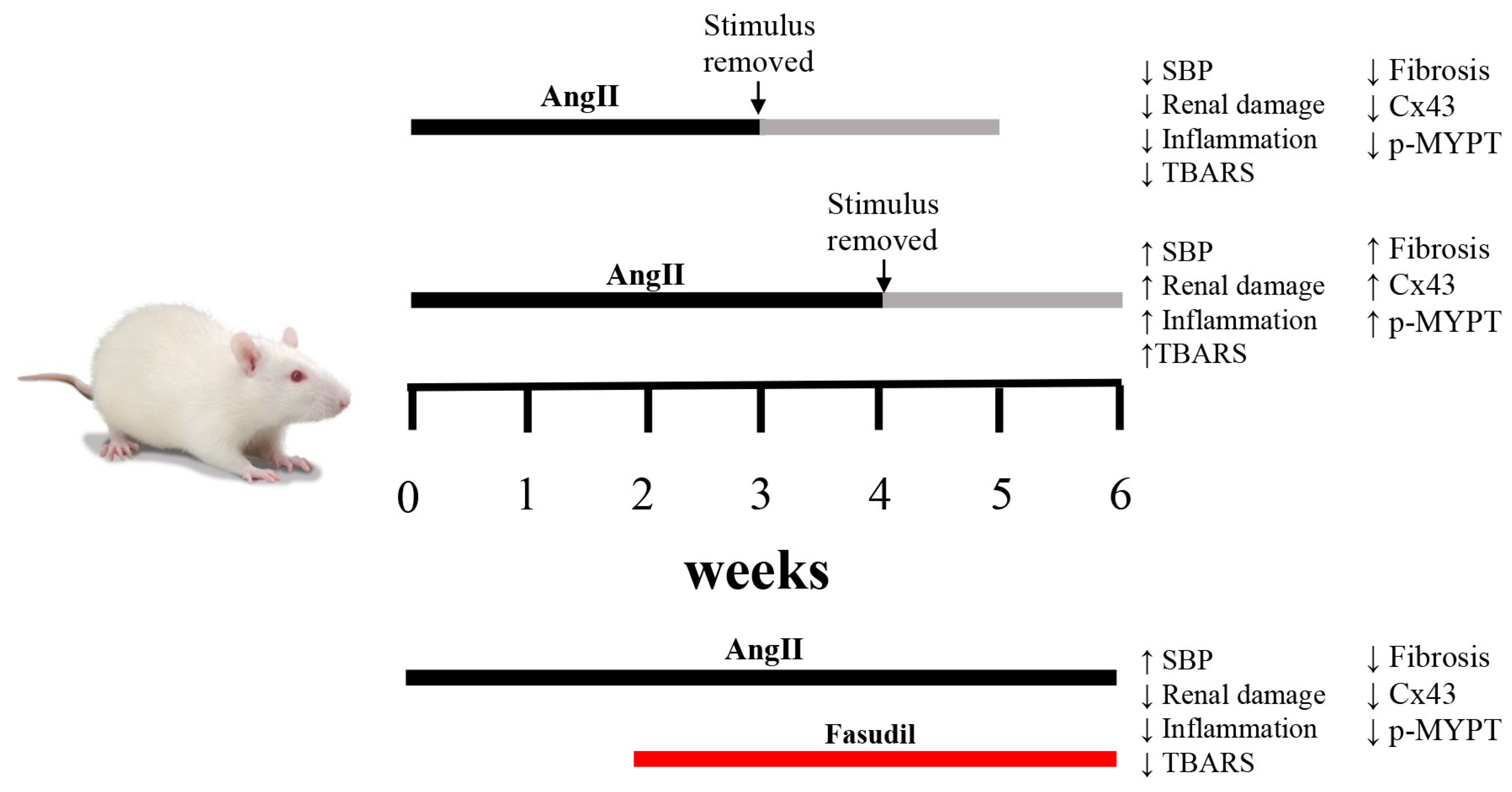
2.1. Four Weeks of Treatment with AngII Causes an Increase in Blood Pressure and Decrease in Renal Function
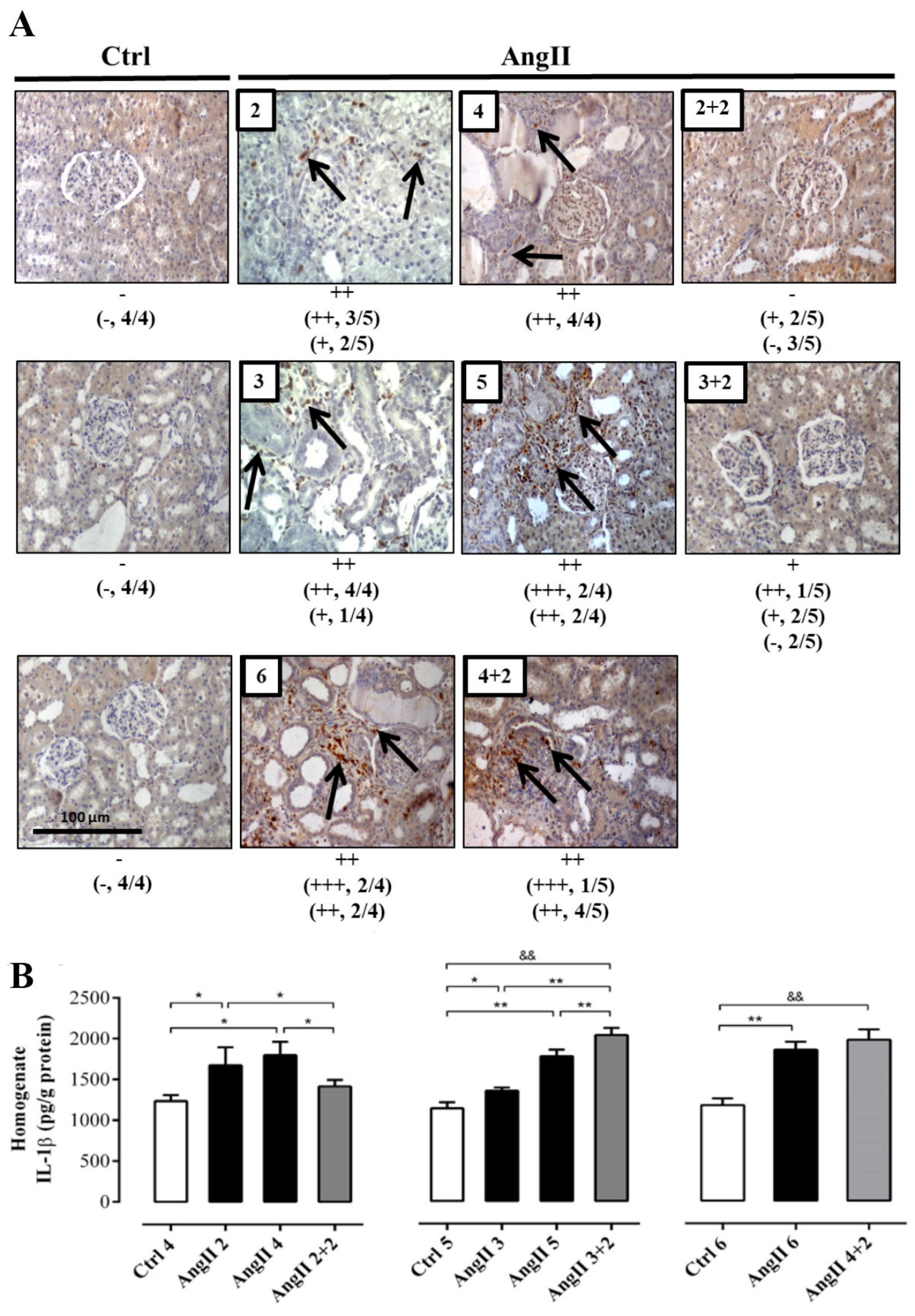
2.2. The Suspension of AngII Infusion does not Reduce OS, Inflammation or Renal Tissue Damage in Rats Infused with AngII for 4 Weeks
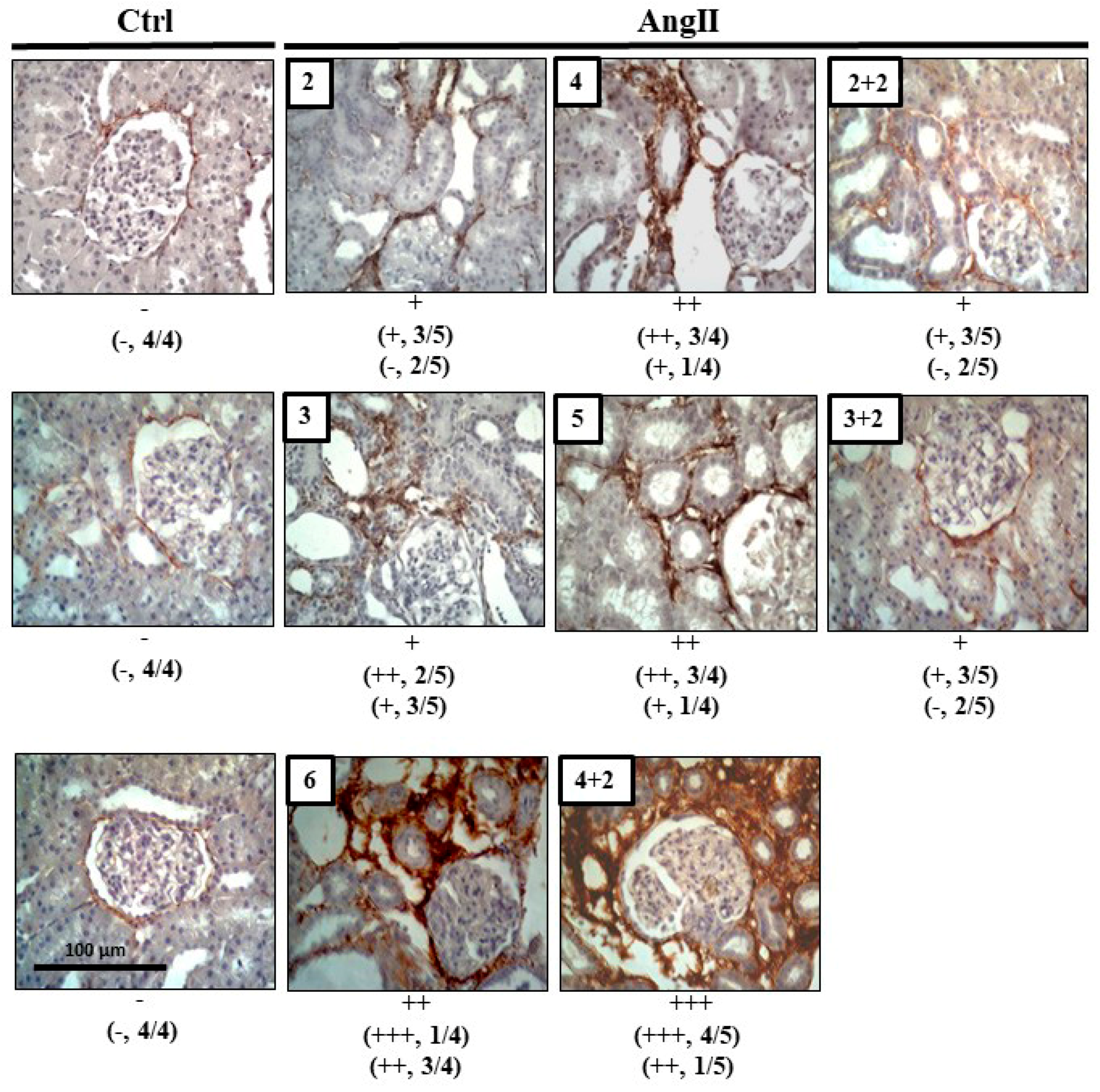
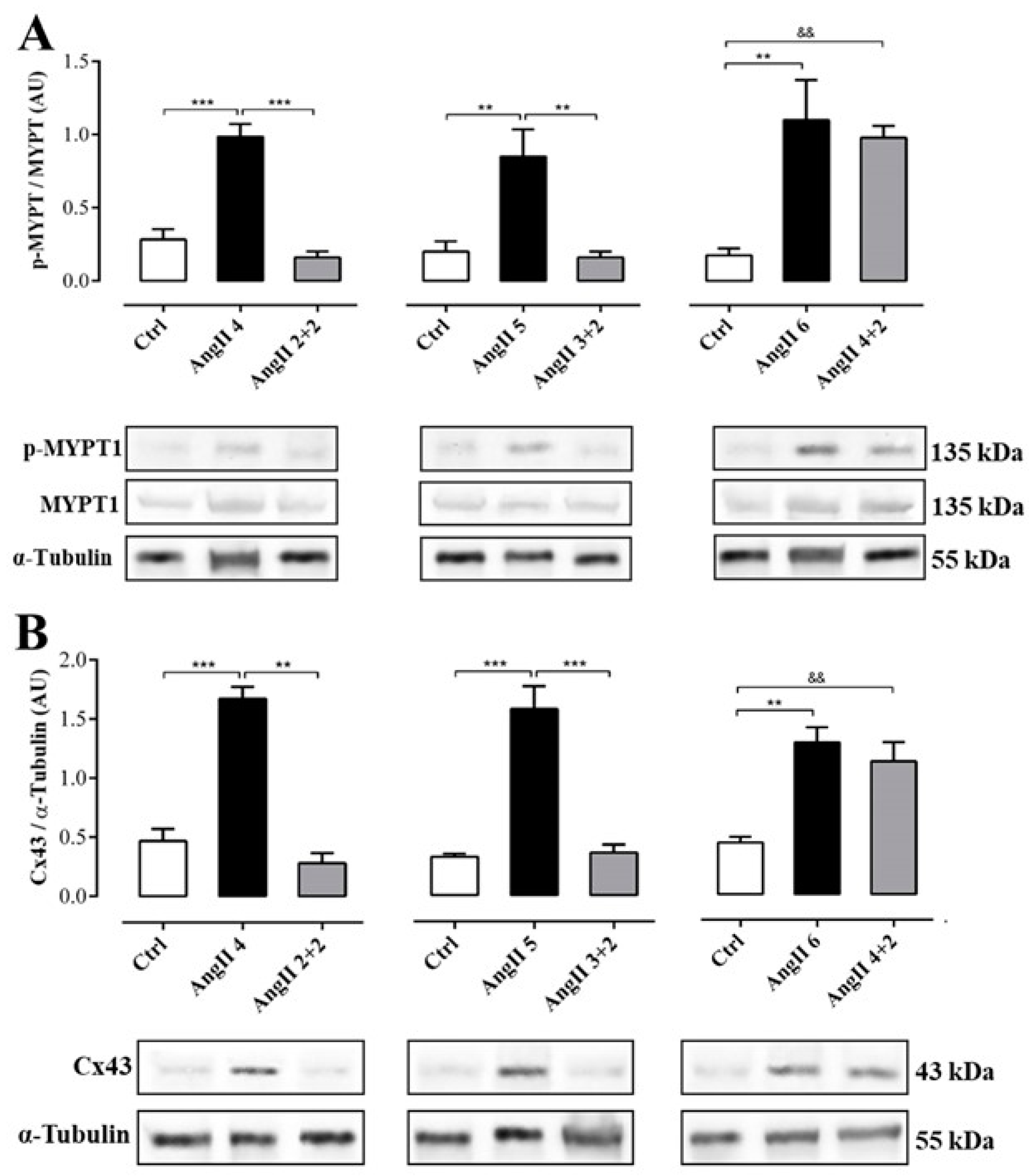
2.3. The Increase in Cx43 and Activation of RhoA/ROCK Become Independent of the Stimulus after 4 Weeks of Treatment with AngII
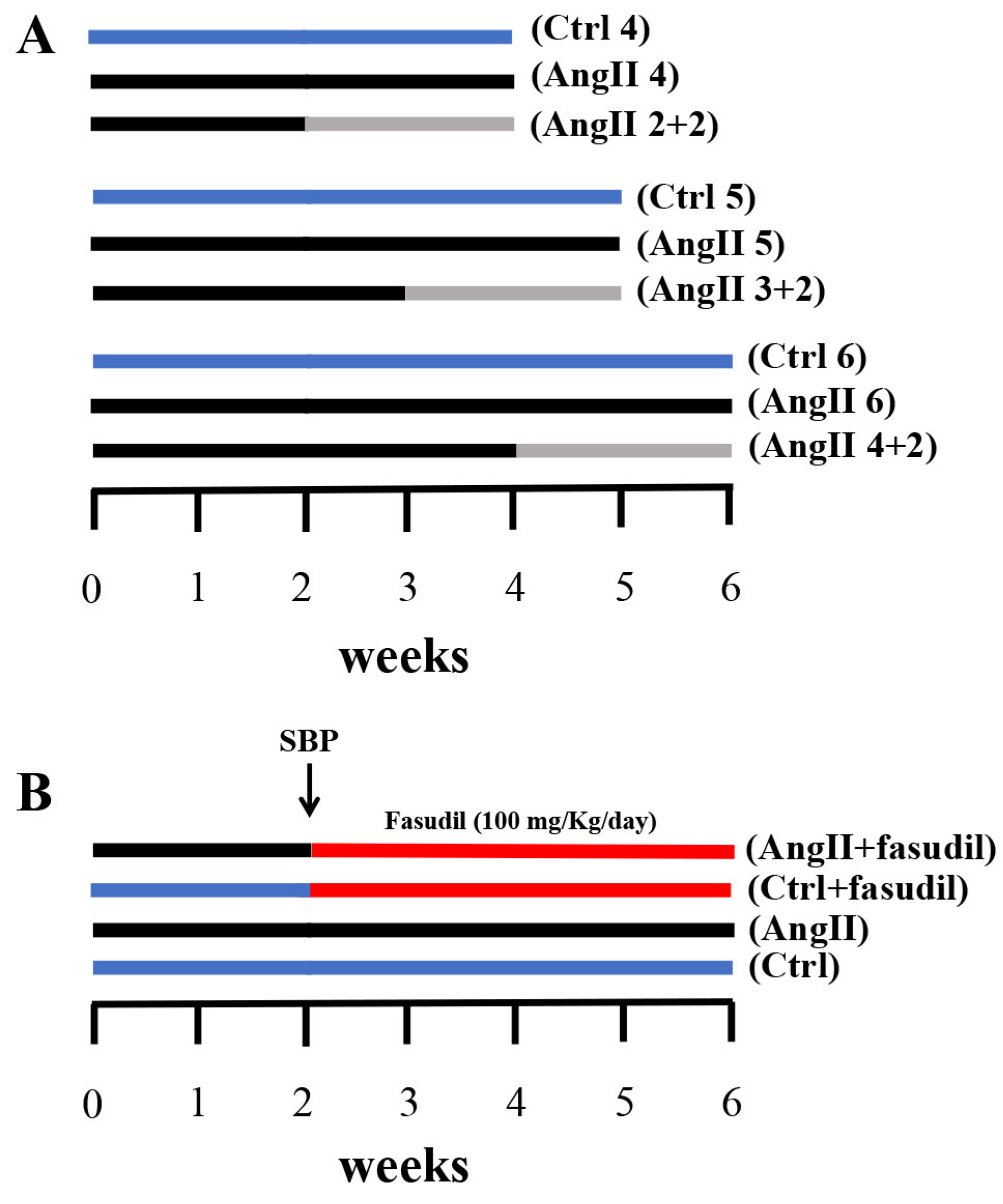
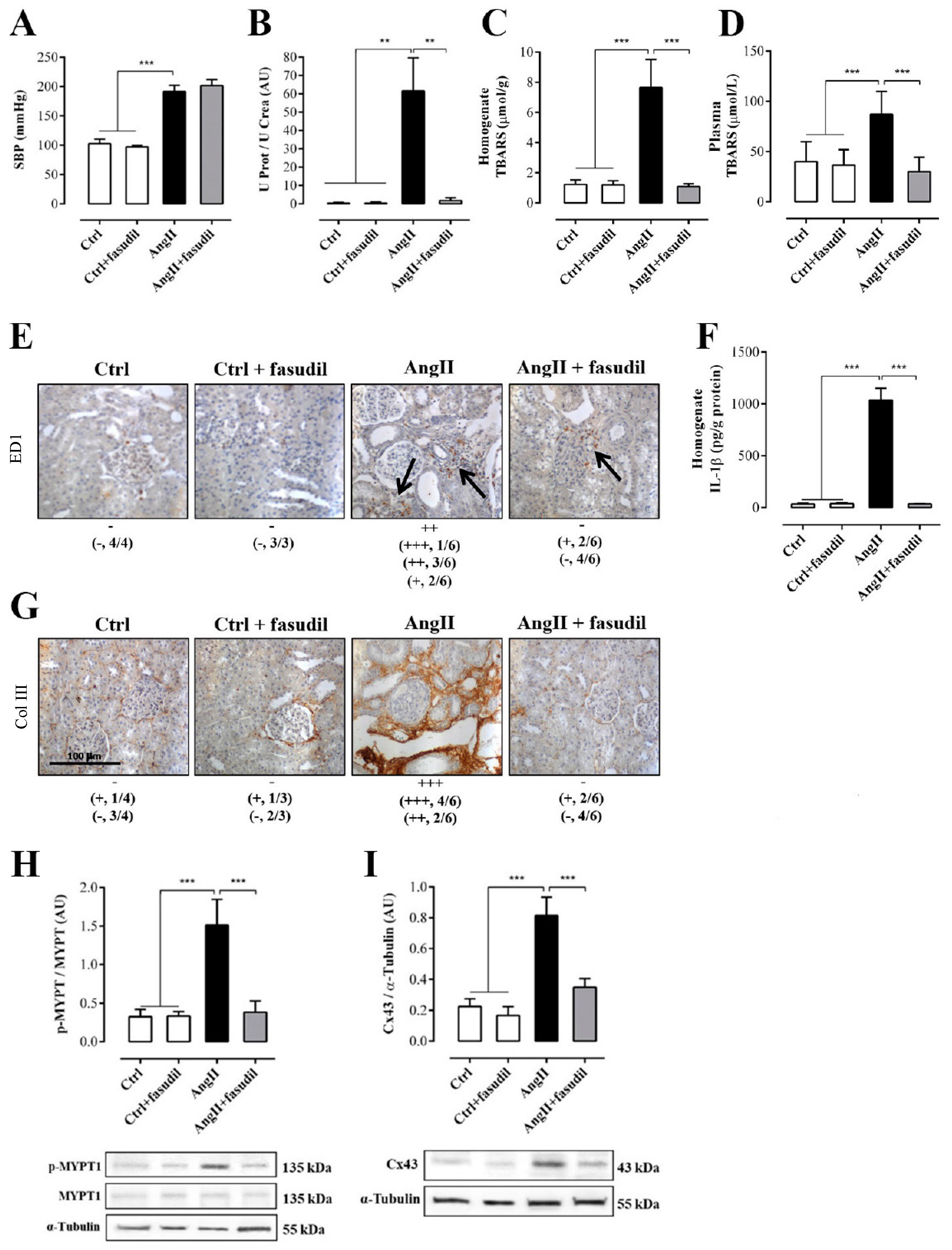
2.4. Fasudil Prevents Kidney Damage, OS, Inflammation, Fibrosis, and the Increase in Protein Amount of Cx43, but does not Decrease SBP in Rats Treated with AngII
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Animals
4.3. AngII Administration and Experimental Procedure
4.4. Blood Pressure Measurements
4.5. Renal Function Measurements
4.6. Thiobarbituric Acid Reactive Substances (TBARS) Measurement
4.7. Histological Damage Assessment
4.8. Tissue Processing and Immunohistochemical Analysis
4.9. Enzyme-linked Immunosorbent Assay (ELISA)
4.10. Western Blot Assays
4.11. Statistical Analysis
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| AngII | Angiotensin II |
| CKD | Chronic kidney disease |
| Col III | Collagen type III |
| Cx GJs | Connexin gap junctions |
| Cx HCs | Connexin hemichannels |
| Cx43 | Connexin 43 |
| ED-1 | Marker of macrophage infiltration |
| GFR | Glomerular filtration rate |
| IL-1β | Interleukin-1β |
| MDA | Malondialdehyde |
| MYPT-1 | Myosin phosphatase target subunit 1 |
| NFκB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NOX | Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase |
| OPN | Osteopontin |
| OS | Oxidative stress |
| RAS | Renin angiotensin system |
| Rho GTPase | Rho family of small GTPases |
| ROCK | Rho kinase |
| ROS | Reactive oxidative species |
| SBP | Systolic blood pressure |
| TBARS | Thiobarbituric reactive species |
| TGF-β | Transforming growth factor beta |
| TNF-α | Tumor necrosis factor-α |
| UProt/UCrea | Ratio urine protein/urine creatinine |
| α-SMA | Alpha-smooth muscle actin |
References
- de Zeeuw, D.; Hillege, H.L.; de Jong, P.E. The kidney, a cardiovascular risk marker, and a new target for therapy. Kidney Int. Suppl. 2005, 68, S25–S29. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; Coresh, J.; Balk, E.; Kausz, A.T.; Levin, A.; Steffes, M.W.; Hogg, R.J.; Perrone, R.D.; Lau, J.; Eknoyan, G.; et al. National Kidney Foundation practice guidelines for chronic kidney disease: Evaluation, classification, and stratification. Ann. Intern. Med. 2003, 139, 137–147. [Google Scholar] [CrossRef] [PubMed]
- López-Novoa, J.M.; Martínez-Salgado, C.; Rodríguez-Peña, A.B.; López-Hernández, F.J. Common pathophysiological mechanisms of chronic kidney disease: Therapeutic perspectives. Pharmacol. Ther. 2010, 128, 61–81. [Google Scholar] [CrossRef] [PubMed]
- Coresh, J.; Byrd-Holt, D.; Astor, B.C.; Briggs, J.P.; Eggers, P.W.; Lacher, D.A.; Hostetter, T.H. Chronic kidney disease awareness, prevalence, and trends among U.S. adults, 1999 to 2000. J. Am. Soc. Nephrol. 2005, 16, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M. Chronic Hypoxia and Tubulointerstitial Injury: A Final Common Pathway to End-Stage Renal Failure. J. Am. Soc. Nephrol. 2005, 17, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Correa-Rotter, R.; Cusumano, A.M. Present, Prevention, and Management of Chronic Kidney Disease in Latin America. Blood Purif. 2008, 26, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.L.; Ma, J.Z.; Louis, T.A.; Collins, A.J. Forecast of the number of patients with end-stage renal disease in the United States to the year 2010. J. Am. Soc. Nephrol. 2001, 12, 2753–2758. [Google Scholar] [PubMed]
- Ozawa, Y.; Kobori, H.; Suzaki, Y.; Navar, L.G. Sustained renal interstitial macrophage infiltration following chronic angiotensin II infusions. Am. J. Physiol. Ren. Physiol. 2007, 292, F330–F339. [Google Scholar] [CrossRef]
- Border, W.A.; Noble, N.A. Interactions of transforming growth factor-beta and angiotensin II in renal fibrosis. Hypertension 1998, 31, 181–188. [Google Scholar] [CrossRef]
- Sánchez-López, E.; Rodriguez-Vita, J.; Cartier, C.; Rupérez, M.; Esteban, V.; Carvajal, G.; Rodrígues-Díez, R.; Plaza, J.J.; Egido, J.; Ruiz-Ortega, M. Inhibitory effect of interleukin-1beta on angiotensin II-induced connective tissue growth factor and type IV collagen production in cultured mesangial cells. Am. J. Physiol. Ren. Physiol. 2008, 294, F149–F160. [Google Scholar] [CrossRef]
- Singh, P.; Bahrami, L.; Castillo, A.; Majid, D.S.A. TNF-α type 2 receptor mediates renal inflammatory response to chronic angiotensin II administration with high salt intake in mice. Am. J. Physiol. Ren. Physiol. 2013, 304, F991–F999. [Google Scholar] [CrossRef] [PubMed]
- Mezzano, S.A.; Ruiz-Ortega, M.; Egido, J. Angiotensin II and renal fibrosis. Hypertension 2001, 38, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Mezzano, S.A.; Aros, C.A.; Droguett, A.; Burgos, M.E.; Ardiles, L.G.; Flores, C.A.; Carpio, D.; Vío, C.P.; Ruiz-Ortega, M.; Egido, J. Renal angiotensin II up-regulation and myofibroblast activation in human membranous nephropathy. Kidney Int. Suppl. 2003, S39–S45. [Google Scholar] [CrossRef]
- Rivera, P.; Ocaranza, M.P.; Lavandero, S.; Jalil, J.E. Rho kinase activation and gene expression related to vascular remodeling in normotensive rats with high angiotensin I converting enzyme levels. Hypertension 2007, 50, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Chen, S.S.; Chen, Y.; Ahokas, R.A.; Sun, Y. Kidney fibrosis in hypertensive rats: Role of oxidative stress. Am. J. Nephrol. 2008, 28, 548–554. [Google Scholar] [CrossRef]
- Clermont, G.; Lecour, S.; Lahet, J.; Siohan, P.; Vergely, C.; Chevet, D.; Rifle, G.; Rochette, L. Alteration in plasma antioxidant capacities in chronic renal failure and hemodialysis patients: A possible explanation for the increased cardiovascular risk in these patients. Cardiovasc. Res. 2000, 47, 618–623. [Google Scholar] [CrossRef]
- Johnson, R.J.; Alpers, C.E.; Yoshimura, A.; Lombardi, D.; Pritzl, P.; Floege, J.; Schwartz, S.M. Renal injury from angiotensin II-mediated hypertension. Hypertension 1992, 19, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Lohmeier, T.E. Angiotensin II Infusion Model of Hypertension. Hypertension 2012, 59, 539–541. [Google Scholar] [CrossRef]
- Rupérez, M.; Sánchez-López, E.; Blanco-Colio, L.M.; Esteban, V.; Rodríguez-Vita, J.; Plaza, J.J.; Egido, J.; Ruiz-Ortega, M. The Rho-kinase pathway regulates angiotensin II-induced renal damage. Kidney Int. Suppl. 2005, 68, S39–S45. [Google Scholar] [CrossRef]
- Guilluy, C.; Rolli-Derkinderen, M.; Loufrani, L.; Bourgé, A.; Henrion, D.; Sabourin, L.; Loirand, G.; Pacaud, P. Ste20-related kinase SLK phosphorylates Ser188 of RhoA to induce vasodilation in response to angiotensin II Type 2 receptor activation. Circ. Res. 2008, 102, 1265–1274. [Google Scholar] [CrossRef]
- Etienne-Manneville, S.; Hall, A. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Kolavennu, V.; Zeng, L.; Peng, H.; Wang, Y.; Danesh, F.R. Targeting of RhoA/ROCK signaling ameliorates progression of diabetic nephropathy independent of glucose control. Diabetes 2008, 57, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Wu, D.; Gao, B.; Ingram, A.J.; Zhang, B.; Chorneyko, K.; McKenzie, R.; Krepinsky, J.C. RhoA/Rho-kinase contribute to the pathogenesis of diabetic renal disease. Diabetes 2008, 57, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Liu, F.; Huang, X.R.; Liu, F.; Chen, H.; Chung, A.C.K.; Shi, J.; Wei, L.; Lan, H.Y.; Fu, P. Amelioration of albuminuria in ROCK1 knockout mice with streptozotocin-induced diabetic kidney disease. Am. J. Nephrol. 2011, 34, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Kushiyama, T.; Oda, T.; Yamamoto, K.; Higashi, K.; Watanabe, A.; Takechi, H.; Uchida, T.; Oshima, N.; Sakurai, Y.; Miura, S.; et al. Protective effects of Rho kinase inhibitor fasudil on rats with chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2013, 304, F1325–F1334. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, A. Renal connexins and blood pressure. Biochim. Biophys. Acta 2012, 1818, 1903–1908. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Retamal, M.A.; Froger, N.; Palacios-Prado, N.; Ezan, P.; Sáez, P.J.; Sáez, J.C.; Giaume, C. C × 43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J. Neurosci. 2007, 27, 13781–13792. [Google Scholar] [CrossRef]
- Orellana, J.A.; Hernández, D.E.; Ezan, P.; Velarde, V.; Bennett, M.V.L.; Giaume, C.; Sáez, J.C. Hypoxia in high glucose followed by reoxygenation in normal glucose reduces the viability of cortical astrocytes through increased permeability of connexin 43 hemichannels. Glia 2010, 58, 329–343. [Google Scholar] [CrossRef]
- Hernández-Salinas, R.; Vielma, A.Z.; Arismendi, M.N.; Boric, M.P.; Sáez, J.C.; Velarde, V. Boldine Prevents Renal Alterations in Diabetic Rats. J. Diabetes Res. 2013, 2013, 1–12. [Google Scholar] [CrossRef]
- Vergara, L.; Bao, X.; Cooper, M.; Bello-Reuss, E.; Reuss, L. Gap-junctional hemichannels are activated by ATP depletion in human renal proximal tubule cells. J. Membr. Biol. 2003, 196, 173–184. [Google Scholar] [CrossRef]
- Gómez, G.; Fernández, P.; Velarde, V.; Sáez, J. Angiotensin II-Induced Mesangial Cell Damage Is Preceded by Cell Membrane Permeabilization Due to Upregulation of Non-Selective Channels. Int. J. Mol. Sci. 2018, 19, 957. [Google Scholar] [CrossRef] [PubMed]
- Haefliger, J.A.; Krattinger, N.; Martin, D.; Pedrazzini, T.; Capponi, A.; Döring, B.; Plum, A.; Charollais, A.; Willecke, K.; Meda, P. Connexin43-dependent mechanism modulates renin secretion and hypertension. J. Clin. Investig. 2006, 116, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Hillis, G.S.; Duthie, L.A.; Mlynski, R.; McKay, N.G.; Mistry, S.; MacLeod, A.M.; Simpson, J.G.; Haites, N.E. The expression of connexin 43 in human kidney and cultured renal cells. Nephron 1997, 75, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Toubas, J.; Beck, S.; Pageaud, A.L.; Huby, A.C.; Mael-Ainin, M.; Dussaule, J.C.; Chatziantoniou, C.; Chadjichristos, C.E. Alteration of connexin expression is an early signal for chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2011, 301, F24–F32. [Google Scholar] [CrossRef] [PubMed]
- Prakoura, N.; Kavvadas, P.; Chadjichristos, C.E. Connexin 43: A new Therapeutic Target Against Chronic Kidney Disease. Cell. Physiol. Biochem. 2018, 49, 985. [Google Scholar] [CrossRef]
- Kao, M.P.C.; Ang, D.S.C.; Pall, A.; Struthers, A.D. Oxidative stress in renal dysfunction: Mechanisms, clinical sequelae and therapeutic options. J. Hum. Hypertens. 2010, 24, 1–8. [Google Scholar] [CrossRef]
- Li, C.; Yang, C.W.; Park, J.H.; Lim, S.W.; Sun, B.K.; Jung, J.Y.; Kim, S.B.; Kim, Y.S.; Kim, J.; Bang, B.K. Pravastatin treatment attenuates interstitial inflammation and fibrosis in a rat model of chronic cyclosporine-induced nephropathy. Am. J. Physiol. Ren. Physiol. 2004, 286, F46–F57. [Google Scholar] [CrossRef]
- Kawai, Y.; Satoh, T.; Hibi, D.; Ohno, Y.; Kohda, Y.; Miura, K.; Gemba, M. The effect of antioxidant on development of fibrosis by cisplatin in rats. J. Pharmacol. Sci. 2009, 111, 433–439. [Google Scholar] [CrossRef]
- Vilayur, E.; Harris, D.C.H. Emerging therapies for chronic kidney disease: What is their role? Nat. Rev. Nephrol. 2009, 5, 375–383. [Google Scholar] [CrossRef]
- Ravarotto, V.; Simioni, F.; Carraro, G.; Bertoldi, G.; Pagnin, E.; Calò, L.A. Oxidative Stress and Cardiovascular-Renal Damage in Fabry Disease: Is There Room for a Pathophysiological Involvement? J. Clin. Med. 2018, 7, 409. [Google Scholar] [CrossRef]
- van Kats, J.P.; de Lannoy, L.M.; Jan Danser, A.H.; van Meegen, J.R.; Verdouw, P.D.; Schalekamp, M.A. Angiotensin II type 1 (AT1) receptor-mediated accumulation of angiotensin II in tissues and its intracellular half-life in vivo. Hypertension 1997, 30, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Hutton, H.L.; Alikhan, M.A.; Kitching, A.R. Inflammasomes in the Kidney. Exp. Suppl. 2018, 8, 177–210. [Google Scholar]
- Yang, F.; Chung, A.C.K.; Huang, X.R.; Lan, H.Y. Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-beta-dependent and -independent Smad pathways: The role of Smad3. Hypertension 2009, 54, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G.; Schneider, A.; Wenzel, U.; Helmchen, U.; Stahl, R.A. Regulation of glomerular TGF-beta expression in the contralateral kidney of two-kidney, one-clip hypertensive rats. J. Am. Soc. Nephrol. 1998, 9, 763–772. [Google Scholar] [PubMed]
- Frenay, A.R.S.; Yazdani, S.; Boersema, M.; van der Graaf, A.M.; Waanders, F.; van den Born, J.; Navis, G.J.; van Goor, H. Incomplete Restoration of Angiotensin II-Induced Renal Extracellular Matrix Deposition and Inflammation Despite Complete Functional Recovery in Rats. PLoS ONE 2015, 10, e0129732. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, S.; Navis, G.; Hillebrands, J.L.; van Goor, H.; van den Born, J. Lymphangiogenesis in renal diseases: Passive bystander or active participant? Expert Rev. Mol. Med. 2014, 16, e15. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, S.; Poosti, F.; Kramer, A.B.; Mirković, K.; Kwakernaak, A.J.; Hovingh, M.; Slagman, M.C.J.; Sjollema, K.A.; de Borst, M.H.; Navis, G.; et al. Proteinuria Triggers Renal Lymphangiogenesis Prior to the Development of Interstitial Fibrosis. PLoS ONE 2012, 7, e50209. [Google Scholar] [CrossRef]
- Papakrivopoulou, E.; Vasilopoulou, E.; Lindenmeyer, M.T.; Pacheco, S.; Brzóska, H.Ł.; Price, K.L.; Kolatsi-Joannou, M.; White, K.E.; Henderson, D.J.; Dean, C.H.; et al. Vangl2, a planar cell polarity molecule, is implicated in irreversible and reversible kidney glomerular injury. J. Pathol. 2018, 246, 485–496. [Google Scholar] [CrossRef]
- Miyata, K.; Satou, R.; Shao, W.; Prieto, M.C.; Urushihara, M.; Kobori, H.; Navar, L.G. ROCK/NF-κB axis-dependent augmentation of angiotensinogen by angiotensin II in primary-cultured preglomerular vascular smooth muscle cells. Am. J. Physiol. Ren. Physiol. 2014, 306, F608–F618. [Google Scholar] [CrossRef]
- Lai, A.; Frishman, W.H. Rho-kinase inhibition in the therapy of cardiovascular disease. Cardiol. Rev. 2005, 13, 285–292. [Google Scholar] [CrossRef]
- Kanda, T.; Wakino, S.; Hayashi, K.; Homma, K.; Ozawa, Y.; Saruta, T. Effect of fasudil on Rho-kinase and nephropathy in subtotally nephrectomized spontaneously hypertensive rats. Kidney Int. 2003, 64, 2009–2019. [Google Scholar] [CrossRef] [PubMed]
- Nishikimi, T.; Akimoto, K.; Wang, X.; Mori, Y.; Tadokoro, K.; Ishikawa, Y.; Shimokawa, H.; Ono, H.; Matsuoka, H. Fasudil, a Rho-kinase inhibitor, attenuates glomerulosclerosis in Dahl salt-sensitive rats. J. Hypertens. 2004, 22, 1787–1796. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Nishikimi, T.; Akimoto, K.; Ishimura, K.; Ono, H.; Matsuoka, H. Long-term administration of rho-kinase inhibitor ameliorates renal damage in malignant hypertensive rats. Hypertension 2006, 47, 1075–1083. [Google Scholar] [CrossRef]
- Suzuki, H.; Yamamoto, T.; Fujigaki, Y.; Eguchi, S.; Hishida, A. Comparison of ROCK and EGFR activation pathways in the progression of glomerular injuries in AngII-infused rats. Ren. Fail. 2011, 33, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Nishikimi, T.; Koshikawa, S.; Ishikawa, Y.; Akimoto, K.; Inaba, C.; Ishimura, K.; Ono, H.; Matsuoka, H. Inhibition of Rho-kinase attenuates nephrosclerosis and improves survival in salt-loaded spontaneously hypertensive stroke-prone rats. J. Hypertens. 2007, 25, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.L.; Yu, J.Z.; Yang, X.W.; Liu, C.Y.; Li, Y.H.; Feng, L.; Chai, Z.; Yang, W.F.; Wang, Q.; Jiang, W.J.; et al. FSD-C10: A more promising novel ROCK inhibitor than Fasudil for treatment of CNS autoimmunity. Biosci. Rep. 2015, 35, e00247. [Google Scholar] [CrossRef] [PubMed]
- Langevin, H.M.; Fujita, T.; Bouffard, N.A.; Takano, T.; Koptiuch, C.; Badger, G.J.; Nedergaard, M. Fibroblast cytoskeletal remodeling induced by tissue stretch involves ATP signaling. J. Cell. Physiol. 2013, 228, 1922–1926. [Google Scholar] [CrossRef]
- Xie, X.; Chen, C.; Huang, K.; Wang, S.; Hao, J.; Huang, J.; Huang, H. RhoA/rho kinase signaling reduces connexin43 expression in high glucose-treated glomerular mesangial cells with zonula occludens-1 involvement. Exp. Cell Res. 2014, 327, 276–286. [Google Scholar] [CrossRef]
- Villanueva, S.; Cespedes, C.; Gonzalez, A.; Vio, C.P. bFGF induces an earlier expression of nephrogenic proteins after ischemic acute renal failure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R1677–R1687. [Google Scholar] [CrossRef]
- Giachelli, C.M.; Pichler, R.; Lombardi, D.; Denhardt, D.T.; Alpers, C.E.; Schwartz, S.M.; Johnson, R.J. Osteopontin expression in angiotensin II-induced tubulointerstitial nephritis. Kidney Int. 1994, 45, 515–524. [Google Scholar] [CrossRef]
- Li, S.; Wang, Y.N.; Niimi, M.; Ning, B.; Chen, Y.; Kang, D.; Waqar, A.B.; Wang, Z.; Yu, Q.; Liu, E.; et al. Angiotensin II Destabilizes Coronary Plaques in Watanabe Heritable Hyperlipidemic Rabbits. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Salas, S.P.; Giacaman, A.; Vío, C.P. Pregnant Rats With 5/6 Nephrectomy Have Normal Volume Expansion Despite Lower Renin and Kallikrein. Hypertension 2003, 42, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, L.; Das, N.P.; Li, Q.T. Studies on lipid oxidation in fish phospholipid liposomes. Biol. Trace Elem. Res. 1994, 40, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Gómez, G.; Velarde, V. Boldine Improves Kidney Damage in the Goldblatt 2K1C Model Avoiding the Increase in TGF-β. Int. J. Mol. Sci. 2018, 19, 1864. [Google Scholar] [CrossRef] [PubMed]
- Lazich, I.; Chang, A.; Watson, S.; Dhar, P.; Madhurapantula, R.S.; Hammes, M. Morphometric and histological parameters in veins of diabetic patients undergoing brachiocephalic fistula placement. Hemodial. Int. 2015, 19, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Greenfeld, Z.; Stillman, I.E.; Brezis, M.; Rosen, S. Medullary injury in the ageing rat kidney: Functional-morphometric correlations. Eur. J. Clin. Investig. 1997, 27, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.R.; Duggal, A.; Thomas, R.; Reeve, R.; Roberts, I.S.D.; Kalra, P.A. Clinicopathological correlation in biopsy-proven atherosclerotic nephropathy: Implications for renal functional outcome in atherosclerotic renovascular disease. Nephrol. Dial. Transplant. 2001, 16, 765–770. [Google Scholar] [CrossRef]
- López-De León, A.; Rojkind, M. A simple micromethod for collagen and total protein determination in formalin-fixed paraffin-embedded sections. J. Histochem. Cytochem. 1985, 33, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Vio, C.P.; An, S.J.; Céspedes, C.; McGiff, J.C.; Ferreri, N.R. Induction of cyclooxygenase-2 in thick ascending limb cells by adrenalectomy. J. Am. Soc. Nephrol. 2001, 12, 649–658. [Google Scholar]
- Vio, C.P.; Quiroz-Munoz, M.; Cuevas, C.A.; Cespedes, C.; Ferreri, N.R. Prostaglandin E 2 EP3 receptor regulates cyclooxygenase-2 expression in the kidney. Am. J. Physiol. Ren. Physiol. 2012, 303, F449–F457. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Weigth (gr) | Proteinuria (mg/day) | Creatinine Clearence (ml/min) | FE Na + (%) | FE K + (%) |
|---|---|---|---|---|---|
| Ctrl | 482 ± 31 | 2.7 ± 1.1 *** | 1.4 ± 0.3 *** | 0.2 ± 0.0 *** | 12.0 ± 2.7 *** |
| Ctrll+fasudil | 480 ± 36 | 3.6 ± 1.1 *** | 2.1± 0.1 *** | 0.1 ± 0.0 *** | 12.5 ± 0.3 *** |
| AngII | 364 ± 42 | 214.0 ± 19.0 | 0.7 ± 0.0 | 2.2 ± 0.4 | 162.0 ± 23.0 |
| AngII+fasudil | 368 ± 17 | 19 ± 7.2 *** | 1.9 ± 0.2 *** | 0.5 ± 0.1 *** | 30 ± 7.2 *** |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez, G.I.; Velarde, V.; Sáez, J.C. Role of a RhoA/ROCK-Dependent Pathway on Renal Connexin43 Regulation in the Angiotensin II-Induced Renal Damage. Int. J. Mol. Sci. 2019, 20, 4408. https://doi.org/10.3390/ijms20184408
Gómez GI, Velarde V, Sáez JC. Role of a RhoA/ROCK-Dependent Pathway on Renal Connexin43 Regulation in the Angiotensin II-Induced Renal Damage. International Journal of Molecular Sciences. 2019; 20(18):4408. https://doi.org/10.3390/ijms20184408
Chicago/Turabian StyleGómez, Gonzalo I., Victoria Velarde, and Juan C. Sáez. 2019. "Role of a RhoA/ROCK-Dependent Pathway on Renal Connexin43 Regulation in the Angiotensin II-Induced Renal Damage" International Journal of Molecular Sciences 20, no. 18: 4408. https://doi.org/10.3390/ijms20184408
APA StyleGómez, G. I., Velarde, V., & Sáez, J. C. (2019). Role of a RhoA/ROCK-Dependent Pathway on Renal Connexin43 Regulation in the Angiotensin II-Induced Renal Damage. International Journal of Molecular Sciences, 20(18), 4408. https://doi.org/10.3390/ijms20184408