Inhibition of Tyrosine-Phosphorylated STAT3 in Human Breast and Lung Cancer Cells by Manuka Honey is Mediated by Selective Antagonism of the IL-6 Receptor

 ,
,
Abstract
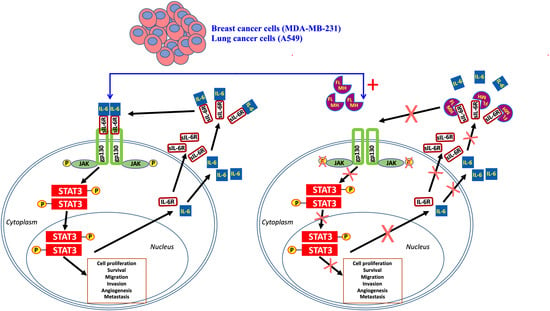
1. Introduction
2. Results
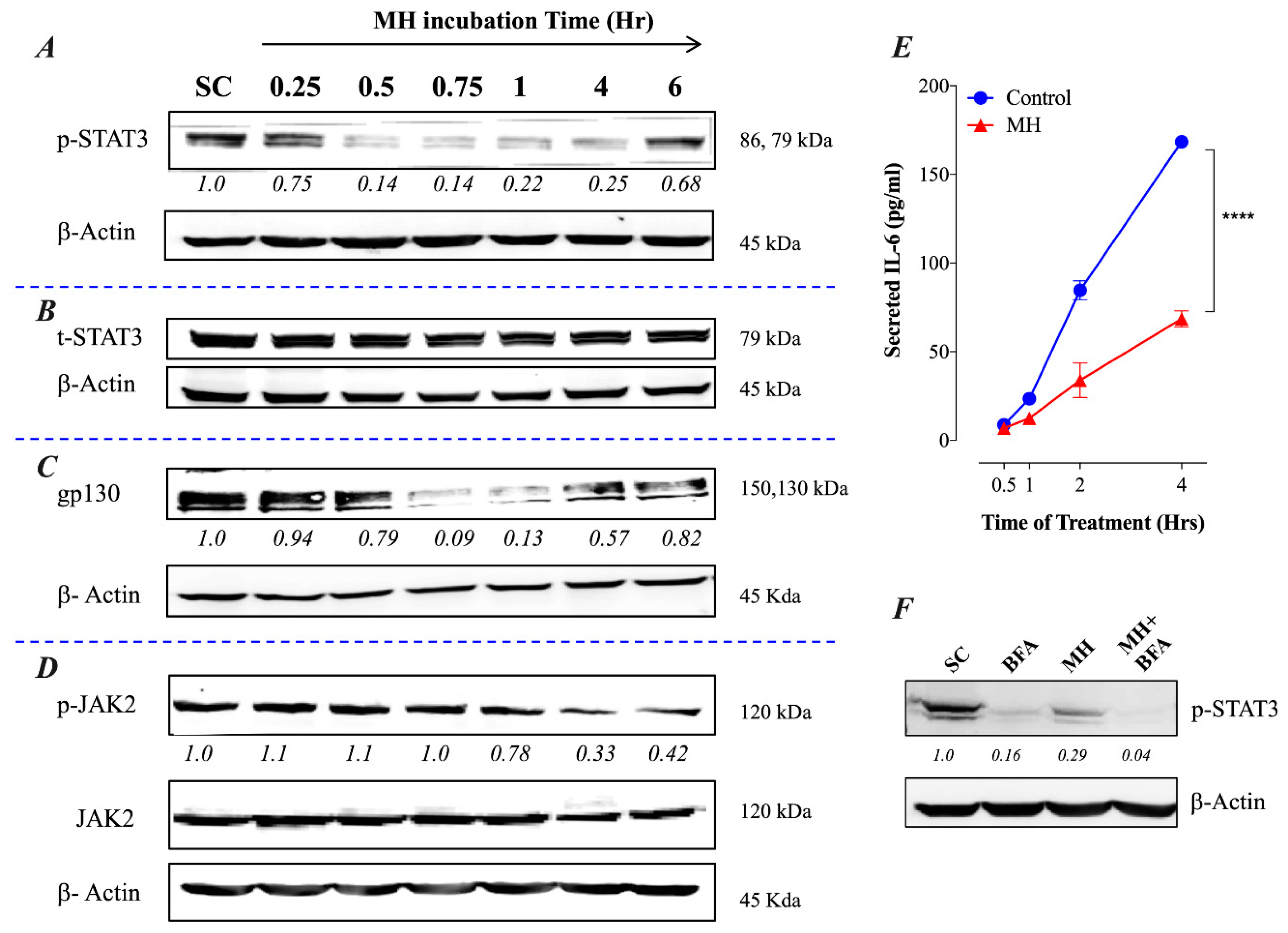
2.1. MH-Induced Loss of p-STAT3 is Associated with Decreased gp130 and p-JAK2
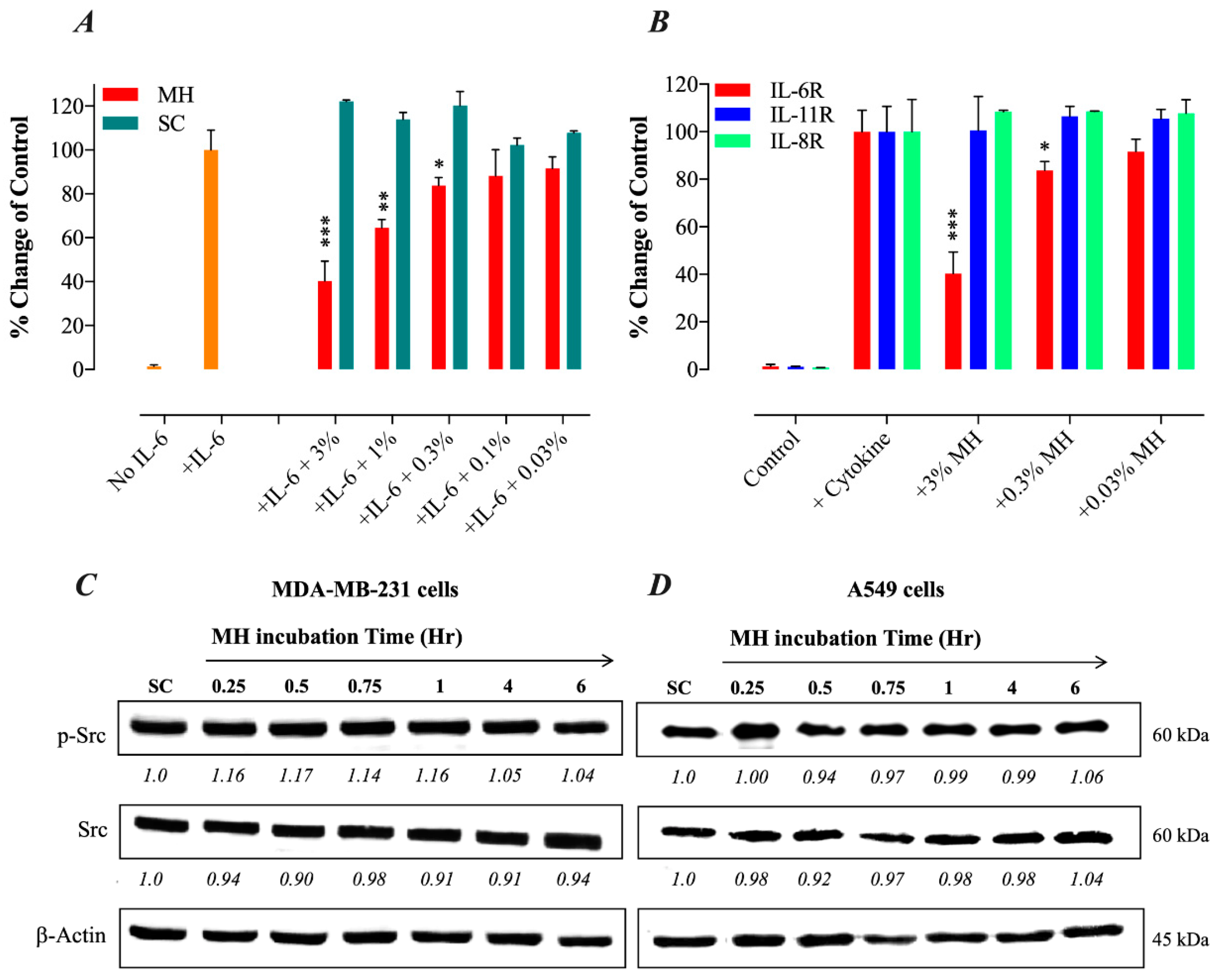
2.2. Constitutive p-STAT3 is Dependent on Autocrine Activation
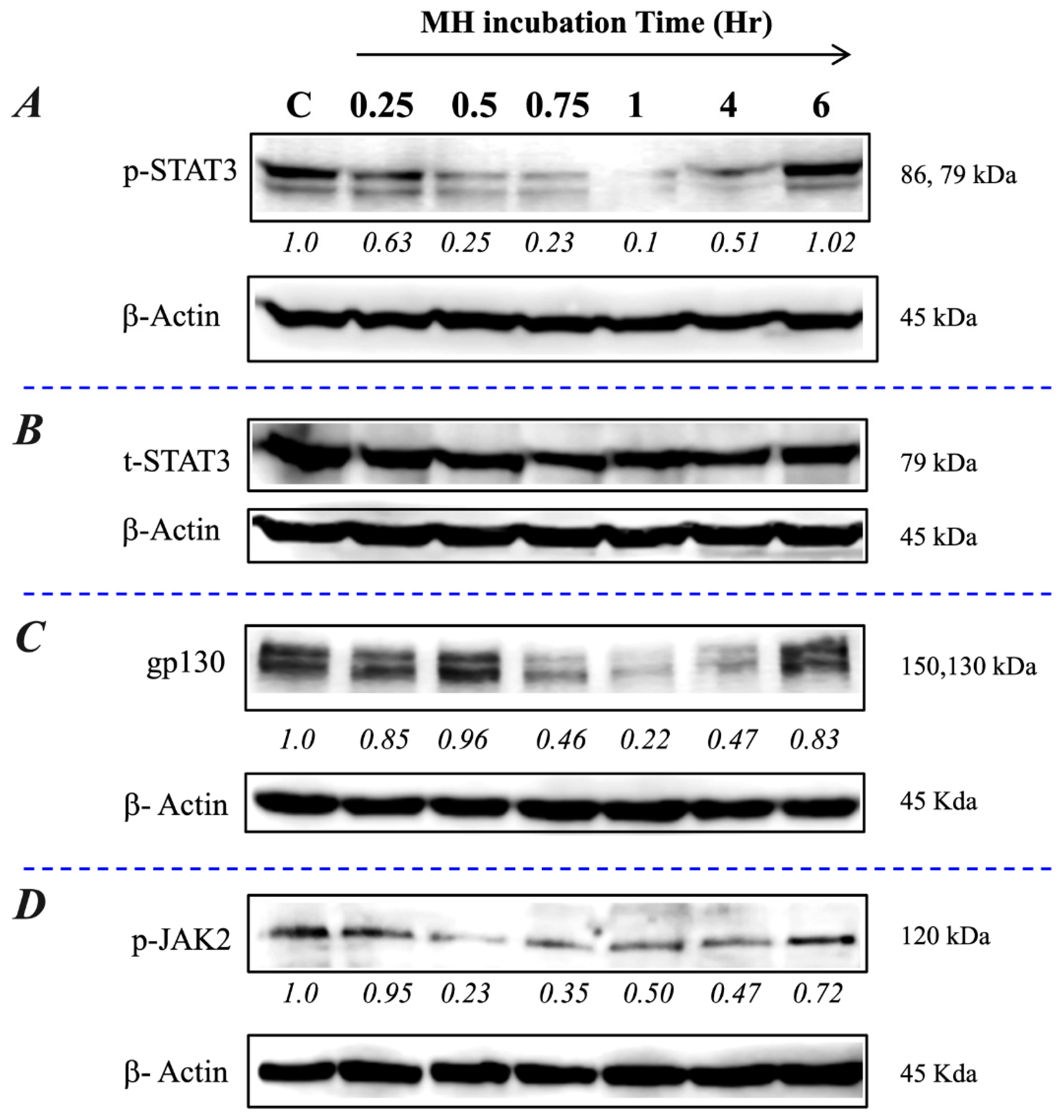
2.3. Exposure to MH Causes a Loss of p-STAT3, gp130, and p-JAK2 in A549 Lung Cancer Cells
2.4. MH Binds Competitively and Specifically to IL-6Rα
MH Flavonoids Bind to IL-6Rα
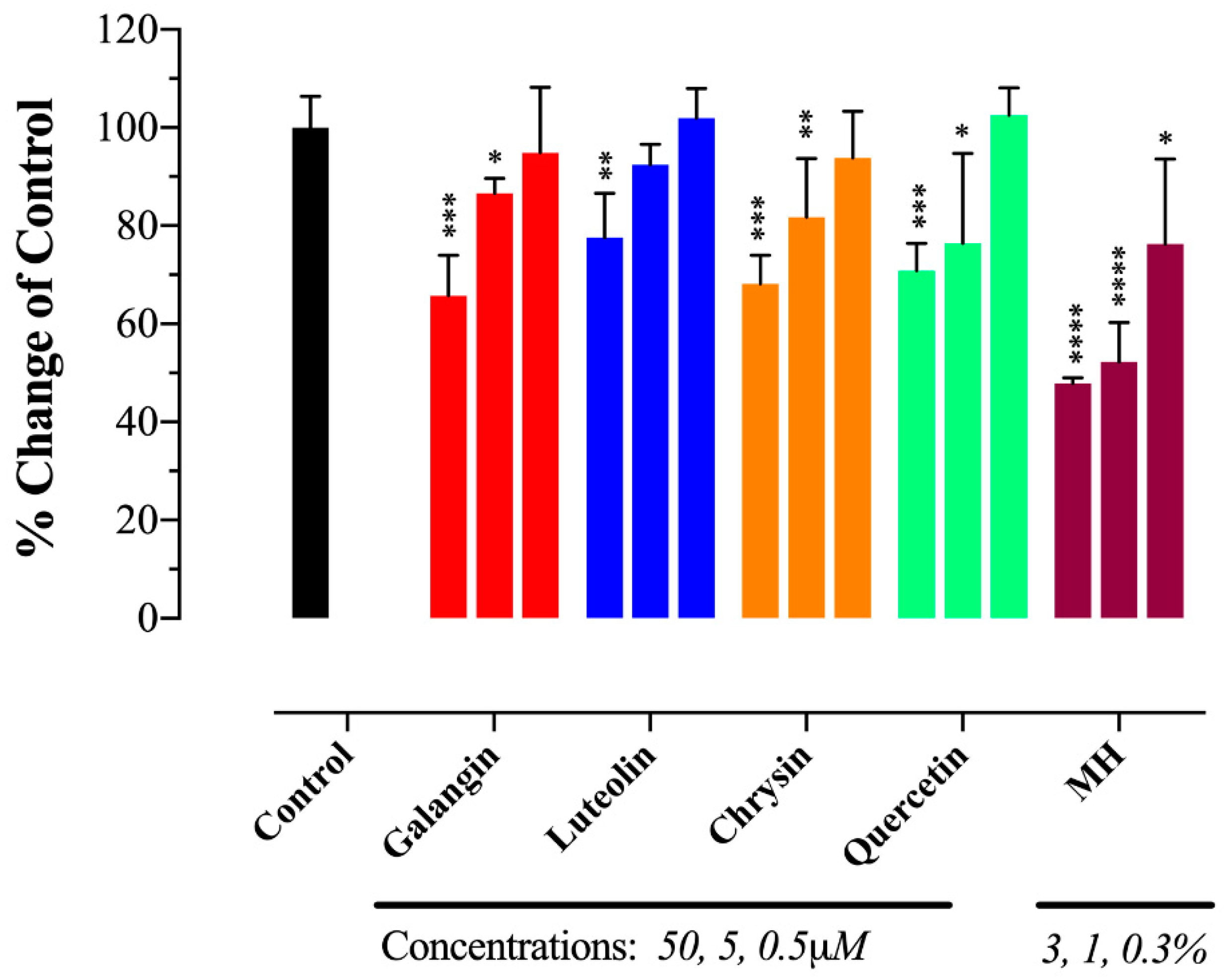
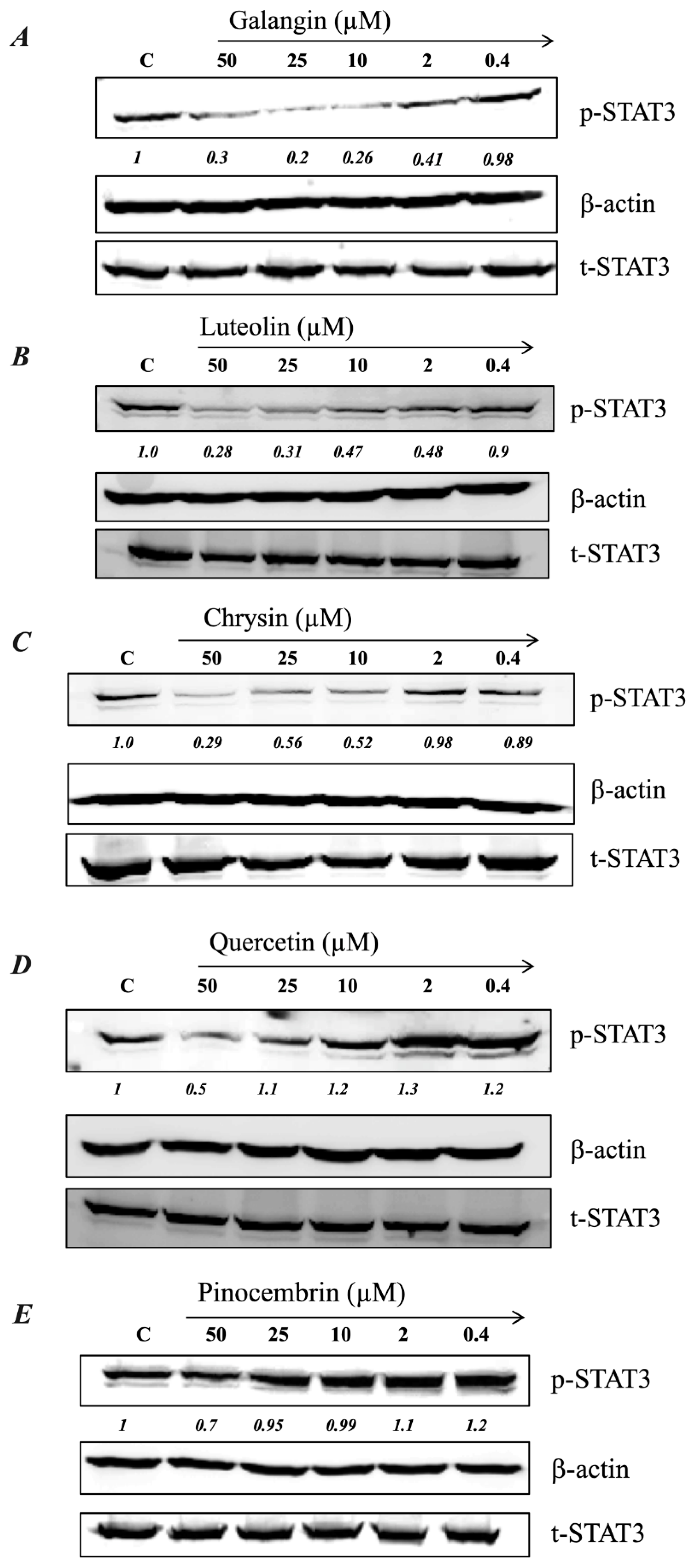
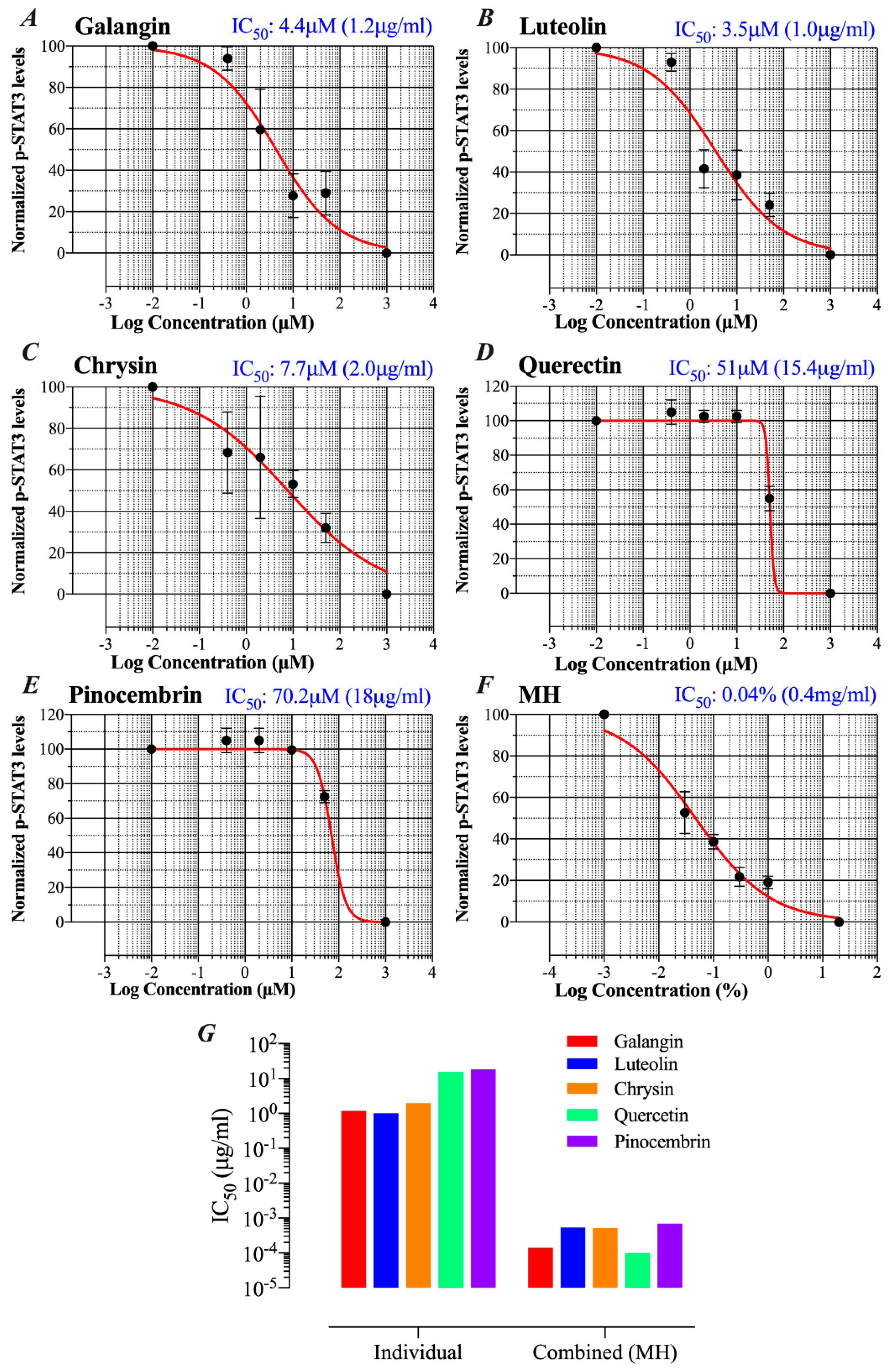
2.5. MH Flavonoids Exhibit Differential Inhibitory Capacity on STAT3 Phosphorylation
2.6. Docking Analysis Reveals Preferential Interaction Between Flavonoid Compounds and IL-6Ra
3. Discussion
4. Materials and Methods
4.1. Cell Line and Reagents
4.2. Western Blot Analysis
4.3. Enzyme-Linked Immunosorbent Assay
4.4. Competitive Cytokine Receptor Binding Assays
4.5. Docking Analysis
5. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MH | Manuka honey |
| TNBC | Triple negative breast cancer |
| NSCLC | Non-small cell lung cancer |
| IL-6 | Interleukin-6 |
| IL-6Rα | IL-6 receptor α chain |
| IL-8R | IL-8 receptor |
| IL-11Rα | IL-11 receptor α chain |
| STAT3 | signal transducer and activator of transcription 3 |
| p-STAT3 | tyrosine-phosphorylated STAT3 |
| gp130 | Glycoprotein 130 |
| JAK1/2 | Janus kinase 1/2 |
| p-JAK2 | tyrosine-phosphorylated JAK2 |
| SC | Sugar control |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Santa-Maria, C.A.; Gradishar, W.J. Changing Treatment Paradigms in Metastatic Breast Cancer: Lessons Learned. JAMA Oncol. 2015, 1, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Knupfer, H.; Preiss, R. Significance of interleukin-6 (IL-6) in breast cancer (review). Breast Cancer Res. Treat. 2007, 102, 129–135. [Google Scholar] [CrossRef]
- Corvinus, F.M.; Orth, C.; Moriggl, R.; Tsareva, S.A.; Wagner, S.; Pfitzner, E.B.; Baus, D.; Kaufmann, R.; Huber, L.A.; Zatloukal, K.; et al. Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia 2005, 7, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Turkson, J.; Karras, J.G.; Jove, R.; Haura, E.B. Activation of Stat3 by receptor tyrosine kinases and cytokines regulates survival in human non-small cell carcinoma cells. Oncogene 2003, 22, 4150–4165. [Google Scholar] [CrossRef]
- Taga, T.; Kishimoto, T. Gp130 and the interleukin-6 family of cytokines. Annu. Rev. Immunol. 1997, 15, 797–819. [Google Scholar] [CrossRef]
- Naugler, W.E.; Karin, M. The wolf in sheep’s clothing: The role of interleukin-6 in immunity, inflammation and cancer. Trends Mol. Med. 2008, 14, 109–119. [Google Scholar] [CrossRef]
- Chiu, J.J.; Sgagias, M.K.; Cowan, K.H. Interleukin 6 acts as a paracrine growth factor in human mammary carcinoma cell lines. Clin. Cancer Res. 1996, 2, 215–221. [Google Scholar]
- Conze, D.; Weiss, L.; Regen, P.S.; Bhushan, A.; Weaver, D.; Johnson, P.; Rincon, M. Autocrine production of interleukin 6 causes multidrug resistance in breast cancer cells. Cancer Res. 2001, 61, 8851–8858. [Google Scholar] [PubMed]
- Berishaj, M.; Gao, S.P.; Ahmed, S.; Leslie, K.; Al-Ahmadie, H.; Gerald, W.L.; Bornmann, W.; Bromberg, J.F. Stat3 is tyrosine-phosphorylated through the interleukin-6/glycoprotein 130/Janus kinase pathway in breast cancer. Breast Cancer Res. 2007, 9, R32. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef] [PubMed]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Bournazou, E.; Sansone, P.; Berishaj, M.; Gao, S.P.; Daly, L.; Wels, J.; Theilen, T.; Granitto, S.; Zhang, X.; et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia 2013, 15, 848–862. [Google Scholar] [CrossRef] [PubMed]
- Yanagawa, H.; Sone, S.; Takahashi, Y.; Haku, T.; Yano, S.; Shinohara, T.; Ogura, T. Serum levels of interleukin 6 in patients with lung cancer. Br. J. Cancer 1995, 71, 1095–1098. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.H.; Lai, W.W.; Chen, H.H.; Liu, H.S.; Su, W.C. Autocrine IL-6-induced Stat3 activation contributes to the pathogenesis of lung adenocarcinoma and malignant pleural effusion. Oncogene 2006, 25, 4300–4309. [Google Scholar] [CrossRef] [PubMed]
- Haura, E.B.; Livingston, S.; Coppola, D. Autocrine interleukin-6/interleukin-6 receptor stimulation in non-small-cell lung cancer. Clin. Lung Cancer 2006, 7, 273–275. [Google Scholar] [CrossRef]
- Banerjee, K.; Resat, H. Constitutive activation of STAT3 in breast cancer cells: A review. Int. J. Cancer 2016, 138, 2570–2578. [Google Scholar] [CrossRef]
- Harada, D.; Takigawa, N.; Kiura, K. The Role of STAT3 in Non-Small Cell Lung Cancer. Cancers (Basel) 2014, 6, 708–722. [Google Scholar] [CrossRef]
- Rokavec, M.; Oner, M.G.; Li, H.; Jackstadt, R.; Jiang, L.; Lodygin, D.; Kaller, M.; Horst, D.; Ziegler, P.K.; Schwitalla, S.; et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J. Clin. Investig. 2014, 124, 1853–1867. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Cabezudo, M.J.; El-Kharrag, R.; Torab, F.; Bashir, G.; George, J.A.; El-Taji, H.; al-Ramadi, B.K. Intravenous administration of manuka honey inhibits tumor growth and improves host survival when used in combination with chemotherapy in a melanoma mouse model. PLoS ONE 2013, 8, e55993. [Google Scholar] [CrossRef] [PubMed]
- Aryappalli, P.; Al-Qubaisi, S.S.; Attoub, S.; George, J.A.; Arafat, K.; Ramadi, K.B.; Mohamed, Y.A.; Al-Dhaheri, M.M.; Al-Sbiei, A.; Fernandez-Cabezudo, M.J.; et al. The IL-6/STAT3 Signaling Pathway Is an Early Target of Manuka Honey-Induced Suppression of Human Breast Cancer Cells. Front. Oncol. 2017, 7, 167. [Google Scholar] [CrossRef] [PubMed]
- Bromann, P.A.; Korkaya, H.; Courtneidge, S.A. The interplay between Src family kinases and receptor tyrosine kinases. Oncogene 2004, 23, 7957–7968. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Huang, W.C.; Li, P.; Guo, H.; Poh, S.B.; Brady, S.W.; Xiong, Y.; Tseng, L.M.; Li, S.H.; Ding, Z.; et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat. Med. 2011, 17, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Playford, M.P.; Schaller, M.D. The interplay between Src and integrins in normal and tumor biology. Oncogene 2004, 23, 7928–7946. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Huang, W.C.; Zhang, L.; Zhang, C.; Lowery, F.J.; Ding, Z.; Guo, H.; Wang, H.; Huang, S.; Sahin, A.A.; et al. SRC family kinases as novel therapeutic targets to treat breast cancer brain metastases. Cancer Res. 2013, 73, 5764–5774. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.M. Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigenesis. Oncogene 2004, 23, 8017–8023. [Google Scholar] [CrossRef]
- Huynh, J.; Etemadi, N.; Hollande, F.; Ernst, M.; Buchert, M. The JAK/STAT3 axis: A comprehensive drug target for solid malignancies. Semin. Cancer Biol. 2017, 45, 13–22. [Google Scholar] [CrossRef]
- Hartman, Z.C.; Poage, G.M.; den Hollander, P.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef]
- Johnstone, C.N.; Chand, A.; Putoczki, T.L.; Ernst, M. Emerging roles for IL-11 signaling in cancer development and progression: Focus on breast cancer. Cytokine Growth Factor. Rev. 2015, 26, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Liu, Y.; Liu, R.; Qi, J.; Hou, Y.; Chang, J.; Ren, L. Upregulation of IL-11, an IL-6 Family Cytokine, Promotes Tumor Progression and Correlates with Poor Prognosis in Non-Small Cell Lung Cancer. Cell. Physiol. Biochem. 2018, 45, 2213–2224. [Google Scholar] [CrossRef] [PubMed]
- Waugh, D.J.; Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef] [PubMed]
- Parsons, S.J.; Parsons, J.T. Src family kinases, key regulators of signal transduction. Oncogene 2004, 23, 7906–7909. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.W.; Deadman, B.J.; Manley-Harris, M.; Wilkins, A.L.; Alber, D.G.; Harry, E. Analysis of the flavonoid component of bioactive New Zealand manuka (Leptospermum scoparium) honey and the isolation, characterisation and synthesis of an unusual pyrrole. Food Chem. 2013, 141, 1772–1781. [Google Scholar] [CrossRef]
- Putoczki, T.L.; Dobson, R.C.; Griffin, M.D. The structure of human interleukin-11 reveals receptor-binding site features and structural differences from interleukin-6. Acta Crystallogr. D Biol. Crystallogr. 2014, 70 Pt 9, 2277–2285. [Google Scholar] [CrossRef]
- Varghese, J.N.; Moritz, R.L.; Lou, M.Z.; Van Donkelaar, A.; Ji, H.; Ivancic, N.; Branson, K.M.; Hall, N.E.; Simpson, R.J. Structure of the extracellular domains of the human interleukin-6 receptor alpha -chain. Proc. Natl. Acad. Sci. USA 2002, 99, 15959–15964. [Google Scholar] [CrossRef]
- Yawata, H.; Yasukawa, K.; Natsuka, S.; Murakami, M.; Yamasaki, K.; Hibi, M.; Taga, T.; Kishimoto, T. Structure-function analysis of human IL-6 receptor: Dissociation of amino acid residues required for IL-6-binding and for IL-6 signal transduction through gp130. EMBO J. 1993, 12, 1705–1712. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin. Immunol. 2014, 26, 54–74. [Google Scholar] [CrossRef]
- Salgado, R.; Junius, S.; Benoy, I.; Van Dam, P.; Vermeulen, P.; Van Marck, E.; Huget, P.; Dirix, L.Y. Circulating interleukin-6 predicts survival in patients with metastatic breast cancer. Int. J. Cancer 2003, 103, 642–646. [Google Scholar] [CrossRef]
- Heikkila, K.; Ebrahim, S.; Lawlor, D.A. Systematic review of the association between circulating interleukin-6 (IL-6) and cancer. Eur. J. Cancer 2008, 44, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Lippitz, B.E. Cytokine patterns in patients with cancer: A systematic review. Lancet Oncol. 2013, 14, e218–e228. [Google Scholar] [CrossRef]
- Boulanger, M.J.; Chow, D.C.; Brevnova, E.E.; Garcia, K.C. Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130 complex. Science 2003, 300, 2101–2104. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Fantini, M.C.; Schramm, C.; Lehr, H.A.; Wirtz, S.; Nikolaev, A.; Burg, J.; Strand, S.; Kiesslich, R.; Huber, S.; et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity 2004, 21, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Kloppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef] [PubMed]
- Scheller, J.; Garbers, C.; Rose-John, S. Interleukin-6: From basic biology to selective blockade of pro-inflammatory activities. Semin. Immunol. 2014, 26, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.T.; Chen, Q.; Skitzki, J.J.; Muhitch, J.B.; Zhou, L.; Appenheimer, M.M.; Vardam, T.D.; Weis, E.L.; Passanese, J.; Wang, W.C.; et al. IL-6 trans-signaling licenses mouse and human tumor microvascular gateways for trafficking of cytotoxic T cells. J. Clin. Investig. 2011, 121, 3846–3859. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef]
- Kang, S.; Tanaka, T.; Narazaki, M.; Kishimoto, T. Targeting Interleukin-6 Signaling in Clinic. Immunity 2019, 50, 1007–1023. [Google Scholar] [CrossRef]
- Oguro, T.; Ishibashi, K.; Sugino, T.; Hashimoto, K.; Tomita, S.; Takahashi, N.; Yanagida, T.; Haga, N.; Aikawa, K.; Suzutani, T.; et al. Humanised antihuman IL-6R antibody with interferon inhibits renal cell carcinoma cell growth in vitro and in vivo through suppressed SOCS3 expression. Eur. J. Cancer 2013, 49, 1715–1724. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Takahashi, F.; Motojima, S.; Nakashima, K.; Kaneko, N.; Hoshi, K.; Takahashi, K. Possible role for tocilizumab, an anti-interleukin-6 receptor antibody, in treating cancer cachexia. J. Clin. Oncol. 2013, 31, e69–e72. [Google Scholar] [CrossRef] [PubMed]
- Nagasaki, T.; Hara, M.; Nakanishi, H.; Takahashi, H.; Sato, M.; Takeyama, H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: Anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour-stroma interaction. Br. J. Cancer 2014, 110, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, H.; Hamaguchi, T.; Nagao, N.; Kato, S.; Iino, T.; Nakamura, T.; Sudo, A. Interleukin-6 receptor inhibitor suppresses bone metastases in a breast cancer cell line. Breast Cancer 2018, 25, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Niedzwiecki, A.; Roomi, M.W.; Kalinovsky, T.; Rath, M. Anticancer Efficacy of Polyphenols and Their Combinations. Nutrients 2016, 8, 552. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Khuda-Bukhsh, A.R. Quercetin Down-regulates IL-6/STAT-3 Signals to Induce Mitochondrial-mediated Apoptosis in a Nonsmall- cell Lung-cancer Cell Line, A549. J. Pharmacopunct. 2015, 18, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tong, Y.; Ying, J.; Lei, Z.; Wan, L.; Zhu, X.; Ye, F.; Mao, P.; Wu, X.; Pan, R.; et al. Chrysin induces cell growth arrest, apoptosis, and ER stress and inhibits the activation of STAT3 through the generation of ROS in bladder cancer cells. Oncol. Lett. 2018, 15, 9117–9125. [Google Scholar] [CrossRef] [PubMed]
- Selvendiran, K.; Koga, H.; Ueno, T.; Yoshida, T.; Maeyama, M.; Torimura, T.; Yano, H.; Kojiro, M.; Sata, M. Luteolin promotes degradation in signal transducer and activator of transcription 3 in human hepatoma cells: An implication for the antitumor potential of flavonoids. Cancer Res. 2006, 66, 4826–4834. [Google Scholar] [CrossRef]
- Fu, J.; Chen, D.; Zhao, B.; Zhao, Z.; Zhou, J.; Xu, Y.; Xin, Y.; Liu, C.; Luo, L.; Yin, Z. Luteolin induces carcinoma cell apoptosis through binding Hsp90 to suppress constitutive activation of STAT3. PLoS ONE 2012, 7, e49194. [Google Scholar] [CrossRef]
- Yang, M.Y.; Wang, C.J.; Chen, N.F.; Ho, W.H.; Lu, F.J.; Tseng, T.H. Luteolin enhances paclitaxel-induced apoptosis in human breast cancer MDA-MB-231 cells by blocking STAT3. Chem. Biol. Interact. 2014, 213, 60–68. [Google Scholar] [CrossRef]
- Aneknan, P.; Kukongviriyapan, V.; Prawan, A.; Kongpetch, S.; Sripa, B.; Senggunprai, L. Luteolin arrests cell cycling, induces apoptosis and inhibits the JAK/STAT3 pathway in human cholangiocarcinoma cells. Asian Pac. J. Cancer Prev. 2014, 15, 5071–5076. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Dai, S.; Dai, J.; Xiao, Y.; Bai, Y.; Chen, B.; Zhou, M. Luteolin decreases invasiveness, deactivates STAT3 signaling, and reverses interleukin-6 induced epithelial-mesenchymal transition and matrix metalloproteinase secretion of pancreatic cancer cells. Oncol. Targets Ther. 2015, 8, 2989–3001. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Su, Z.; Xu, H.; Niu, M.; Chen, X.; Min, H.; Zhang, B.; Sun, G.; Xie, S.; Wang, H.; et al. Luteolin selectively kills STAT3 highly activated gastric cancer cells through enhancing the binding of STAT3 to SHP-1. Cell Death Dis. 2017, 8, e2612. [Google Scholar] [CrossRef] [PubMed]
- Sonoki, H.; Tanimae, A.; Endo, S.; Matsunaga, T.; Furuta, T.; Ichihara, K.; Ikari, A. Kaempherol and Luteolin Decrease Claudin-2 Expression Mediated by Inhibition of STAT3 in Lung Adenocarcinoma A549 Cells. Nutrients 2017, 9, 597. [Google Scholar] [CrossRef] [PubMed]
- Matadeen, R.; Hon, W.C.; Heath, J.K.; Jones, E.Y.; Fuller, S. The dynamics of signal triggering in a gp130-receptor complex. Structure 2007, 15, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Crichton, M.B.; Nichols, J.E.; Zhao, Y.; Bulun, S.E.; Simpson, E.R. Expression of transcripts of interleukin-6 and related cytokines by human breast tumors, breast cancer cells, and adipose stromal cells. Mol. Cell Endocrinol. 1996, 118, 215–220. [Google Scholar] [CrossRef]
- Lacroix, M.; Siwek, B.; Marie, P.J.; Body, J.J. Production and regulation of interleukin-11 by breast cancer cells. Cancer Lett. 1998, 127, 29–35. [Google Scholar] [CrossRef]
- Afrin, S.; Haneefa, S.M.; Fernandez-Cabezudo, M.J.; Giampieri, F.; al-Ramadi, B.K.; Battino, M. Therapeutic and preventive properties of honey and its major bioactive compounds in cancer: An evidence-based review. Nutr. Res. Rev. 2019, in press. [Google Scholar]
- Al-Ramadi, B.K.; Zhang, H.; Bothwell, A.L. Cell-cycle arrest and apoptosis hypersusceptibility as a consequence of Lck deficiency in nontransformed T lymphocytes. Proc. Natl. Acad. Sci. USA 1998, 95, 12498–12503. [Google Scholar] [CrossRef]
- Fernandez-Cabezudo, M.J.; Lorke, D.E.; Azimullah, S.; Mechkarska, M.; Hasan, M.Y.; Petroianu, G.A.; al-Ramadi, B.K. Cholinergic stimulation of the immune system protects against lethal infection by Salmonella enterica serovar Typhimurium. Immunology 2010, 130, 388–398. [Google Scholar] [CrossRef]
- Su, J.L.; Lai, K.P.; Chen, C.A.; Yang, C.Y.; Chen, P.S.; Chang, C.C.; Chou, C.H.; Hu, C.L.; Kuo, M.L.; Hsieh, C.Y.; et al. A novel peptide specifically binding to interleukin-6 receptor (gp80) inhibits angiogenesis and tumor growth. Cancer Res. 2005, 65, 4827–4835. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W2777. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Flavonoid | Cluster | Estimated ∆G (kcal/mol) | Hydrogen Bonds |
|---|---|---|---|
| Luteolin (Lut) | 5 | −6.72 | Lut H7−−Pro7 O (1.851 Å) Lut H9−−Ala127 O (2.100 Å) Cys146 HN−−Lut O5 (2.341 Å) Cys174 SG−−Lut O3 (3.205 Å) |
| Quercetin (Que) | 8 | −6.79 | Que H6−−Pro7 O (2.323 Å) Que H10−−Cys146 O (2.437 Å) Cys174 SG−−Que O3 (3.495 Å) |
| Chrysin (Chr) | 6 | −6.42 | Chr H9−−Pro7 O (2.072 Å) Cys174 SG−−Chr O3 (3.307 Å) |
| Pinocembrin (Pin) | 26 | −6.25 | Pin H11−−Lys126 O (2. 396 Å) Cys174SG−−Pin O3 (3.472Å) |
| Galangin (Gal) | 14 | −6.83 | Gal H10−−Arg5 O (2.572 Å) Gal H8−−Glu10 OE2 (1.983 Å) Gln143 HE21−−Gal O1 (2.651 Å) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aryappalli, P.; Shabbiri, K.; Masad, R.J.; Al-Marri, R.H.; Haneefa, S.M.; Mohamed, Y.A.; Arafat, K.; Attoub, S.; Cabral-Marques, O.; Ramadi, K.B.; et al. Inhibition of Tyrosine-Phosphorylated STAT3 in Human Breast and Lung Cancer Cells by Manuka Honey is Mediated by Selective Antagonism of the IL-6 Receptor. Int. J. Mol. Sci. 2019, 20, 4340. https://doi.org/10.3390/ijms20184340
Aryappalli P, Shabbiri K, Masad RJ, Al-Marri RH, Haneefa SM, Mohamed YA, Arafat K, Attoub S, Cabral-Marques O, Ramadi KB, et al. Inhibition of Tyrosine-Phosphorylated STAT3 in Human Breast and Lung Cancer Cells by Manuka Honey is Mediated by Selective Antagonism of the IL-6 Receptor. International Journal of Molecular Sciences. 2019; 20(18):4340. https://doi.org/10.3390/ijms20184340
Chicago/Turabian StyleAryappalli, Priyanka, Khadija Shabbiri, Razan J. Masad, Roadha H. Al-Marri, Shoja M. Haneefa, Yassir A. Mohamed, Kholoud Arafat, Samir Attoub, Otavio Cabral-Marques, Khalil B. Ramadi, and et al. 2019. "Inhibition of Tyrosine-Phosphorylated STAT3 in Human Breast and Lung Cancer Cells by Manuka Honey is Mediated by Selective Antagonism of the IL-6 Receptor" International Journal of Molecular Sciences 20, no. 18: 4340. https://doi.org/10.3390/ijms20184340
APA StyleAryappalli, P., Shabbiri, K., Masad, R. J., Al-Marri, R. H., Haneefa, S. M., Mohamed, Y. A., Arafat, K., Attoub, S., Cabral-Marques, O., Ramadi, K. B., Fernandez-Cabezudo, M. J., & al-Ramadi, B. K. (2019). Inhibition of Tyrosine-Phosphorylated STAT3 in Human Breast and Lung Cancer Cells by Manuka Honey is Mediated by Selective Antagonism of the IL-6 Receptor. International Journal of Molecular Sciences, 20(18), 4340. https://doi.org/10.3390/ijms20184340