Clinical Trials with Combination of Cytokine-Induced Killer Cells and Dendritic Cells for Cancer Therapy

Abstract
1. Introduction
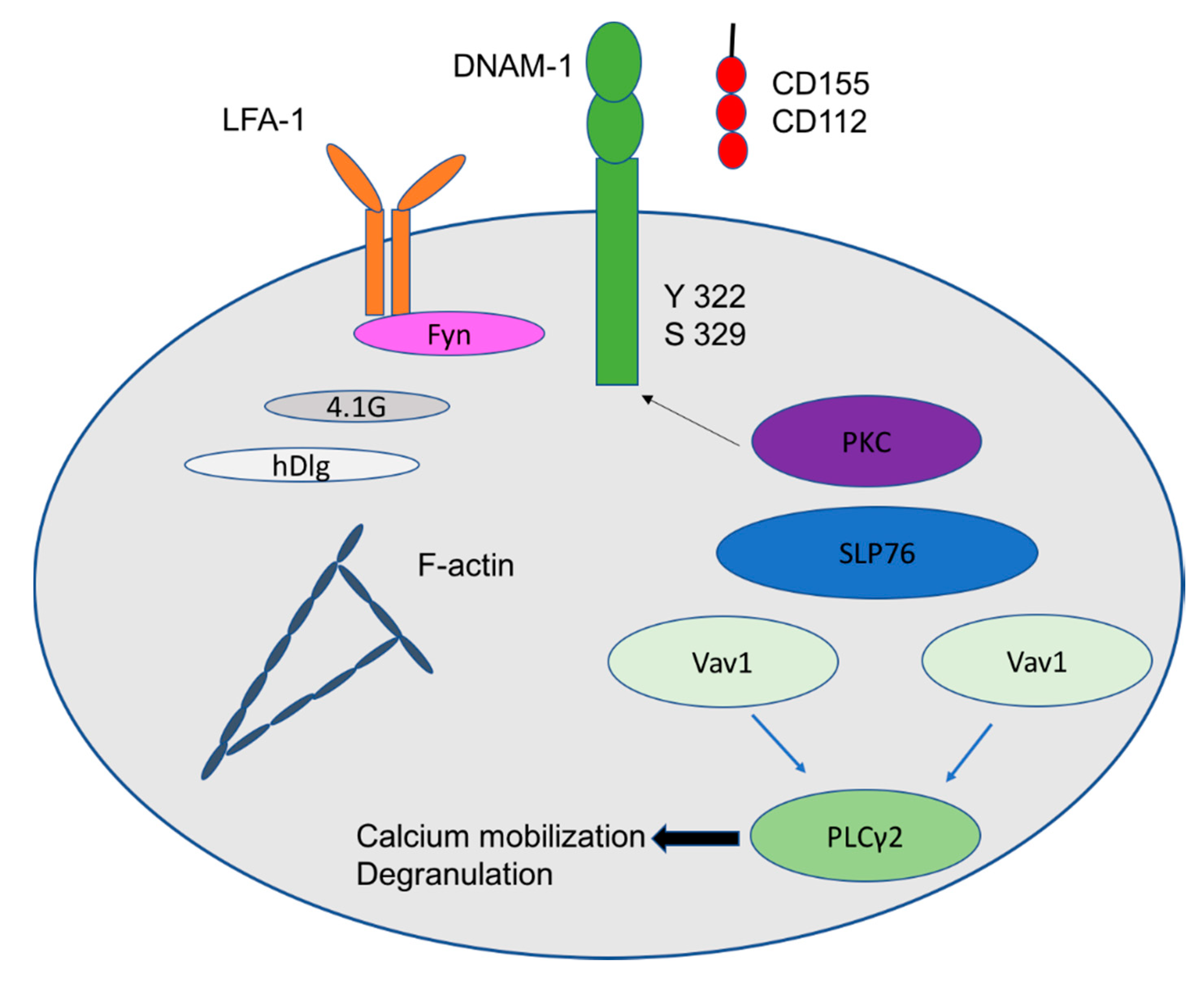
1.1. Activation Receptors: NKG2D, DNAM-1, CD56 and NKp30
1.2. Inhibitory Receptor
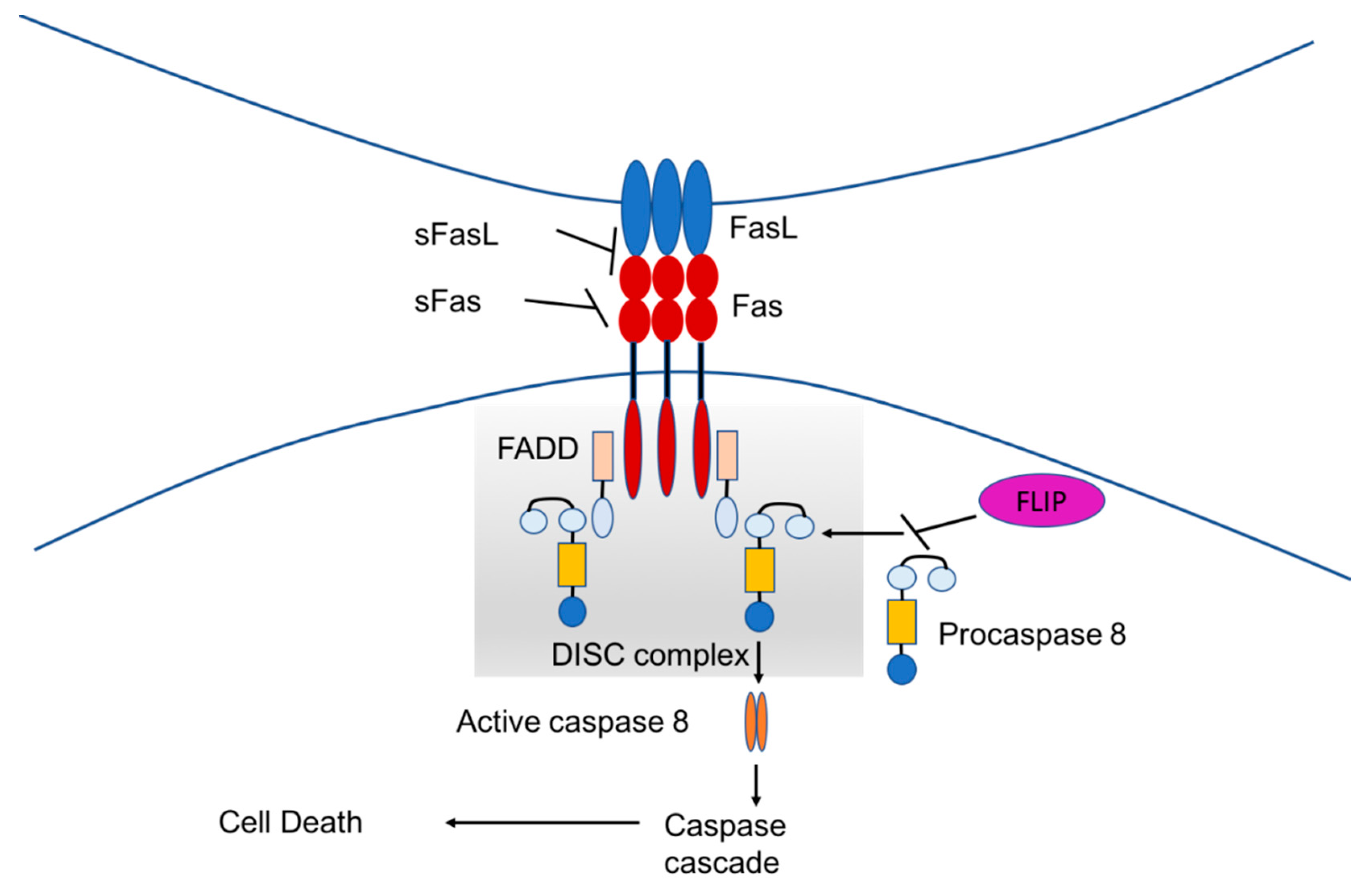
1.3. Fas-Mediated Apoptosis
2. Gastrointestinal Tumors
2.1. Hepatocellular Carcinoma
2.2. Immunotherapy with CIK Alone or in Combination with DC as Adjuvant Therapy After Resection or RFA
2.3. Immunotherapy with CIK Alone or in Combination with DC in Combination with TACE (Palliative)
2.4. Gastric Cancer
2.5. Colorectal Liver Metastases
2.6. Pancreatic Cancer
3. Lung Cancer
4. Breast Cancer
4.1. Classification and Therapy of Breast cancer
4.2. Meta-Analysis and Stage IV Breast Cancer with the Follow-up Time Up to 10 Years
5. Glioblastoma
6. Renal Cell Carcinoma
7. Hematological Diseases
8. Future Perspectives
9. Conclusions
Funding
Conflicts of Interest
References
- Schmidt-Wolf, I.G.; Negrin, R.S.; Kiem, H.P.; Blume, K.G.; Weissman, I.L. Use of a SCID mouse/human lymphoma model to evaluate cytokine-induced killer cells with potent antitumor activity. Exp. Med. 1991, 174, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Sangiolo, D.; Martinuzzi, E.; Todorovic, M.; Vitaggio, K.; Vallario, A.; Jordaney, N.; Carnevale-Schianca, F.; Capaldi, A.; Geuna, M.; Casorzo, L.; et al. Alloreactivity and anti-tumor activity segregate within two distinct subsets of cytokine-induced killer (CIK) cells: Implications for their infusion across major HLA barriers. Int. Immunol. 2008, 20, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Lopez, R.D.; Waller, E.K.; Lu, P.H.; Negrin, R.S. CD58/LFA-3 and IL-12 provided by activated monocytes are critical in the in vitro expansion of CD56+ T cells. Cancer Immunol. Immunother. 2000, 49, 629–640. [Google Scholar] [CrossRef]
- Verneris, M.R.; Karami, M.; Baker, J.; Jayaswal, A.; Negrin, R.S. Role of NKG2D signaling in the cytotoxicity of activated and expanded CD8+T cells. Blood 2004, 103, 3065–3072. [Google Scholar] [CrossRef] [PubMed]
- Groh, V.; Rhinehart, R.; Secrist, H.; Bauer, S.; Grabstein, K.H.; Spies, T. Broad tumor-associated expression and recognition by tumor-derived γδ T cells of MICA and MICB. Proc. Natl. Acad. Sci. USA 1999, 96, 6879–6884. [Google Scholar] [CrossRef] [PubMed]
- Iudicone, P.; Fioravanti, D.; Cicchetti, E.; Zizzari, I.G.; Pandolfi, A.; Scocchera, R.; Fazzina, R.; Pierelli, L. Interleukin-15 enhances cytokine induced killer (CIK) cytotoxic potential against epithelial cancer cell lines via an innate pathway. Hum. Immunol. 2016, 77, 1239–1247. [Google Scholar] [CrossRef]
- Rettinger, E.; Kuçi, S.; Naumann, I.; Becker, P.; Kreyenberg, H.; Anzaghe, M.; Willasch, A.; Koehl, U.; Bug, G.; Ruthardt, M.; et al. The cytotoxic potential of interleukin-15-stimulated cytokine-induced killer cells against leukemia cells. Cytotherapy 2012, 14, 91–103. [Google Scholar] [CrossRef]
- Rajbhandary, S.; Zhao, M.F.; Zhao, N.; Lu, W.Y.; Zhu, H.B.; Xiao, X.; Deng, Q.; Li, Y.M. Multiple cytotoxic factors involved in IL-21 enhanced antitumor function of CIK cells signaled through STAT-3 and STAT5b pathways. Asian Pac. J. Cancer Prev. 2013, 14, 5825–5831. [Google Scholar] [CrossRef]
- Cappuzzello, E.; Tosi, A.; Zanovello, P.; Sommaggio, R.; Rosato, A. Retargeting cytokine-induced killer cell activity by CD16 engagement with clinical-grade antibodies. Oncoimmunology 2016, 5, e1199311. [Google Scholar] [CrossRef]
- Schmidt-Wolf, G.D.; Negrin, R.S.; Schmidt-Wolf, I.G. Activated T cells and cytokine-induced CD3+CD56+killer cells. Ann. Hematol. 1997, 74, 51–56. [Google Scholar] [CrossRef]
- Thorne, S.H.; Negrin, R.S.; Contag, C.H. Synergistic antitumor effects of immune cell-viral biotherapy. Science 2006, 311, 1780–1784. [Google Scholar] [CrossRef] [PubMed]
- Marten, A.; Ziske, C.; Schottker, B.; Renoth, S.; Weineck, S.; Buttgereit, P.; Schakowski, F.; Von Rücker, A.; Sauerbruch, T.; Schmidt-Wolf, I.G.H. Interactions between dendritic cells and cytokine-induced killer cells lead to an activation of both populations. J. Immunother. 2001, 24, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Franceschetti, M.; Pievani, A.; Borleri, G.; Vago, L.; Fleischhauer, K.; Golay, J.; Introna, M. Cytokine-induced killer cells are terminally differentiated activated CD8 cytotoxic T-EMRA lymphocytes. Exp. Hematol. 2009, 37, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Jelenčić, V.; Lenartić, M.; Wensveen, F.M.; Polić, B. NKG2D: A versatile player in the immune system. Immunol. Lett. 2017, 189, 48–53. [Google Scholar] [CrossRef]
- Salih, H.R.; Antropius, H.; Gieseke, F.; Lutz, S.Z.; Kanz, L.; Rammensee, H.G.; Steinle, A. Functional expression and release of ligands for the activating immunoreceptor NKG2D in leukemia. Blood 2003, 102, 1389–1396. [Google Scholar] [CrossRef]
- Pende, D.; Rivera, P.; Marcenaro, S.; Chang, C.C.; Biassoni, R.; Conte, R.; Kubin, M.; Cosman, D.; Ferrone, S.; Moretta, L.; et al. Major histocompatibility complex class I-related chain A and UL16-binding protein expression on tumor cell lines of different histotypes: Analysis of tumor susceptibility to NKG2D-dependent natural killer cell cytotoxicity. Cancer Res. 2002, 62, 6178–6186. [Google Scholar] [PubMed]
- De Andrade, L.F.; Smyth, M.J.; Martinet, L. DNAM-1 control of natural killer cells functions through nectin and nectin-like proteins. Immunol. Cell Biol. 2014, 92, 237–244. [Google Scholar] [CrossRef]
- Shibuya, A.; Lanier, L.L.; Phillips, J.H. Protein Kinase C Is Involved in the Regulation of Both Signaling and Adhesion Mediated by DNAX Accessory Molecule-1 Receptor. J. Immunol. 1998, 161, 1671–1676. [Google Scholar]
- Valgardsdottir, R.; Capitanio, C.; Texido, G.; Pende, D.; Cantoni, C.; Pesenti, E.; Rambaldi, A.; Golay, J.; Introna, M. Direct involvement of CD56 in cytokine-induced killer-mediated lysis of CD56+ hematopoietic target cells. Exp. Hematol. 2014, 42, 1013–1021. [Google Scholar] [CrossRef]
- Dai, C.; Lin, F.; Geng, R.; Ge, X.; Tang, W.; Chang, J.; Wu, Z.; Liu, X.; Lin, Y.; Zhang, Z.; et al. Implication of combined PD-L1/PD-1 blockade with cytokine-induced killer cells as a synergistic immunotherapy for gastrointestinal cancer. Oncotarget 2016, 7, 10332–10344. [Google Scholar] [CrossRef]
- O’Brien, D.I.; Nally, K.; Kelly, R.G.; O’Connor, T.M.; Shanahan, F.; O’Connell, J. Targeting the Fas/Fas ligand pathway in cancer. Expert Opin. Ther. Targets 2005, 9, 1031–1044. [Google Scholar] [CrossRef] [PubMed]
- Waring, P.; Müllbacher, A. Cell death induced by the Fas/Fas ligand pathway and its role in pathology. Immunol. Cell Biol. 1999. [Google Scholar] [CrossRef] [PubMed]
- Verneris, M.R.; Kornacker, M.; Mailänder, V.; Negrin, R.S. Resistance of ex vivo expanded CD3+CD56+ T cells to Fas-mediated apoptosis. Cancer Immunol. Immunother. 2000, 49, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Durrieu, L.; Dieng, M.M.; Le Deist, F.; Haddad, E. Cord blood-derived and peripheral blood-derived cytokine-induced killer cells are sensitive to Fas-mediated apoptosis. Biol. Blood Marrow Transplant. 2013, 9, 1407–1411. [Google Scholar] [CrossRef] [PubMed]
- Ladhams, A.; Schmidt, C.; Sing, G.; Butterworth, L.; Fielding, G.; Tesar, P.; Strong, R.; Leggett, B.; Powell, L.; Maddern, G.; et al. Treatment of non-resectable hepatocellular carcinoma with autologous tumor-pulsed dendritic cells. J. Gastroenterol. Hepatol. 2002, 17, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.; Briux, J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: Chemoembolization improves survival. Hepatology 2003, 37, 429–442. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Zhu, A.X.; Park, J.O.; Ryoo, B.Y.; Yen, C.J.; Poon, R.; Pastorelli, D.; Blanc, J.F.; Chung, H.C.; Baron, A.D.; Pfiffer, T.E.; et al. Ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib (REACH): A randomised, double-blind, multicentre, phase 3 trial. Lancet 2015. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Takayama, T.; Sekine, T.; Makuuchi, M.; Yamasaki, S.; Kosuge, T.; Yamamoto, J.; Shimada, K.; Sakamoto, M.; Hirohashi, S.; Ohashi, Y.; et al. Adoptive immunotherapy to lower postsurgical recurrence rates of hepatocellular carcinoma: A randomized trial. Lancet 2000, 356, 802–807. [Google Scholar] [CrossRef]
- Xie, F.; Zhang, X.; Li, H.; Zheng, T.; Xu, F.; Shen, R.; Yan, L.; Yang, J.; He, J. Adoptive Immunotherapy in Postoperative Hepatocellular Carcinoma: A Systemic Review. PLoS ONE 2012, 7, e42879. [Google Scholar] [CrossRef]
- Zhou, W.; Wu, M.; Yao, X.; Yang, J. The Effects of Combined Hepatectomy and Immunochemotherapy on Postoperative Recurrence of Primary Liver Cancer. Chin. Ger. J. Clin. Oncol. 2002, 1, 163–165. [Google Scholar] [CrossRef]
- Xie, L.; Pang, R.; Jin, Y.; Xiang, S.; Li, H. Effects of hepatic artery chemotherapeutic embolization combined with perfusing LAK cells into hepatic artery after radical operation of liver cancer. Chin. J. Hepatol. 2000, 8, 142–143. [Google Scholar]
- Wang, H.; Liu, A.; Bo, W.; Feng, X.; Hu, Y.; Tian, L.; Zhang, H.; Tang, X. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma patients after curative resection, a systematic review and meta-analysis. Dig. Liver Dis. 2016, 48, 1275–1282. [Google Scholar] [CrossRef]
- Hao, M.Z.; Lin, H.L.; Chen, Q.; Ye, Y.B.; Chen, Q.Z.; Chen, M.S. Efficacy of transcatheter arterial chemoembolization combined with cytokine-induced killer cell therapy on hepatocellular carcinoma: A comparative study. Chin. J. Cancer 2010, 29, 172–177. [Google Scholar] [CrossRef]
- Wang, J.P.; Li, W.; Huang, Z.L.; Wu, P.H.; Li, X.S.; Wei, Y.D.; Zhou, Q.M.; Pan, C.C.; Xia, J.C.; Zhao, M. Value of CIK in the treatment of TACE combined with RFA for HCC in long-term survival and prognostic analysis. Chin. Med J. 2012, 92, 3062–3066. [Google Scholar]
- Lee, J.H.; Lee, J.H.; Lim, Y.S.; Yeon, J.E.; Song, T.J.; Yu, S.J.; Gwak, G.Y.; Kim, K.M.; Kim, Y.J.; Lee, J.W.; et al. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology 2015, 148, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Zheng, C.; Huo, H.; Zhang, H.; Zhu, Z.; Li, J.; Zhang, H. TACE combined with dendritic cells and cytokine-induced killer cells in the treatment of hepatocellular carcinoma: A meta-analysis. Int. Immunopharmacol. 2016, 40, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.R.; Li, X.; Lin, J.X.; Wang, T.T.; Dong, M.; Chen, Z.H.; Jia, C.C.; Hong, Y.F.; Lin, Q.; Wu, X.Y. Autologous transplantation of cytokine-induced killer cells as an adjuvant therapy for hepatocellular carcinoma in Asia: An update meta-analysis and systematic review. Oncotarget 2017, 8, 31318–31328. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Global Cancer Facts & Figures, 2nd ed.; American Cancer Society: Atlanta, GA, USA, 2011. [Google Scholar]
- Liu, K.; Song, G.; Hu, X.; Zhou, Y.; Li, Y.; Chen, Q.; Feng, G. A Positive Role of Cytokine-Induced Killer Cell Therapy on Gastric Cancer Therapy in a Chinese Population: A Systematic Meta-Analysis. Med. Sci. Monit. 2015, 21, 3363–3370. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, V.; von Weikersthal, L.F.; Decker, T.; Kiani, A.; Vehling-Kaiser, U.; Al-Batran, S.E.; Heintges, T.; Lerchenmüller, C.; Kahl, C.; Seipelt, G.; et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1065–1075. [Google Scholar] [CrossRef]
- Mizukoshi, E.; Yamashita, T.; Arai, K.; Sunagozaka, H.; Ueda, T.; Arihara, F.; Kagaya, T.; Fushimi, K.; Kaneko, S. Enhancement of tumor-associated antigen-specific t cell responses by radiofrequency ablation of hepatocellular carcinoma. Hepatology 2013, 57, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Nakatsura, T. Radiofrequency ablation for hepatocellular carcinoma induces glypican-3 peptide-specific cytotoxic t lymphocytes. Int. J. Oncol. 2012, 40, 63–70. [Google Scholar]
- Li, X.; Dai, X.; Shi, L.; Jiang, Y.; Chen, X.; Chen, L.; Zhao, J.; Qiang, W.; Wu, J.; Ji, M.; et al. Phase II/III Study of Radiofrequency Ablation Combined with Cytokine-Induced Killer Cells Treating Colorectal Liver Metastases. Cell Physiol. Biochem. 2016, 40, 137–145. [Google Scholar] [CrossRef]
- Lowenfels, A.B.; Maisonneuve, P. Epidemiology and risk factors for pancreatic cancer. Best Pract. Res. Clin. Gastroenterol. 2006, 20, 197–209. [Google Scholar] [CrossRef]
- Hidalgo, M. Pancreatic Cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic Adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Christine Cripps, M.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, X. Primary analysis for clinical efficacy of immunotherapy in patients with pancreatic cancer. Immunotherapy 2015, 8, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cao, J.X.; Pan, J.H.; Liu, Y.S.; Xu, B.L.; Li, D.; Zhang, X.Y.; Li, J.L.; Liu, J.L.; Wang, H.B.; et al. Adoptive immunotherapy of cytokine-induced killer cell therapy in the treatment of non-small cell lung cancer. PLoS ONE 2014, 9, e112662. [Google Scholar] [CrossRef]
- Zheng, F.C.; Zhang, X.Y.; Feng, H.Z.; Chen, J.; Sun, Y.; Zhong, G. Clinical Study of Stereotactic Conformal Body c-knife Combined with Adoptive Immunotherapy (Dendritic Cell and Cytokine-induced Killer Cell) in the Treatment for Advanced Non-small Cell Lung Cancer. J. Chin. Oncol. 2012, 18, 815–818. [Google Scholar]
- Wang, Y.Y.; Wang, Y.S.; Liu, T.; Yang, K.; Yang, G.Q.; Liu, H.C.; Wang, S.S.; Yang, J.L. Efficacy study of Cyber Knife stereotactic radio surgery combined with CIK cell immunotherapy for advanced refractory lung cancer. Exp. Ther. Med. 2013, 5, 453–456. [Google Scholar] [CrossRef]
- Mi, D.; Ren, W.; Yang, K. Adoptive immunotherapy with interleukin-2 & induced killer cells in non-small cell lung cancer: A systematic review and meta-analysis. Indian J. Med. Res. 2016, 143, S1–S10. [Google Scholar] [CrossRef]
- Lange, J.R.; Raubitschek, A.A.; Pockaj, B.A.; Spencer, W.F.; Lotze, M.T.; Topalian, S.L.; Yang, J.C.; Rosenberg, S.A. A pilot study of the combination of interleukin-2-based immunotherapy and radiation therapy. J. Immunother. 1992, 12, 265–271. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Lotze, M.T.; Yang, J.C.; Aebersold, P.M.; Linehan, W.M.; Seipp, C.A.; White, D.E. Experience with the use of high-dose interleukin-2 in the treatment of 652 cancer patients. Ann. Surg. 1989, 210, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Treisman, J.; Hwu, P.; Minamoto, S.; Shafer, G.E.; Cowherd, R.; Morgan, R.A.; Rosenberg, S.A. Interleukin-2-transduced lymphocytes grow in an autocrine fashion and remain responsive to antigen. Blood 1995, 85, 139–145. [Google Scholar] [PubMed]
- Xiao, Z.; Wang, C.Q.; Zhou, M.H.; Li, N.N.; Liu, S.Y.; He, Y.J.; Wang, Y.Z.; Feng, J.H.; Yao, X.S.; Chen, L.; et al. Clinical efficacy and safety of CIK plus radiotherapy for lung cancer: A meta-analysis of 16 randomized controlled trials. Int. Immunopharmacol. 2018, 61, 363–375. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef]
- Gralow, J.R. Breast cancer 2004: Progress and promise on the clinical front. Phys. Med. 2006, 21, 2. [Google Scholar] [CrossRef]
- American Cancer Society. Breast Cancer Facts & Figures 2017–18; American Cancer Society: Atlanta, GA, USA, 2017. [Google Scholar]
- Wang, Z.X.; Cao, J.X.; Wang, M.; Li, D.; Cui, Y.X.; Zhang, X.Y.; Liu, J.L.; Li, J.L. Adoptive cellular immunotherapy for the treatment of patients with breast cancer: A meta-analysis. Cytotherapy 2014, 16, 934–945. [Google Scholar] [CrossRef]
- Lin, M.; Liang, S.; Jiang, F.; Xu, J.; Zhu, W.; Qian, W.; Hu, Y.; Zhou, Z.; Chen, J.; Niu, L.; et al. 2003-2013, A valuable study: Autologous tumor lysate-pulsed dendritic cell immunotherapy with cytokine-induced killer cells improves survival in stage IV breast cancer. Immunol. Lett. 2017, 183, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Hu, J.; Liu, X.; Hu, C.; Li, M.; Han, W. Effect and safety of cytokine-induced killer (CIK) cell immunotherapy in patients with breast cancer: A meta-analysis. Medicine 2017, 96, e8310. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2008–2012. Neuro Oncol. 2015, 17, iv1–iv62. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The brain tumor microenvironment. Glia 2012, 60, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Szulzewsky, F.; Yerevanian, A.; Chen, Z.; Heinzmann, D.; Rasmussen, R.D.; Alvarez-Garcia, V.; Kim, Y.; Wang, B.; Tamagno, I. Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget 2015, 6, 15077–15094. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.S.; Nam, D.H.; Kang, S.H.; Lee, J.W.; Chang, J.H.; Kim, J.H.; Lim, Y.J.; Koh, Y.C.; Chung, Y.G.; Kim, J.M.; et al. Phase III randomized trial of autologous cytokine-induced killer cell immunotherapy for newly diagnosed glioblastoma in Korea. Oncotarget 2017, 8, 7003–7013. [Google Scholar] [CrossRef] [PubMed]
- Muglia, V.F.; Prando, A. Renal cell carcinoma: Histological classification and correlation with imaging findings. Radiol. Bras. 2015, 48, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.X.; Li, J.L.; Cao, J.X.; Liu, Y.S.; Li, D.; Zhang, X.Y.; Wang, M.; Wu, M.; Xu, B.L.; Liu, J.L.; et al. Cytokine-induced killer cells in the treatment of patients with renal cell carcinoma: A pooled meta-analysis. Immunotherapy 2014, 6, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, Z.; Li, H.; Huang, J.; Yang, S.; Xie, T.; Huang, L.; Yue, D.; Xu, L.; Wang, L.; et al. Cytokine induced killer cell-based immunotherapies in patients with different stages of renal cell carcinoma. Cancer Lett. 2015, 362, 192–198. [Google Scholar] [CrossRef]
- Vercauteren, S.M.; Starczynowski, D.T.; Sung, S.; McNeil, K.; Salski, C.; Jensen, C.L.; Bruyere, H.; Lam, W.L.; Karsan, A. T cells of patients with myelodysplastic syndrome are frequently derived from the malignant clone. Br. J. Haematol. 2012, 156, 409–412. [Google Scholar] [CrossRef]
- Jung, S.H.; Lee, H.J.; Vo, M.C.; Kim, H.J.; Lee, J.J. Immunotherapy for the treatment of multiple myeloma. Crit. Rev. Oncol. Hematol. 2017, 111, 87–93. [Google Scholar] [CrossRef]
- Wang, Y.; Lv, B.; Li, K.; Zhang, A.; Liu, H. Adjuvant immunotherapy of dendritic cells and cytokine-induced killer cells is safe and enhances chemotherapy efficacy for multiple myeloma in China: A meta-analysis of clinical trials. Drug Des. Dev. Ther. 2017, 11, 3245–3256. [Google Scholar] [CrossRef][Green Version]
- Bader, P. CIK-Cells in Relapsing Patients With Acute Leukemia or Myelodysplastic Syndromes After SCT. Available online: https://clinicaltrials.gov/ct2/show/NCT02752243 (accessed on 2 September 2019).
- Yu, S.; Li, A.; Liu, Q.; Li, T.; Yuan, X.; Han, X.; Wu, K. Chimeric antigen receptor T cells: A novel therapy for solid tumors. J. Hematol. Oncol. 2017. [Google Scholar] [CrossRef]
- Liu, B.; Song, Y.; Liu, D. Clinical trials of CAR-T cells in China. J. Hematol. Oncol. 2017, 10, 166. [Google Scholar] [CrossRef] [PubMed]
- Schmeel, L.C.; Schmeel, F.C.; Coch, C.; Schmidt-Wolf, I.G.H. Cytokine-induced killer (CIK) cells in cancer immunotherapy: Report of the international registry on CIK cells (IRCC). J. Cancer Res. Clin. Oncol. 2015, 141, 839–849. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Study Reference | Patients (n) Total | Patients (n) Treated with CIK Cells | Therapy | Results | Conclusions |
|---|---|---|---|---|---|
| Takayama et al. (2000) [34] | 150 | 76 | Resection; Immunotherapy group: additional infusions of lymphocytes activated in vitro with rIL-2 and anti-CD3 | Better recurrence-free in immunotherapy group; no differences in OS between the two groups | CIK cell therapy can improve recurrence-free outcomes after surgery |
| Hao et al. (2010) [39] | 146 | 72 | TACE; Immunotherapy group: additional i.v. CIK cell transfusions | Improved PFS and OS in TACE plus CIK cell therapy group | Adjuvant immunotherapy with CIK cells improve the efficacy of TACE in HCC patients |
| Wang et al. (2012) [40] | 95 | 48 | TACE/RFA; Immunotherapy group: additional CIK cell transfusions | Significantly longer DFS in CIK cell plus TACE with RFA group; no significant difference for OS | Recurrence can be controlled by CIK infusion combined with TACE and RFA |
| Lee et al. (2015) [41] | 230 | 115 | TACE/RFA or percutaneous ethanol injection PEI Immunotherapy group: additional i.v. CIK cell transfusions | RFS and OS significantly better in immunotherapy group; no significant for severe adverse events between the two groups | CIK cells can prevent recurrence and improve survival in combination with surgical resection, RFA or percutaneous ethanol injection |
| Event | No. of Trials | No. pts | Before-CIK | Mean Difference | 95% CI | p Value | Heterogeneity |
|---|---|---|---|---|---|---|---|
| CD3+ | 9 | 359 | 359 | 8.21 | 5.79 to 10.64 | <0.00001 | 67% |
| CD4+ | 5 | 174 | 174 | 5.59 | 4.10 to 7.07 | <0.0001 | 0% |
| CD3+CD8+ | 4 | 174 | 174 | 2.55 | −2.46 to 7.56 | 0.32 | 89% |
| CD4+CD8+ | 4 | 144 | 144 | 0.49 | 0.37 to 0.61 | <0.00001 | 53% |
| CD3+CD56+ | 6 | 222 | 222 | 7.80 | 2.61 to 12.98 | 0.003 | 99% |
| NK | 4 | 154 | 154 | 6.21 | 2.25 to 10.17 | 0.002 | 90% |
| CD8+ | 5 | 174 | 174 | -2.75 | −3.88 to −1.63 | <0.00001 | 0% |
| Treg | 3 | 153 | 153 | -1.26 | −1.94 to −0.58 | 0.0003 | 58% |
| Study Reference | RCTs (n) Total | Patients (n) Total | Therapy |
|---|---|---|---|
| Wang et al. (2014) [57] | 17 | 1172 | Chemotherapy; Immunotherapy group: additional CIK cell and DC cell transfusions |
| Mi et al. (2016) [60] | 10 | Chemotherapy; Immunotherapy group: additional CIK cell transfusions and IL-2 | |
| Xiao et al. (2018) [64] | 16 | 1197 | Radiotherapy; Immunotherapy group: additional CIK cell and DC cell transfusions |
| Total | 43 | 2369 |
| Study Reference | RCTs (n) Total | Patients (n) Total | Therapy |
|---|---|---|---|
| Wang et al. (2014) [68] | 633 | CIK cell therapy alone, DC cell therapy alone and CIK and DC combination | |
| Hu et al. (2017) [70] | 11 | 914 | Chemotherapy; Immunotherapy group: additional CIK cell and DC cell transfusions |
| Total | 11 | 1547 |
| Study Reference | RCTs (n) Total | Patients (n) Total | Therapy | CR | PR | ORR | DCR | PD |
|---|---|---|---|---|---|---|---|---|
| Wang et al., 2017, [81] | 12 | 594 | Chemotherapy; | OR = 2.71 CI = 1.60–4.58 p = 0.0002 | OR = 1.49 CI = 1.01–2.20 p = 0.04 | OR = 2.77 CI = 1.88–4.10 p < 0.00001 | OR = 2.90 CI = 1.72–4.90 p < 0.0001 | OR = 0.34 CI = 0.20–0.58 p < 0.0001 |
| Immunotherapy group: additional CIK cell and DC cell transfusions | ||||||||
| Bader, 2016, [82] | 1 | 40 | Chemotherapy; | n.a | n.a | n.a. | n.a. | n.a. |
| Immunotherapy group: additional IL-15 activated CIK cell transfusions | ||||||||
| Total | 13 | 634 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garofano, F.; Gonzalez-Carmona, M.A.; Skowasch, D.; Schmidt-Wolf, R.; Abramian, A.; Hauser, S.; Strassburg, C.P.; Schmidt-Wolf, I.G.H. Clinical Trials with Combination of Cytokine-Induced Killer Cells and Dendritic Cells for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 4307. https://doi.org/10.3390/ijms20174307
Garofano F, Gonzalez-Carmona MA, Skowasch D, Schmidt-Wolf R, Abramian A, Hauser S, Strassburg CP, Schmidt-Wolf IGH. Clinical Trials with Combination of Cytokine-Induced Killer Cells and Dendritic Cells for Cancer Therapy. International Journal of Molecular Sciences. 2019; 20(17):4307. https://doi.org/10.3390/ijms20174307
Chicago/Turabian StyleGarofano, Francesca, Maria A. Gonzalez-Carmona, Dirk Skowasch, Roland Schmidt-Wolf, Alina Abramian, Stefan Hauser, Christian P. Strassburg, and Ingo G. H. Schmidt-Wolf. 2019. "Clinical Trials with Combination of Cytokine-Induced Killer Cells and Dendritic Cells for Cancer Therapy" International Journal of Molecular Sciences 20, no. 17: 4307. https://doi.org/10.3390/ijms20174307
APA StyleGarofano, F., Gonzalez-Carmona, M. A., Skowasch, D., Schmidt-Wolf, R., Abramian, A., Hauser, S., Strassburg, C. P., & Schmidt-Wolf, I. G. H. (2019). Clinical Trials with Combination of Cytokine-Induced Killer Cells and Dendritic Cells for Cancer Therapy. International Journal of Molecular Sciences, 20(17), 4307. https://doi.org/10.3390/ijms20174307