Ginsenoside Compound K Induces Ros-Mediated Apoptosis and Autophagic Inhibition in Human Neuroblastoma Cells In Vitro and In Vivo


Abstract

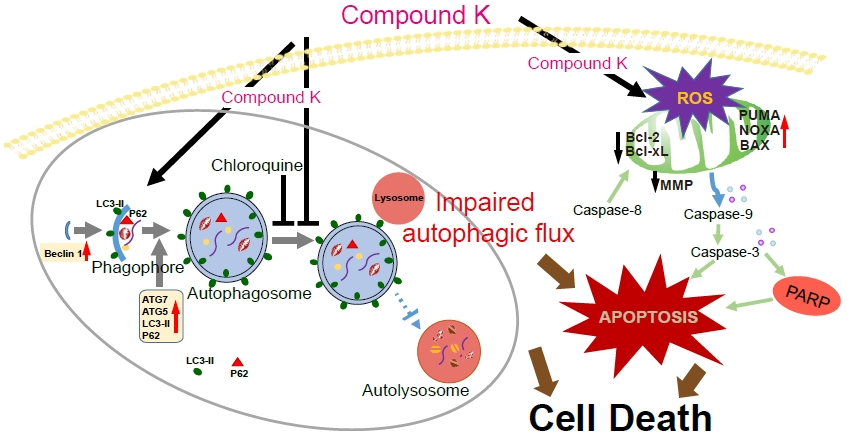{kind=link}
{kind=link}
{kind=link}
{kind=link}
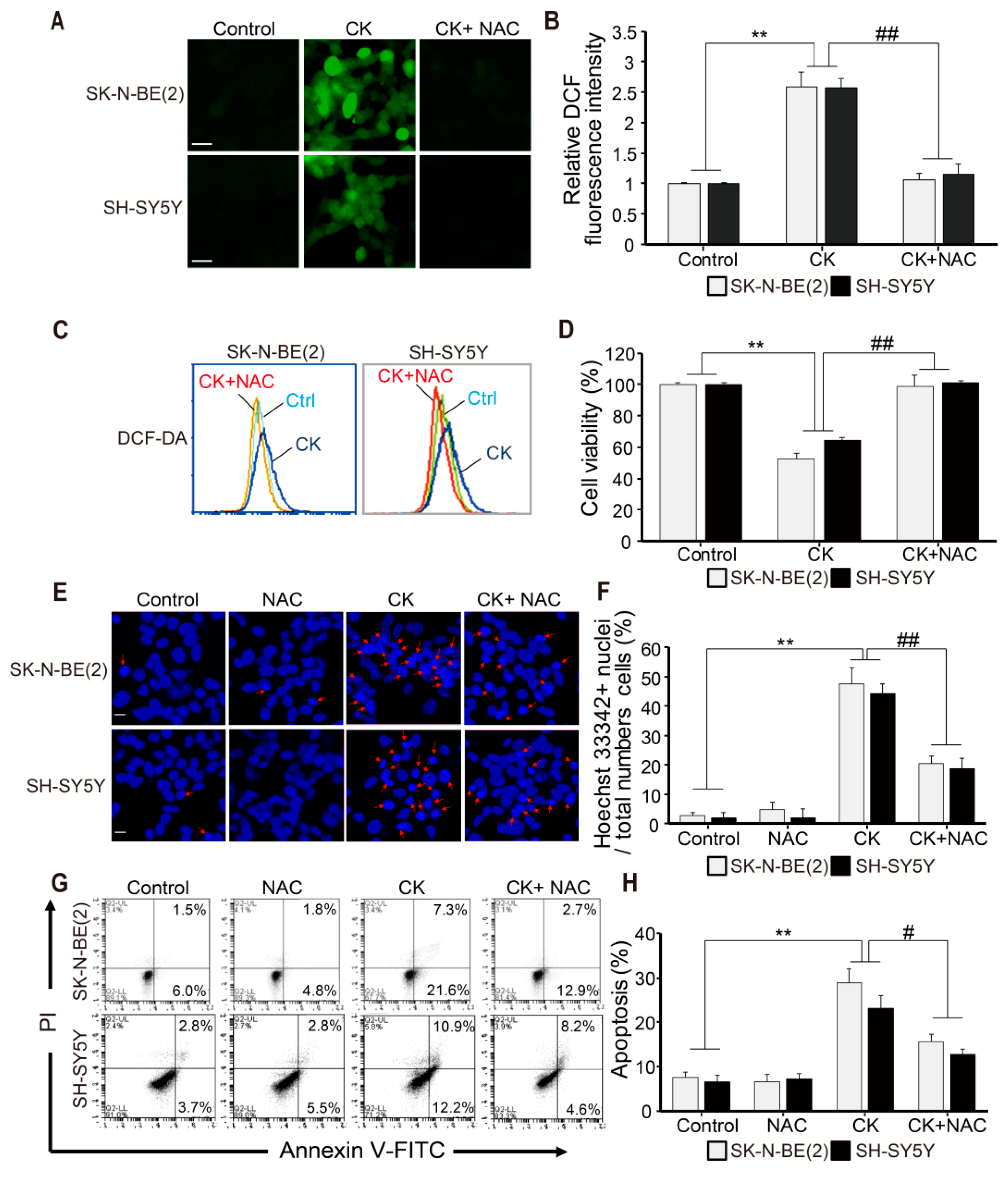{kind=link}
{kind=link}
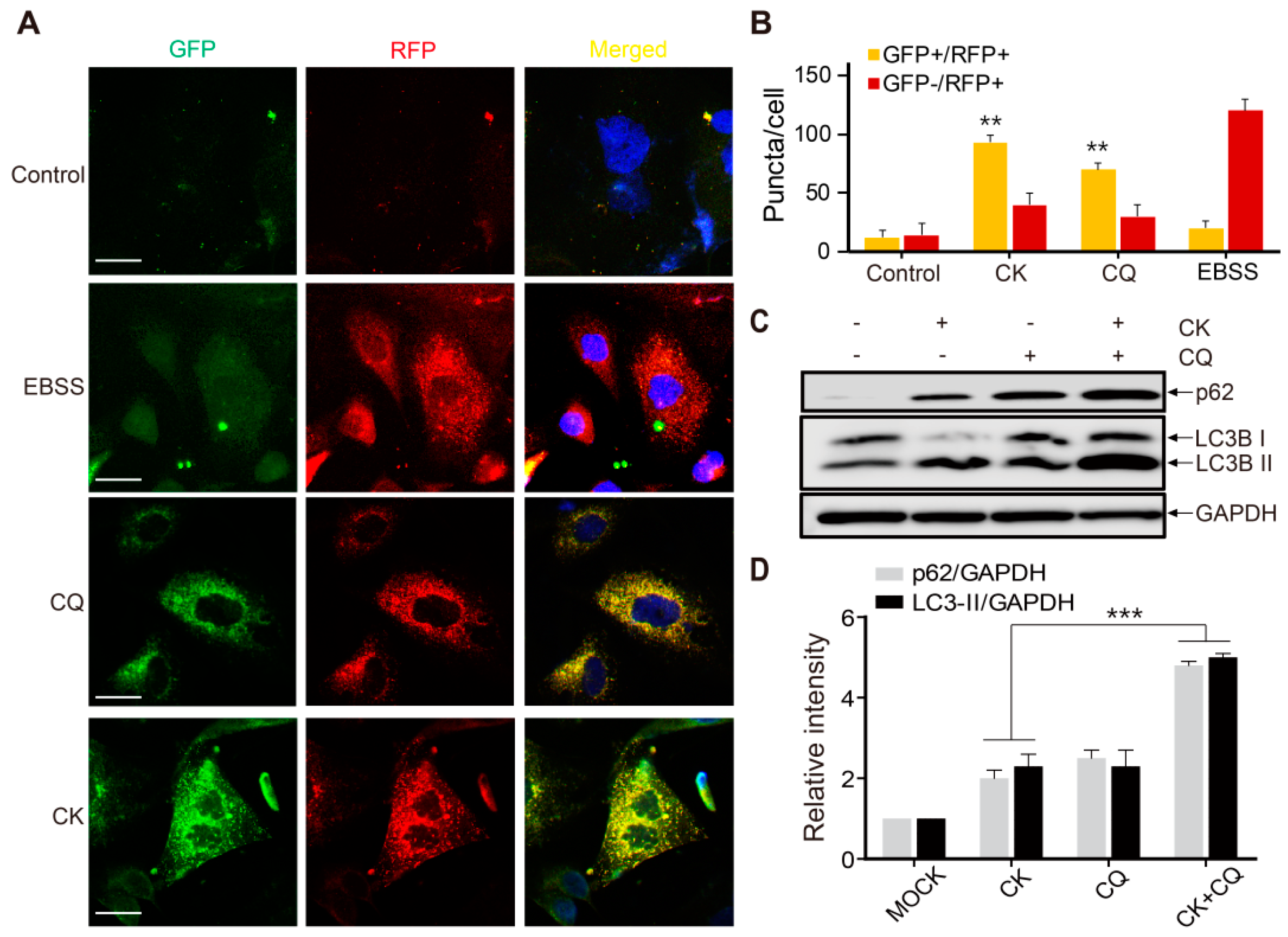{kind=link}
{kind=link}
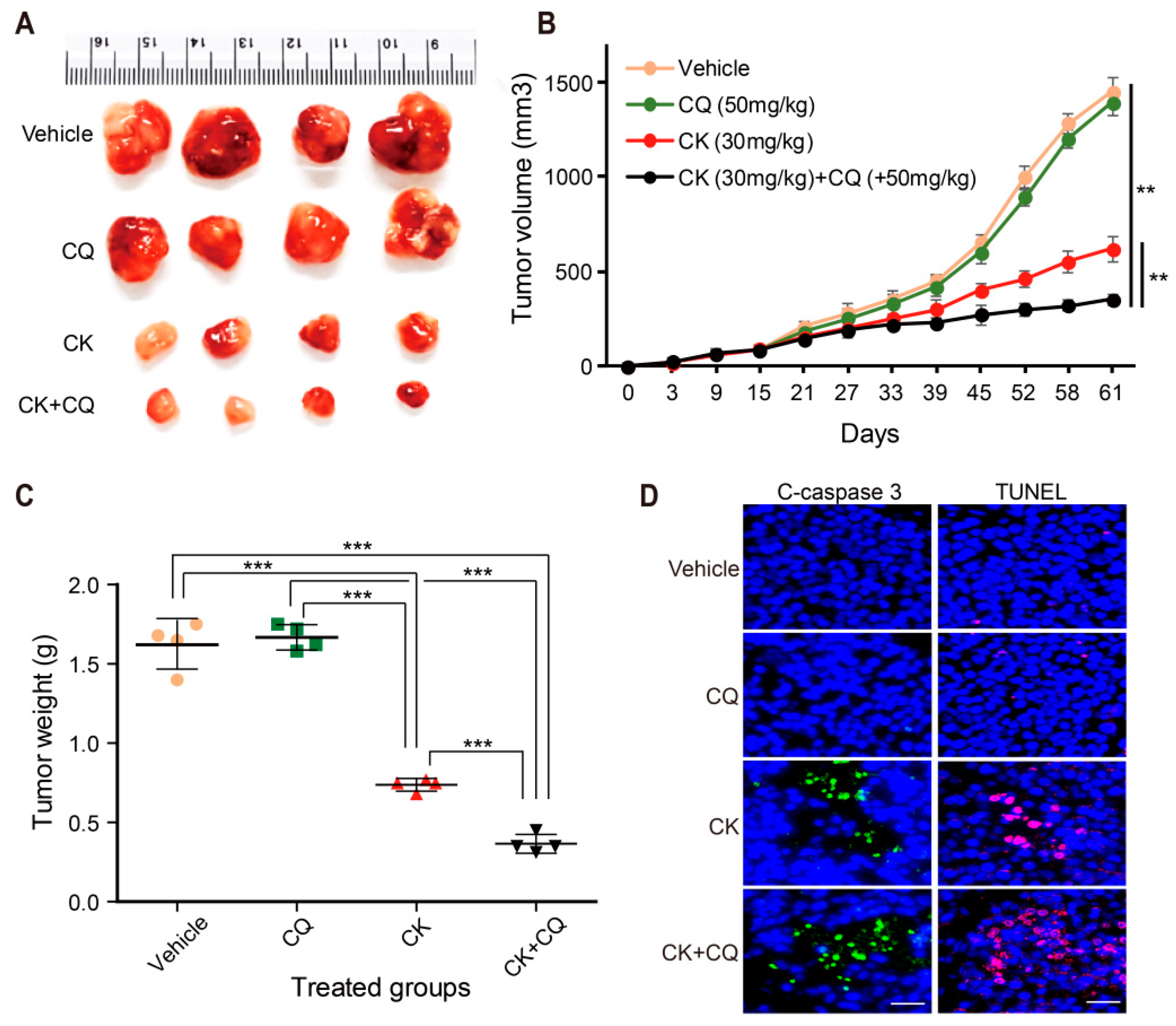{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. CK Inhibits the Growth of Human Neuroblastoma Cells
2.2. CK Induces Cell Cycle Arrest and Apoptotic Cell Death in Neuroblastoma Cells
2.3. CK Induces Apoptotic Cell Death in Neuroblastoma Cells via Caspase Dependent- Pathways
2.4. CK-induced Apoptosis Involves ROS Generation in Neuroblastoma Cells
2.5. CK Induces Early-Stage Autophagy and Autophagosome Accumulation in Human Neuroblastoma Cells
2.6. CK Inhibits Late-Phase Autophagy and the Fusion of Autophagosomes with Autolysosomes
2.7. CK and CQ Combination Therapy Enhances CK-Induced Apoptosis in Human Neuroblastoma Cells
2.8. CQ and CK Combination Treatment Improves the Anti-tumor Effect of CK in Neuroblastoma Xenograft Models
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cells and Culture Conditions
4.3. Cell Viability Assay
4.4. Colony Formation Assay
4.5. Cell Cycle Analysis
4.6. Hoechst 33342 Staining
4.7. Apoptosis Assay with Annexin V and PI Staining
4.8. Mitochondrial Membrane Potential (∆ψm) Assay
4.9. Hoechst 33342 and PI Staining
4.10. RNA Isolation and Reverse Transcription (RT)-PCR
4.11. Western Blot Analysis
4.12. Measurement of Intracellular ROS Generation
4.13. Immunofluorescence Staining
4.14. Detection of Autophagic Vacuoles by Acridine Orange Staining (AO)
4.15. Plasmids Construction
4.16. Cell Transfection and Stable Cell Screening
4.17. Detection of GFP-LC3 or mRFP-GFP-LC3B Assay
4.18. Measurement of Mitochondrial ROS Generation
4.19. Tumor Xenograft Studies
4.20. Histology and TdT-Mediated dUTP Nick end Labelling (TUNEL)
4.21. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CK | Compound K |
| ROS | Reactive Oxygen Species |
| PPD | ProtoPanaxaDiol |
| AO | Acridine Orange |
| PCNA | Proliferating Cell Nuclear Antigen |
| CCK-8 | Cell Count Kit-8 |
| CQ | Chloroquine |
| RAPA | Rapamycin |
| AVOs | Acidic Vesicle Organelles |
| MMP | Mitochondria Membrane Potential |
References
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Apel, A.; Herr, I.; Schwarz, H.; Rodemann, H.P.; Mayer, A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008, 68, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Park, J.R.; Eggert, A.; Caron, H. Neuroblastoma: Biology, prognosis, and treatment. Pediatr. Clin. N. Am. 2008, 55, 97–120. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.L.; Katzenstein, H.M.; Cohn, S.L. Advances in the diagnosis and treatment of neuroblastoma. Oncologist 2003, 8, 278–292. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- Dey, P.; Kundu, A.; Chakraborty, H.J.; Kar, B.; Choi, W.S.; Lee, B.M.; Bhakta, T.; Atanasov, A.G.; Kim, H.S. Therapeutic value of steroidal alkaloids in cancer: Current trends and future perspectives. Int. J. Cancer 2018, 145, 1731–1744. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef]
- Hasima, N.; Ozpolat, B. Regulation of autophagy by polyphenolic compounds as a potential therapeutic strategy for cancer. Cell Death Dis. 2014, 5, e1509. [Google Scholar] [CrossRef]
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies within. Science 1998, 281, 1312–1316. [Google Scholar] [CrossRef]
- Liu, G.; Pei, F.; Yang, F.; Li, L.; Amin, A.D.; Liu, S.; Buchan, J.R.; Cho, W.C. Role of Autophagy and Apoptosis in Non-Small-Cell Lung Cancer. Int. J. Mol. Sci. 2017, 18, 367. [Google Scholar] [CrossRef]
- Radogna, F.; Dicato, M.; Diederich, M. Cancer-type-specific crosstalk between autophagy, necroptosis and apoptosis as a pharmacological target. Biochem. Pharm. 2015, 94, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef]
- Attele, A.S.; Wu, J.A.; Yuan, C.S. Ginseng pharmacology: Multiple constituents and multiple actions. Biochem. Pharm. 1999, 58, 1685–1693. [Google Scholar] [CrossRef]
- Tode, T.; Kikuchi, Y.; Kita, T.; Hirata, J.; Imaizumi, E.; Nagata, I. Inhibitory effects by oral administration of ginsenoside Rh2 on the growth of human ovarian cancer cells in nude mice. J. Cancer Res. Clin. Oncol. 1993, 120, 24–26. [Google Scholar] [CrossRef]
- Oh, G.S.; Pae, H.O.; Choi, B.M.; Seo, E.A.; Kim, D.H.; Shin, M.K.; Kim, J.D.; Kim, J.B.; Chung, H.T. 20(S)-Protopanaxatriol, one of ginsenoside metabolites, inhibits inducible nitric oxide synthase and cyclooxygenase-2 expressions through inactivation of nuclear factor-kappaB in RAW 264.7 macrophages stimulated with lipopolysaccharide. Cancer Lett. 2004, 205, 23–29. [Google Scholar] [CrossRef]
- Leung, K.W.; Yung, K.K.; Mak, N.K.; Chan, Y.S.; Fan, T.P.; Wong, R.N. Neuroprotective effects of ginsenoside-Rg1 in primary nigral neurons against rotenone toxicity. Neuropharmacology 2007, 52, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.Y.; Shang, W.Q.; Yu, J.J.; Sun, Q.; Li, M.Q.; Sun, J.S. The antitumor activity study of ginsenosides and metabolites in lung cancer cell. Am. J. Transl. Res. 2016, 8, 1708–1718. [Google Scholar] [PubMed]
- Hasegawa, H.; Sung, J.H.; Matsumiya, S.; Uchiyama, M. Main ginseng saponin metabolites formed by intestinal bacteria. Planta Med. 1996, 62, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Sung, J.H.; Huh, J.H. Ginseng intestinal bacterial metabolite IH901 as a new anti-metastatic agent. Arch. Pharm. Res. 1997, 20, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Chae, S.; Kang, K.A.; Chang, W.Y.; Kim, M.J.; Lee, S.J.; Lee, Y.S.; Kim, H.S.; Kim, D.H.; Hyun, J.W. Effect of compound K, a metabolite of ginseng saponin, combined with gamma-ray radiation in human lung cancer cells in vitro and in vivo. J. Agric. Food Chem. 2009, 57, 5777–5782. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.D.; Kang, K.A.; Zhang, R.; Lim, C.M.; Kim, H.S.; Kim, D.H.; Jeon, Y.J.; Lee, C.H.; Park, J.; Chang, W.Y.; et al. Ginseng saponin metabolite induces apoptosis in MCF-7 breast cancer cells through the modulation of AMP-activated protein kinase. Environ. Toxicol. Pharm. 2010, 30, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.K.; Kang, K.A.; Lim, C.M.; Kim, K.C.; Kim, H.S.; Kim, D.H.; Kim, B.J.; Chang, W.Y.; Choi, J.H.; Hyun, J.W. Compound K, a metabolite of ginseng saponin, induces mitochondria-dependent and caspase-dependent apoptosis via the generation of reactive oxygen species in human colon cancer cells. Int. J. Mol. Sci. 2010, 11, 4916–4931. [Google Scholar] [CrossRef]
- Kim, A.D.; Kang, K.A.; Kim, H.S.; Kim, D.H.; Choi, Y.H.; Lee, S.J.; Kim, H.S.; Hyun, J.W. A ginseng metabolite, compound K, induces autophagy and apoptosis via generation of reactive oxygen species and activation of JNK in human colon cancer cells. Cell Death Dis. 2013, 4, e750. [Google Scholar] [CrossRef]
- Espada, L.; Meo-Evoli, N.; Sancho, P.; Real, S.; Fabregat, I.; Ambrosio, S.; Tauler, A. ROS production is essential for the apoptotic function of E2F1 in pheochromocytoma and neuroblastoma cell lines. PLoS ONE 2012, 7, e51544. [Google Scholar] [CrossRef]
- Herrera, B.; Alvarez, A.M.; Sanchez, A.; Fernandez, M.; Roncero, C.; Benito, M.; Fabregat, I. Reactive oxygen species (ROS) mediates the mitochondrial-dependent apoptosis induced by transforming growth factor (beta) in fetal hepatocytes. FASEB J. 2001, 15, 741–751. [Google Scholar] [CrossRef]
- Eruslanov, E.; Kusmartsev, S. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol. Biol. 2010, 594, 57–72. [Google Scholar] [PubMed]
- Kanzawa, T.; Germano, I.M.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004, 11, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, G.; Wada, K.; Okuno, M.; Kurosawa, M.; Nukina, N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol. Cell 2011, 44, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Gonzalez-Polo, R.A.; Casares, N.; Perfettini, J.L.; Dessen, P.; Larochette, N.; Metivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell Biol. 2005, 25, 1025–1040. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Jaattela, M. Lysosomes and autophagy in cell death control. Nat. Rev. Cancer 2005, 5, 886–897. [Google Scholar] [CrossRef]
- Pan, T.; Rawal, P.; Wu, Y.; Xie, W.; Jankovic, J.; Le, W. Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience 2009, 164, 541–551. [Google Scholar] [CrossRef]
- Gavilan, E.; Giraldez, S.; Sanchez-Aguayo, I.; Romero, F.; Ruano, D.; Daza, P. Breast cancer cell line MCF7 escapes from G1/S arrest induced by proteasome inhibition through a GSK-3beta dependent mechanism. Sci. Rep. 2015, 5, 10027. [Google Scholar] [CrossRef]
- Han, Y.; Sun, B.; Hu, X.; Zhang, H.; Jiang, B.; Spranger, M.I.; Zhao, Y. Transformation of bioactive compounds by Fusarium sacchari fungus isolated from the soil-cultivated ginseng. J. Agric. Food Chem. 2007, 55, 9373–9379. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Sung, J.H.; Lee, S.J.; Moon, C.K.; Lee, B.H. Antitumor activity of a novel ginseng saponin metabolite in human pulmonary adenocarcinoma cells resistant to cisplatin. Cancer Lett. 1999, 144, 39–43. [Google Scholar] [CrossRef]
- Wang, C.Z.; Du, G.J.; Zhang, Z.; Wen, X.D.; Calway, T.; Zhen, Z.; Musch, M.W.; Bissonnette, M.; Chang, E.B.; Yuan, C.S. Ginsenoside compound K, not Rb1, possesses potential chemopreventive activities in human colorectal cancer. Int. J. Oncol. 2012, 40, 1970–1976. [Google Scholar]
- Zhou, W.; Feng, M.Q.; Li, J.Y.; Zhou, P. Studies on the preparation, crystal structure and bioactivity of ginsenoside compound K. J. Asian Nat. Prod. Res. 2006, 8, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.H.; Woo, M.S.; Kim, S.Y.; Kim, W.K.; Hyun, J.W.; Kim, E.J.; Kim, D.H.; Kim, H.S. Ginseng saponin metabolite suppresses phorbol ester-induced matrix metalloproteinase-9 expression through inhibition of activator protein-1 and mitogen-activated protein kinase signaling pathways in human astroglioma cells. Int. J. Cancer 2006, 118, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Dong, Y.; Wang, L.; Xu, G.; Yang, Q.; Tang, X.; Qiao, Y.; Cong, Z. Ginsenoside metabolite compound K induces apoptosis and autophagy in non-small cell lung cancer cells via AMPK-mTOR and JNK pathways. Biochem. Cell Biol. 2019, 97, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Huang, Q.; Wang, M.; Yan, X.; Song, X.; Ma, R.; Jiang, R.; Zhao, D.; Sun, L. Compound K Inhibits Autophagy-Mediated Apoptosis Through Activation of the PI3K-Akt Signaling Pathway Thus Protecting Against Ischemia/Reperfusion Injury. Cell Physiol. Biochem. 2018, 47, 2589–2601. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.P.; Lee, C.H.; Ying, T.H.; Lin, C.L.; Lin, C.L.; Hsueh, J.T.; Hsieh, Y.H. Licochalcone A induces autophagy through PI3K/Akt/mTOR inactivation and autophagy suppression enhances Licochalcone A-induced apoptosis of human cervical cancer cells. Oncotarget 2015, 6, 28851–28866. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Loos, B.; du Toit, A.; Hofmeyr, J.H. Defining and measuring autophagosome flux-concept and reality. Autophagy 2014, 10, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, N.; Steele, R.; Isbell, T.S.; Philips, N.; Ray, R.B. Bitter melon extract inhibits breast cancer growth in preclinical model by inducing autophagic cell death. Oncotarget 2017, 8, 66226–66236. [Google Scholar] [CrossRef] [PubMed]
- Go, D.H.; Lee, Y.G.; Lee, D.H.; Kim, J.A.; Jo, I.H.; Han, Y.S.; Jo, Y.H.; Kim, K.Y.; Seo, Y.K.; Moon, J.H.; et al. 3-Decylcatechol induces autophagy-mediated cell death through the IRE1alpha/JNK/p62 in hepatocellular carcinoma cells. Oncotarget 2017, 8, 58790–58800. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Li, Y.; Wang, S.; Wang, X.; Liao, C.; Hu, X.; Fan, L.; Kang, Q.; Zeng, Y.; Wu, X.; et al. Inhibition of autophagosome-lysosome fusion by ginsenoside Ro via the ESR2-NCF1-ROS pathway sensitizes esophageal cancer cells to 5-fluorouracil-induced cell death via the CHEK1-mediated DNA damage checkpoint. Autophagy 2016, 12, 1593–1613. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Jung, J.Y.; Choi, S.; Lee, H.; Morales, L.D.; Koh, J.T.; Kim, S.H.; Choi, Y.D.; Choi, C.; Slaga, T.J.; et al. GFRA1 promotes cisplatin-induced chemoresistance in osteosarcoma by inducing autophagy. Autophagy 2017, 13, 149–168. [Google Scholar] [CrossRef] [PubMed]
- Brech, A.; Ahlquist, T.; Lothe, R.A.; Stenmark, H. Autophagy in tumour suppression and promotion. Mol. Oncol. 2009, 3, 366–375. [Google Scholar] [CrossRef]
- Fujiwara, K.; Iwado, E.; Mills, G.B.; Sawaya, R.; Kondo, S.; Kondo, Y. Akt inhibitor shows anticancer and radiosensitizing effects in malignant glioma cells by inducing autophagy. Int. J. Oncol. 2007, 31, 753–760. [Google Scholar] [CrossRef]
- Kim, E.H.; Sohn, S.; Kwon, H.J.; Kim, S.U.; Kim, M.J.; Lee, S.J.; Choi, K.S. Sodium selenite induces superoxide-mediated mitochondrial damage and subsequent autophagic cell death in malignant glioma cells. Cancer Res. 2007, 67, 6314–6324. [Google Scholar] [CrossRef]
- Chang, C.H.; Lee, C.Y.; Lu, C.C.; Tsai, F.J.; Hsu, Y.M.; Tsao, J.W.; Juan, Y.N.; Chiu, H.Y.; Yang, J.S.; Wang, C.C. Resveratrol-induced autophagy and apoptosis in cisplatin-resistant human oral cancer CAR cells: A key role of AMPK and Akt/mTOR signaling. Int. J. Oncol. 2017, 50, 873–882. [Google Scholar] [CrossRef]
- Tong, L.; Chuang, C.C.; Wu, S.; Zuo, L. Reactive oxygen species in redox cancer therapy. Cancer Lett. 2015, 367, 18–25. [Google Scholar] [CrossRef]
- Radogna, F.; Cerella, C.; Gaigneaux, A.; Christov, C.; Dicato, M.; Diederich, M. Cell type-dependent ROS and mitophagy response leads to apoptosis or necroptosis in neuroblastoma. Oncogene 2016, 35, 3839–3853. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Liu, H.; Luo, X.J.; Zhao, Z.; Zou, Z.Y.; Li, J.; Lin, X.J.; Liang, Y. The roles of reactive oxygen species (ROS) and autophagy in the survival and death of leukemia cells. Crit. Rev. Oncol. Hematol. 2017, 112, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Poillet-Perez, L.; Despouy, G.; Delage-Mourroux, R.; Boyer-Guittaut, M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015, 4, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Wang, B.; Yang, L.; Zhang, Y. The role of ROS-induced autophagy in hepatocellular carcinoma. Clin. Res. Hepatol. Gastroenterol. 2018, 42, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Roper, M.G. Measurement of DCF fluorescence as a measure of reactive oxygen species in murine islets of Langerhans. Anal. Methods 2014, 6, 3019–3024. [Google Scholar] [CrossRef]
- Degtyarev, M.; Reichelt, M.; Lin, K. Novel quantitative autophagy analysis by organelle flow cytometry after cell sonication. PLoS ONE 2014, 9, e87707. [Google Scholar] [CrossRef] [PubMed]
- Paglin, S.; Hollister, T.; Delohery, T.; Hackett, N.; McMahill, M.; Sphicas, E.; Domingo, D.; Yahalom, J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001, 61, 439–444. [Google Scholar]
- Kauffman, M.E.; Kauffman, M.K.; Traore, K.; Zhu, H.; Trush, M.A.; Jia, Z.; Li, Y.R. MitoSOX-Based Flow Cytometry for Detecting Mitochondrial ROS. React. Oxyg. Species (Apex) 2016, 2, 361–370. [Google Scholar] [CrossRef]
- Oh, J.M.; Lee, J.; Im, W.T.; Chun, S. Ginsenoside Rk1 Induces Apoptosis in Neuroblastoma Cells Through Loss of Mitochondrial Membrane Potential and Activation of Caspases. Int. J. Mol. Sci. 2019, 20, 1213. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, J.-M.; Kim, E.; Chun, S. Ginsenoside Compound K Induces Ros-Mediated Apoptosis and Autophagic Inhibition in Human Neuroblastoma Cells In Vitro and In Vivo. Int. J. Mol. Sci. 2019, 20, 4279. https://doi.org/10.3390/ijms20174279
Oh J-M, Kim E, Chun S. Ginsenoside Compound K Induces Ros-Mediated Apoptosis and Autophagic Inhibition in Human Neuroblastoma Cells In Vitro and In Vivo. International Journal of Molecular Sciences. 2019; 20(17):4279. https://doi.org/10.3390/ijms20174279
Chicago/Turabian StyleOh, Jung-Mi, Eunhee Kim, and Sungkun Chun. 2019. "Ginsenoside Compound K Induces Ros-Mediated Apoptosis and Autophagic Inhibition in Human Neuroblastoma Cells In Vitro and In Vivo" International Journal of Molecular Sciences 20, no. 17: 4279. https://doi.org/10.3390/ijms20174279
APA StyleOh, J.-M., Kim, E., & Chun, S. (2019). Ginsenoside Compound K Induces Ros-Mediated Apoptosis and Autophagic Inhibition in Human Neuroblastoma Cells In Vitro and In Vivo. International Journal of Molecular Sciences, 20(17), 4279. https://doi.org/10.3390/ijms20174279