Cyclo-Oxygenase (COX) Inhibitors and Cardiovascular Risk: Are Non-Steroidal Anti-Inflammatory Drugs Really Anti-Inflammatory?

{kind=link}
Abstract
1. Introduction
2. COX Inhibitors and Cardiovascular Disease Risk
3. COX Inhibitors and Hypertension Risk: Preclinical and Clinical Studies
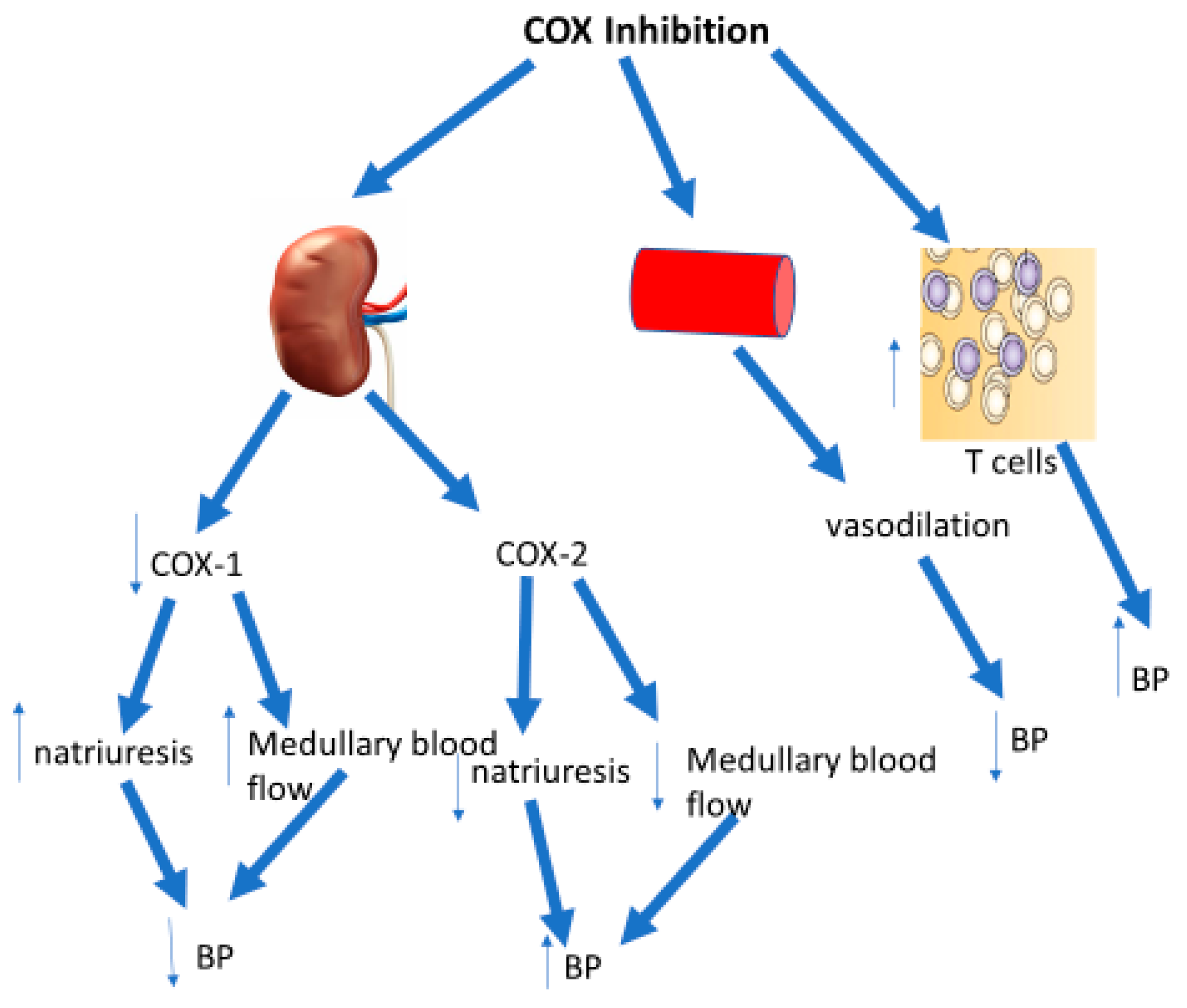
4. COX Inhibitors and Renal Function
5. COX Inhibitors and Vascular Function
6. COX Inhibition and Cardiac Tissue: Preclinical Studies
7. Prostaglandins and T Cells
8. Conclusions and Perspectives
Conflicts of Interest
References
- Qi, Z.; Hao, C.M.; Langenbach, R.I.; Breyer, R.M.; Redha, R.; Morrow, J.D.; Breyer, M.D. Opposite effects of cyclooxygenase-1 and -2 activity on the pressor response to angiotensin II. J. Clin. Investig. 2002, 110, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Bally, M.; Dendukuri, N.; Rich, B.; Nadeau, L.; Helin-Salmivaara, A.; Garbe, E.; Brophy, J.M. Risk of acute myocardial infarction with NSAIDs in real world use: Bayesian meta-analysis of individual patient data. BMJ 2017, 357, j1909. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.I.; Shihata, W.A.; Andrews, K.L.; Lee, M.K.S.; Moore, X.L.; Jefferis, A.M.; Vinh, A.; Gaspari, T.; Dragoljevic, D.; Jennings, G.L.; et al. Effects of high- and low-dose aspirin on adaptive immunity and hypertension in the stroke-prone spontaneously hypertensive rat. FASEB J. 2019, 33, 1510–1521. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.; Weyand, C.; Harrison, D.G. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 2007, 204, 2449–2460. [Google Scholar] [CrossRef] [PubMed]
- Bombardier, C.; Laine, L.; Reicin, A.; Shapiro, D.; Burgos-Vargas, R.; Davis, B.; Day, R.; Ferraz, M.B.; Hawkey, C.J.; Hochberg, M.C.; et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N. Engl. J. Med. 2000, 343, 1520–1528. [Google Scholar] [CrossRef] [PubMed]
- Van Hecken, A.; Schwartz, J.I.; Depre, M.; De Lepeleire, I.; Dallob, A.; Tanaka, W.; Wynants, K.; Buntinx, A.; Arnout, J.; Wong, P.H.; et al. Comparative inhibitory activity of rofecoxib, meloxicam, diclofenac, ibuprofen, and naproxen on COX-2 versus COX-1 in healthy volunteers. J. Clin. Pharm. 2000, 40, 1109–1120. [Google Scholar]
- Justice, E.; Carruthers, D.M. Cardiovascular risk and COX-2 inhibition in rheumatological practice. J. Hum. Hypertens. 2005, 19, 1–5. [Google Scholar] [CrossRef]
- Fitzgerald, G.A. Coxibs and cardiovascular disease. N. Engl. J. Med. 2004, 351, 1709–1711. [Google Scholar] [CrossRef]
- Marcus, A.J.; Broekman, M.J.; Pinsky, D.J. COX inhibitors and thromboregulation. N. Engl. J. Med. 2002, 347, 1025–1026. [Google Scholar] [CrossRef]
- Cheng, Y.; Austin, S.C.; Rocca, B.; Koller, B.H.; Coffman, T.M.; Grosser, T.; Lawson, J.A.; FitzGerald, G.A. Role of prostacyclin in the cardiovascular response to thromboxane A2. Science 2002, 296, 539–541. [Google Scholar] [CrossRef]
- Silverstein, F.E.; Faich, G.; Goldstein, J.L.; Simon, L.S.; Pincus, T.; Whelton, A.; Makuch, R.; Eisen, G.; Agrawal, N.M.; Stenson, W.F.; et al. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: The CLASS study: A randomized controlled trial. Celecoxib Long-term Arthritis Safety Study. JAMA 2000, 284, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Ray, W.A.; Stein, C.M.; Daugherty, J.R.; Hall, K.; Arbogast, P.G.; Griffin, M.R. COX-2 selective non-steroidal anti-inflammatory drugs and risk of serious coronary heart disease. Lancet 2002, 360, 1071–1073. [Google Scholar] [CrossRef]
- Dilger, K.; Herrlinger, C.; Peters, J.; Seyberth, H.W.; Schweer, H.; Klotz, U. Effects of celecoxib and diclofenac on blood pressure, renal function, and vasoactive prostanoids in young and elderly subjects. J. Clin. Pharm. 2002, 42, 985–994. [Google Scholar] [CrossRef]
- White, W.B.; Faich, G.; Borer, J.S.; Makuch, R.W. Cardiovascular thrombotic events in arthritis trials of the cyclooxygenase-2 inhibitor celecoxib. Am. J. Cardiol. 2003, 92, 411–418. [Google Scholar] [CrossRef]
- Nissen, S.E.; Yeomans, N.D.; Solomon, D.H.; Lüscher, T.F.; Libby, P.; Husni, M.E.; Graham, D.Y.; Borer, J.S.; Wisniewski, L.M.; Wolski, K.E.; et al. Cardiovascular Safety of Celecoxib, Naproxen, or Ibuprofen for Arthritis. N. Engl. J. Med. 2016, 375, 2519–2529. [Google Scholar] [CrossRef] [PubMed]
- Coxib traditional, N.T.C.; Bhala, N.; Emberson, J.; Merhi, A.; Abramson, S.; Arber, N.; Baron, J.A.; Bombardier, C.; Cannon, C.; Farkouh, M.E.; et al. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: Meta-analyses of individual participant data from randomised trials. Lancet 2013, 382, 769–779. [Google Scholar] [CrossRef]
- Arfe, A.; Scotti, L.; Varas-Lorenzo, C.; Nicotra, F.; Zambon, A.; Kollhorst, B.; Schink, T.; Garbe, E.; Herings, R.; Straatman, H.; et al. Non-steroidal anti-inflammatory drugs and risk of heart failure in four European countries: Nested case-control study. BMJ 2016, 354, i4857. [Google Scholar] [CrossRef]
- Bally, M.; Beauchamp, M.E.; Abrahamowicz, M.; Nadeau, L.; Brophy, J.M. Risk of acute myocardial infarction with real-world NSAIDs depends on dose and timing of exposure. Pharm. Drug Saf. 2018, 27, 69–77. [Google Scholar] [CrossRef]
- Hocherl, K.; Endemann, D.; Kammerl, M.C.; Grobecker, H.F.; Kurtz, A. Cyclo-oxygenase-2 inhibition increases blood pressure in rats. Br. J. Pharm. 2002, 136, 1117–1126. [Google Scholar] [CrossRef]
- Wu, R.; Lamontagne, D.; de Champlain, J. Antioxidative properties of acetylsalicylic Acid on vascular tissues from normotensive and spontaneously hypertensive rats. Circulation 2002, 105, 387–392. [Google Scholar] [CrossRef]
- Hermann, M.; Shaw, S.; Kiss, E.; Camici, G.; Buhler, N.; Chenevard, R.; Luscher, T.F.; Grone, H.J.; Ruschitzka, F. Selective COX-2 inhibitors and renal injury in salt-sensitive hypertension. Hypertension 2005, 45, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Hermann, M.; Camici, G.; Fratton, A.; Hurlimann, D.; Tanner, F.C.; Hellermann, J.P.; Fiedler, M.; Thiery, J.; Neidhart, M.; Gay, R.E.; et al. Differential effects of selective cyclooxygenase-2 inhibitors on endothelial function in salt-induced hypertension. Circulation 2003, 108, 2308–2311. [Google Scholar] [CrossRef] [PubMed]
- Chenevard, R.; Hurlimann, D.; Bechir, M.; Enseleit, F.; Spieker, L.; Hermann, M.; Riesen, W.; Gay, S.; Gay, R.E.; Neidhart, M.; et al. Selective COX-2 inhibition improves endothelial function in coronary artery disease. Circulation 2003, 107, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Widlansky, M.E.; Price, D.T.; Gokce, N.; Eberhardt, R.T.; Duffy, S.J.; Holbrook, M.; Maxwell, C.; Palmisano, J.; Keaney, J.F., Jr.; Morrow, J.D.; et al. Short- and long-term COX-2 inhibition reverses endothelial dysfunction in patients with hypertension. Hypertension 2003, 42, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.F.; Harris, R.C. Renal effects of non-steroidal anti-inflammatory drugs and selective cyclooxygenase-2 inhibitors. Curr. Pharm. Des. 2005, 11, 1795–1804. [Google Scholar] [CrossRef] [PubMed]
- Whelton, A. Cyclooxygenase-2 inhibition and renal function. Ann. Intern. Med. 2001, 134, 1077–1078. [Google Scholar] [CrossRef] [PubMed]
- Whelton, A.; White, W.B.; Bello, A.E.; Puma, J.A.; Fort, J.G.; Investigators, S.-V. Effects of celecoxib and rofecoxib on blood pressure and edema in patients > or = 65 years of age with systemic hypertension and osteoarthritis. Am. J. Cardiol. 2002, 90, 959–963. [Google Scholar] [CrossRef]
- Whelton, A.; Fort, J.G.; Puma, J.A.; Normandin, D.; Bello, A.E.; Verburg, K.M.; Group, S.V.S. Cyclooxygenase-2--specific inhibitors and cardiorenal function: A randomized, controlled trial of celecoxib and rofecoxib in older hypertensive osteoarthritis patients. Am. J. 2001, 8, 85–95. [Google Scholar] [CrossRef]
- White, W.B.; Kent, J.; Taylor, A.; Verburg, K.M.; Lefkowith, J.B.; Whelton, A. Effects of celecoxib on ambulatory blood pressure in hypertensive patients on ACE inhibitors. Hypertension 2002, 39, 929–934. [Google Scholar] [CrossRef]
- Izhar, M.; Alausa, T.; Folker, A.; Hung, E.; Bakris, G.L. Effects of COX inhibition on blood pressure and kidney function in ACE inhibitor-treated blacks and hispanics. Hypertension 2004, 43, 573–577. [Google Scholar] [CrossRef]
- Ichihara, A.; Imig, J.D.; Inscho, E.W.; Navar, L.G. Cyclooxygenase-2 participates in tubular flow-dependent afferent arteriolar tone: Interaction with neuronal NOS. Am. J. Physiol. 1998, 275, F605–F612. [Google Scholar] [CrossRef] [PubMed]
- Ichihara, A.; Imig, J.D.; Navar, L.G. Cyclooxygenase-2 modulates afferent arteriolar responses to increases in pressure. Hypertension 1999, 34, 843–847. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Cai, H.; Morrow, J.D.; Breyer, M.D. Differentiation of cyclooxygenase 1- and 2-derived prostanoids in mouse kidney and aorta. Hypertension 2006, 48, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Gross, J.M.; Dwyer, J.E.; Knox, F.G. Natriuretic response to increased pressure is preserved with COX-2 inhibitors. Hypertension 1999, 34, 1163–1167. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lopez-Parra, M.; Claria, J.; Planaguma, A.; Titos, E.; Masferrer, J.L.; Woerner, B.M.; Koki, A.T.; Jimenez, W.; Altuna, R.; Arroyo, V.; et al. Cyclooxygenase-1 derived prostaglandins are involved in the maintenance of renal function in rats with cirrhosis and ascites. Br. J. Pharm. 2002, 135, 891–900. [Google Scholar] [CrossRef]
- Yang, T.; Singh, I.; Pham, H.; Sun, D.; Smart, A.; Schnermann, J.B.; Briggs, J.P. Regulation of cyclooxygenase expression in the kidney by dietary salt intake. Am. J. Physiol. 1998, 274, F481–F489. [Google Scholar] [CrossRef]
- Rodriguez, F.; Llinas, M.T.; Gonzalez, J.D.; Rivera, J.; Salazar, F.J. Renal changes induced by a cyclooxygenase-2 inhibitor during normal and low sodium intake. Hypertension 2000, 36, 276–281. [Google Scholar] [CrossRef]
- Catella-Lawson, F.; McAdam, B.; Morrison, B.W.; Kapoor, S.; Kujubu, D.; Antes, L.; Lasseter, K.C.; Quan, H.; Gertz, B.J.; FitzGerald, G.A. Effects of specific inhibition of cyclooxygenase-2 on sodium balance, hemodynamics, and vasoactive eicosanoids. J. Pharm. Exp. 1999, 289, 735–741. [Google Scholar]
- Whelton, A.; Schulman, G.; Wallemark, C.; Drower, E.J.; Isakson, P.C.; Verburg, K.M.; Geis, G.S. Effects of celecoxib and naproxen on renal function in the elderly. Arch. Intern. Med. 2000, 160, 1465–1470. [Google Scholar] [CrossRef]
- Rossat, J.; Maillard, M.; Nussberger, J.; Brunner, H.R.; Burnier, M. Renal effects of selective cyclooxygenase-2 inhibition in normotensive salt-depleted subjects. Clin. Pharm. 1999, 66, 76–84. [Google Scholar] [CrossRef]
- Simon, L.S.; Weaver, A.L.; Graham, D.Y.; Kivitz, A.J.; Lipsky, P.E.; Hubbard, R.C.; Isakson, P.C.; Verburg, K.M.; Yu, S.S.; Zhao, W.W.; et al. Anti-inflammatory and upper gastrointestinal effects of celecoxib in rheumatoid arthritis: A randomized controlled trial. JAMA 1999, 282, 1921–1928. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.C. The macula densa: Recent developments. J. Hypertens. 1996, 14, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.C.; McKanna, J.A.; Akai, Y.; Jacobson, H.R.; Dubois, R.N.; Breyer, M.D. Cyclooxygenase-2 is associated with the macula densa of rat kidney and increases with salt restriction. J. Clin. Investig. 1994, 94, 2504–2510. [Google Scholar] [CrossRef] [PubMed]
- Hartner, A.; Goppelt-Struebe, M.; Hilgers, K.F. Coordinate expression of cyclooxygenase-2 and renin in the rat kidney in renovascular hypertension. Hypertension 1998, 31, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Jensen, B.L.; Kurtz, A. Differential regulation of renal cyclooxygenase mRNA by dietary salt intake. Kidney Int. 1997, 52, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.F.; Wang, J.L.; Zhang, M.Z.; Miyazaki, Y.; Ichikawa, I.; McKanna, J.A.; Harris, R.C. Angiotensin II attenuates renal cortical cyclooxygenase-2 expression. J. Clin. Investig. 1999, 103, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.L.; Cheng, H.F.; Harris, R.C. Cyclooxygenase-2 inhibition decreases renin content and lowers blood pressure in a model of renovascular hypertension. Hypertension 1999, 34, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Traynor, T.R.; Smart, A.; Briggs, J.P.; Schnermann, J. Inhibition of macula densa-stimulated renin secretion by pharmacological blockade of cyclooxygenase-2. Am. J. Physiol. 1999, 277, F706–F710. [Google Scholar] [CrossRef]
- Cheng, H.F.; Wang, J.L.; Zhang, M.Z.; Wang, S.W.; McKanna, J.A.; Harris, R.C. Genetic deletion of COX-2 prevents increased renin expression in response to ACE inhibition. Am. J. Physiol. Ren. Physiol. 2001, 280, F449–F456. [Google Scholar] [CrossRef]
- Kammerl, M.C.; Nusing, R.M.; Schweda, F.; Endemann, D.; Stubanus, M.; Kees, F.; Lackner, K.J.; Fischereder, M.; Kramer, B.K. Low sodium and furosemide-induced stimulation of the renin system in man is mediated by cyclooxygenase 2. Clin. Pharm. 2001, 70, 468–474. [Google Scholar] [CrossRef]
- Kleta, R.; Basoglu, C.; Kuwertz-Broking, E. New treatment options for Bartter’s syndrome. N. Engl. J. Med. 2000, 343, 661–662. [Google Scholar] [CrossRef] [PubMed]
- Roig, F.; Llinas, M.T.; Lopez, R.; Salazar, F.J. Role of cyclooxygenase-2 in the prolonged regulation of renal function. Hypertension 2002, 40, 721–728. [Google Scholar] [CrossRef] [PubMed]
- De Mey, J.G.; Vanhoutte, P.M. Heterogeneous behavior of the canine arterial and venous wall. Importance of the endothelium. Circ. Res. 1982, 51, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Miller, V.M.; Vanhoutte, P.M. Endothelium-dependent contractions to arachidonic acid are mediated by products of cyclooxygenase. Am. J. Physiol. 1985, 248, H432–H437. [Google Scholar] [CrossRef] [PubMed]
- Luscher, T.F.; Vanhoutte, P.M. Endothelium-dependent contractions to acetylcholine in the aorta of the spontaneously hypertensive rat. Hypertension 1986, 8, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Taddei, S.; Virdis, A.; Ghiadoni, L.; Magagna, A.; Salvetti, A. Cyclooxygenase inhibition restores nitric oxide activity in essential hypertension. Hypertension 1997, 29, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Taddei, S.; Virdis, A.; Mattei, P.; Salvetti, A. Vasodilation to acetylcholine in primary and secondary forms of human hypertension. Hypertension 1993, 21, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Luscher, T.F.; Cooke, J.P.; Houston, D.S.; Neves, R.J.; Vanhoutte, P.M. Endothelium-dependent relaxations in human arteries. Mayo Clin. Proc. 1987, 62, 601–606. [Google Scholar] [CrossRef]
- Ge, T.; Hughes, H.; Junquero, D.C.; Wu, K.K.; Vanhoutte, P.M.; Boulanger, C.M. Endothelium-dependent contractions are associated with both augmented expression of prostaglandin H synthase-1 and hypersensitivity to prostaglandin H2 in the SHR aorta. Circ. Res. 1995, 76, 1003–1010. [Google Scholar] [CrossRef]
- Tang, E.H.; Vanhoutte, P.M. Gene expression changes of prostanoid synthases in endothelial cells and prostanoid receptors in vascular smooth muscle cells caused by aging and hypertension. Physiol. Genom. 2008, 32, 409–418. [Google Scholar] [CrossRef]
- Yang, D.; Feletou, M.; Boulanger, C.M.; Wu, H.F.; Levens, N.; Zhang, J.N.; Vanhoutte, P.M. Oxygen-derived free radicals mediate endothelium-dependent contractions to acetylcholine in aortas from spontaneously hypertensive rats. Br. J. Pharmacol. 2002, 136, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Tang, E.H.; Ku, D.D.; Tipoe, G.L.; Feletou, M.; Man, R.Y.; Vanhoutte, P.M. Endothelium-dependent contractions occur in the aorta of wild-type and COX2-/- knockout but not COX1-/- knockout mice. J. Cardiovasc. Pharmacol. 2005, 46, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Bulut, D.; Liaghat, S.; Hanefeld, C.; Koll, R.; Miebach, T.; Mugge, A. Selective cyclo-oxygenase-2 inhibition with parecoxib acutely impairs endothelium-dependent vasodilatation in patients with essential hypertension. J. Hypertens. 2003, 21, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- Virdis, A.; Colucci, R.; Neves, M.F.; Rugani, I.; Aydinoglu, F.; Fornai, M.; Ippolito, C.; Antonioli, L.; Duranti, E.; Solini, A.; et al. Resistance artery mechanics and composition in angiotensin II-infused mice: Effects of cyclooxygenase-1 inhibition. Eur. Heart J. 2012, 33, 2225–2234. [Google Scholar] [CrossRef] [PubMed]
- Feletou, M.; Kohler, R.; Vanhoutte, P.M. Endothelium-derived vasoactive factors and hypertension: Possible roles in pathogenesis and as treatment targets. Curr. Hypertens. Rep. 2010, 12, 267–275. [Google Scholar] [CrossRef]
- Feletou, M.; Verbeuren, T.J.; Vanhoutte, P.M. Endothelium-dependent contractions in SHR: A tale of prostanoid TP and IP receptors. Br. J. Pharm. 2009, 156, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Gomez, E.; Schwendemann, C.; Roger, S.; Simonet, S.; Paysant, J.; Courchay, C.; Verbeuren, T.J.; Feletou, M. Aging and prostacyclin responses in aorta and platelets from WKY and SHR rats. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2198–H2211. [Google Scholar] [CrossRef]
- Gluais, P.; Lonchampt, M.; Morrow, J.D.; Vanhoutte, P.M.; Feletou, M. Acetylcholine-induced endothelium-dependent contractions in the SHR aorta: The Janus face of prostacyclin. Br. J. Pharmacol. 2005, 146, 834–845. [Google Scholar] [CrossRef]
- Xiao, C.Y.; Hara, A.; Yuhki, K.; Fujino, T.; Ma, H.; Okada, Y.; Takahata, O.; Yamada, T.; Murata, T.; Narumiya, S.; et al. Roles of prostaglandin I(2) and thromboxane A(2) in cardiac ischemia-reperfusion injury: A study using mice lacking their respective receptors. Circulation 2001, 104, 2210–2215. [Google Scholar] [CrossRef]
- Arehart, E.; Stitham, J.; Asselbergs, F.W.; Douville, K.; MacKenzie, T.; Fetalvero, K.M.; Gleim, S.; Kasza, Z.; Rao, Y.; Martel, L.; et al. Acceleration of cardiovascular disease by a dysfunctional prostacyclin receptor mutation: Potential implications for cyclooxygenase-2 inhibition. Circ. Res. 2008, 102, 986–993. [Google Scholar] [CrossRef]
- Tesfamariam, B.; Jakubowski, J.A.; Cohen, R.A. Contraction of diabetic rabbit aorta caused by endothelium-derived PGH2-TxA2. Am. J. Physiol. 1989, 257, H1327–H1333. [Google Scholar] [CrossRef] [PubMed]
- Auch-Schwelk, W.; Katusic, Z.S.; Vanhoutte, P.M. Thromboxane A2 receptor antagonists inhibit endothelium-dependent contractions. Hypertension 1990, 15, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Iwama, Y.; Okumura, K.; Hashimoto, H.; Ito, T.; Satake, T. Prostaglandin H2 may be the endothelium-derived contracting factor released by acetylcholine in the aorta of the rat. Hypertension 1990, 15, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Mayhan, W.G. Role of prostaglandin H2-thromboxane A2 in responses of cerebral arterioles during chronic hypertension. Am. J. Physiol. 1992, 262, H539–H543. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Feletou, M.; Levens, N.; Zhang, J.N.; Vanhoutte, P.M. A diffusible substance(s) mediates endothelium-dependent contractions in the aorta of SHR. Hypertension 2003, 41, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Varadharaj, S.; Zhao, X.; Parinandi, N.; Flavahan, N.A.; Zweier, J.L. Acetylcholine causes endothelium-dependent contraction of mouse arteries. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1027–H1032. [Google Scholar] [CrossRef] [PubMed]
- Okon, E.B.; Golbabaie, A.; van Breemen, C. In the presence of L-NAME SERCA blockade induces endothelium-dependent contraction of mouse aorta through activation of smooth muscle prostaglandin H2/thromboxane A2 receptors. Br. J. Pharmacol. 2002, 137, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.S.; Ramamurthy, S.K.; Lin, X.; Le Breton, G.C. Cell signalling through thromboxane A2 receptors. Cell. Signal. 2004, 16, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, R.M.; Williams, S.P. Role of prostaglandins in acetylcholine-induced contraction of aorta from spontaneously hypertensive and Wistar-Kyoto rats. Hypertension 1996, 28, 64–75. [Google Scholar] [CrossRef]
- Nakahata, N. Thromboxane A2: Physiology/pathophysiology, cellular signal transduction and pharmacology. Pharmacol. Ther. 2008, 118, 18–35. [Google Scholar] [CrossRef]
- Liu, C.Q.; Leung, F.P.; Wong, S.L.; Wong, W.T.; Lau, C.W.; Lu, L.; Yao, X.; Yao, T.; Huang, Y. Thromboxane prostanoid receptor activation impairs endothelial nitric oxide-dependent vasorelaxations: The role of Rho kinase. Biochem. Pharmacol. 2009, 78, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Cayatte, A.J.; Du, Y.; Oliver-Krasinski, J.; Lavielle, G.; Verbeuren, T.J.; Cohen, R.A. The thromboxane receptor antagonist S18886 but not aspirin inhibits atherogenesis in apo E-deficient mice: Evidence that eicosanoids other than thromboxane contribute to atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1724–1728. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.G.; Nguyen, T.V.; Day, R.O. Do nonsteroidal anti-inflammatory drugs affect blood pressure? A meta-analysis. Ann. Intern. Med. 1994, 121, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Belhassen, L.; Pelle, G.; Dubois-Rande, J.L.; Adnot, S. Improved endothelial function by the thromboxane A2 receptor antagonist S 18886 in patients with coronary artery disease treated with aspirin. J. Am. Coll. Cardiol. 2003, 41, 1198–1204. [Google Scholar] [CrossRef]
- Koga, T.; Takata, Y.; Kobayashi, K.; Takishita, S.; Yamashita, Y.; Fujishima, M. Age and hypertension promote endothelium-dependent contractions to acetylcholine in the aorta of the rat. Hypertension 1989, 14, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Gluais, P.; Paysant, J.; Badier-Commander, C.; Verbeuren, T.; Vanhoutte, P.M.; Feletou, M. In SHR aorta, calcium ionophore A-23187 releases prostacyclin and thromboxane A2 as endothelium-derived contracting factors. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2255–H2264. [Google Scholar] [CrossRef] [PubMed]
- Arikawa, E.; Cheung, C.; Sekirov, I.; Battell, M.L.; Yuen, V.G.; McNeill, J.H. Effects of endothelin receptor blockade on hypervasoreactivity in streptozotocin-diabetic rats: Vessel-specific involvement of thromboxane A2. Can. J. Physiol. Pharmacol. 2006, 84, 823–833. [Google Scholar] [CrossRef]
- Yang, D.; Gluais, P.; Zhang, J.N.; Vanhoutte, P.M.; Feletou, M. Endothelium-dependent contractions to acetylcholine, ATP and the calcium ionophore A 23187 in aortas from spontaneously hypertensive and normotensive rats. Fundam. Clin. Pharmacol. 2004, 18, 321–326. [Google Scholar] [CrossRef]
- Carter, T.D.; Hallam, T.J.; Cusack, N.J.; Pearson, J.D. Regulation of P2y-purinoceptor-mediated prostacyclin release from human endothelial cells by cytoplasmic calcium concentration. Br. J. Pharmacol. 1988, 95, 1181–1190. [Google Scholar] [CrossRef]
- Wong, S.L.; Leung, F.P.; Lau, C.W.; Au, C.L.; Yung, L.M.; Yao, X.; Chen, Z.Y.; Vanhoutte, P.M.; Gollasch, M.; Huang, Y. Cyclooxygenase-2-derived prostaglandin F2alpha mediates endothelium-dependent contractions in the aortae of hamsters with increased impact during aging. Circ. Res. 2009, 104, 228–235. [Google Scholar] [CrossRef]
- Tang, E.H.; Jensen, B.L.; Skott, O.; Leung, G.P.; Feletou, M.; Man, R.Y.; Vanhoutte, P.M. The role of prostaglandin E and thromboxane-prostanoid receptors in the response to prostaglandin E2 in the aorta of Wistar Kyoto rats and spontaneously hypertensive rats. Cardiovasc. Res. 2008, 78, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Watkins, M.T.; Patton, G.M.; Soler, H.M.; Albadawi, H.; Humphries, D.E.; Evans, J.E.; Kadowaki, H. Synthesis of 8-epi-prostaglandin F2alpha by human endothelial cells: Role of prostaglandin H2 synthase. Biochem. J. 1999, 344 Pt 3, 747–754. [Google Scholar] [CrossRef]
- Zou, M.H.; Shi, C.; Cohen, R.A. High glucose via peroxynitrite causes tyrosine nitration and inactivation of prostacyclin synthase that is associated with thromboxane/prostaglandin H(2) receptor-mediated apoptosis and adhesion molecule expression in cultured human aortic endothelial cells. Diabetes 2002, 51, 198–203. [Google Scholar] [PubMed]
- Dinchuk, J.E.; Car, B.D.; Focht, R.J.; Johnston, J.J.; Jaffee, B.D.; Covington, M.B.; Contel, N.R.; Eng, V.M.; Collins, R.J.; Czerniak, P.M.; et al. Renal abnormalities and an altered inflammatory response in mice lacking cyclooxygenase II. Nature 1995, 378, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Patel, V.V.; Ricciotti, E.; Zhou, R.; Levin, M.D.; Gao, E.; Yu, Z.; Ferrari, V.A.; Lu, M.M.; Xu, J.; et al. Cardiomyocyte cyclooxygenase-2 influences cardiac rhythm and function. Proc. Natl. Acad. Sci. USA 2009, 106, 7548–7552. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.H.; Bertucci, M.C.; Ma, J.Y.; Adrahtas, A.; Cheung, R.Y.; Krum, H. Celecoxib, but not rofecoxib or naproxen, attenuates cardiac hypertrophy and fibrosis induced in vitro by angiotensin and aldosterone. Clin. Exp. Pharm. Physiol. 2010, 37, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Delgado, R.M., 3rd; Nawar, M.A.; Zewail, A.M.; Kar, B.; Vaughn, W.K.; Wu, K.K.; Aleksic, N.; Sivasubramanian, N.; McKay, K.; Mann, D.L.; et al. Cyclooxygenase-2 inhibitor treatment improves left ventricular function and mortality in a murine model of doxorubicin-induced heart failure. Circulation 2004, 109, 1428–1433. [Google Scholar] [CrossRef] [PubMed]
- Straino, S.; Salloum, F.N.; Baldi, A.; Ockaili, R.A.; Piro, M.; Das, A.; Qureshi, I.Z.; Biasucci, L.M.; Capogrossi, M.C.; Biondi-Zoccai, G.G.; et al. Protective effects of parecoxib, a cyclo-oxygenase-2 inhibitor, in postinfarction remodeling in the rat. J. Cardiovasc. Pharm. 2007, 50, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Such, L.; Morcillo, E.J.; Fortana, A.; Alberola, A.; Vina, J. Effects of antiinflammatory drugs in a model of acute transmural infarction in the dog. J. De Pharmacol. 1983, 14, 283–293. [Google Scholar]
- Wu, R.; Laplante, M.A.; de Champlain, J. Cyclooxygenase-2 inhibitors attenuate angiotensin II-induced oxidative stress, hypertension, and cardiac hypertrophy in rats. Hypertension 2005, 45, 1139–1144. [Google Scholar] [CrossRef]
- Francois, H.; Athirakul, K.; Howell, D.; Dash, R.; Mao, L.; Kim, H.S.; Rockman, H.A.; Fitzgerald, G.A.; Koller, B.H.; Coffman, T.M. Prostacyclin protects against elevated blood pressure and cardiac fibrosis. Cell Metab. 2005, 2, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Hara, A.; Yuhki, K.-i.; Fujino, T.; Yamada, T.; Takayama, K.; Kuriyama, S.; Takahata, O.; Karibe, H.; Okada, Y.; Xiao, C.-Y.; et al. Augmented Cardiac Hypertrophy in Response to Pressure Overload in Mice Lacking the Prostaglandin I2 Receptor. Circulation 2005, 112, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Fu, S.L.; Zhang, Y.P.; Qiao, M.M.; Chen, Y. Cyclooxygenase-2 inhibitors suppress angiogenesis and growth of gastric cancer xenografts. Biomed. Pharm. 2005, 59 (Suppl. 2), S289–S292. [Google Scholar] [CrossRef]
- Adamek, A.; Hu, K.; Bayer, B.; Wagner, H.; Ertl, G.; Bauersachs, J.; Frantz, S. High dose aspirin and left ventricular remodeling after myocardial infarction: Aspirin and myocardial infarction. Basic Res. Cardiol. 2007, 102, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.G.; Guzik, T.J.; Lob, H.E.; Madhur, M.S.; Marvar, P.J.; Thabet, S.R.; Vinh, A.; Weyand, C.M. Inflammation, immunity, and hypertension. Hypertension 2011, 57, 132–140. [Google Scholar] [CrossRef] [PubMed]
- McMaster, W.G.; Kirabo, A.; Madhur, M.S.; Harrison, D.G. Inflammation, immunity, and hypertensive end-organ damage. Circ. Res. 2015, 116, 1022–1033. [Google Scholar] [CrossRef]
- Rojo, J.M.; Portoles, M.P.; Barasoain, I.; Portoles, A. Exogenous additions of prostaglandins variably alter the blastogenic response of B and T lymphocytes from different mice lymphoid organs. Immunopharmacology 1982, 4, 95–104. [Google Scholar] [CrossRef]
- Kaufmann, S.H.; Simon, M.M.; Hahn, H. Regulatory interactions between macrophages and T-cell subsets in Listeria monocytogenes-specific T-cell activation. Infect. Immun. 1982, 38, 907–913. [Google Scholar]
- Gemsa, D.; Leser, H.G.; Deimann, W.; Resch, K. Suppression of T lymphocyte proliferation during lymphoma growth in mice: Role of PGE2-producing suppressor macrophages. Immunobiology 1982, 161, 385–391. [Google Scholar] [CrossRef]
- Betz, M.; Fox, B.S. Prostaglandin E2 inhibits production of Th1 lymphokines but not of Th2 lymphokines. J. Immunol. 1991, 146, 108–113. [Google Scholar]
- Bryn, T.; Yaqub, S.; Mahic, M.; Henjum, K.; Aandahl, E.M.; Tasken, K. LPS-activated monocytes suppress T-cell immune responses and induce FOXP3+ T cells through a COX-2-PGE2-dependent mechanism. Int. Immunol. 2008, 20, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Mahic, M.; Yaqub, S.; Johansson, C.C.; Tasken, K.; Aandahl, E.M. FOXP3+CD4+CD25+ adaptive regulatory T cells express cyclooxygenase-2 and suppress effector T cells by a prostaglandin E2-dependent mechanism. J. Immunol. 2006, 177, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Yang, S.C.; Zhu, L.; Reckamp, K.; Gardner, B.; Baratelli, F.; Huang, M.; Batra, R.K.; Dubinett, S.M. Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of FOXP3 expression and CD4+CD25+ T regulatory cell activities in lung cancer. Cancer Res. 2005, 65, 5211–5220. [Google Scholar] [CrossRef] [PubMed]
- Farrar, W.L.; Evans, S.W.; Rapp, U.R.; Cleveland, J.L. Effects of anti-proliferative cyclic AMP on interleukin 2-stimulated gene expression. J. Immunol. 1987, 139, 2075–2080. [Google Scholar] [PubMed]
- Mary, D.; Aussel, C.; Ferrua, B.; Fehlmann, M. Regulation of interleukin 2 synthesis by cAMP in human T cells. J. Immunol. 1987, 139, 1179–1184. [Google Scholar] [PubMed]
- Chouaib, S.; Welte, K.; Mertelsmann, R.; Dupont, B. Prostaglandin E2 acts at two distinct pathways of T lymphocyte activation: Inhibition of interleukin 2 production and down-regulation of transferrin receptor expression. J. Immunol. 1985, 135, 1172–1179. [Google Scholar]
- Lerner, A.; Jacobson, B.; Miller, R.A. Cyclic AMP concentrations modulate both calcium flux and hydrolysis of phosphatidylinositol phosphates in mouse T lymphocytes. J. Immunol. 1988, 140, 936–940. [Google Scholar]
- Liang, S.; Ledbetter, J.; Goodwin, J.S. Phosphatidyl inositol hydrolysis after CD3 binding in human peripheral blood T cells: Inhibition by prostaglandin E2. Int. J. Immunopharmacol. 1989, 11, 809–816. [Google Scholar] [CrossRef]
- Kalinski, P.; Hilkens, C.M.; Snijders, A.; Snijdewint, F.G.; Kapsenberg, M.L. IL-12-deficient dendritic cells, generated in the presence of prostaglandin E2, promote type 2 cytokine production in maturing human naive T helper cells. J. Immunol. 1997, 159, 28–35. [Google Scholar]
- Sharma, S.; Stolina, M.; Yang, S.C.; Baratelli, F.; Lin, J.F.; Atianzar, K.; Luo, J.; Zhu, L.; Lin, Y.; Huang, M.; et al. Tumor cyclooxygenase 2-dependent suppression of dendritic cell function. Clin. Cancer Res. J. Am. Assoc. Cancer Res. 2003, 9, 961–968. [Google Scholar]
- Sombroek, C.C.; Stam, A.G.; Masterson, A.J.; Lougheed, S.M.; Schakel, M.J.; Meijer, C.J.; Pinedo, H.M.; van den Eertwegh, A.J.; Scheper, R.J.; de Gruijl, T.D. Prostanoids play a major role in the primary tumor-induced inhibition of dendritic cell differentiation. J. Immunol. 2002, 168, 4333–4343. [Google Scholar] [CrossRef] [PubMed]
- Heusinkveld, M.; de Vos van Steenwijk, P.J.; Goedemans, R.; Ramwadhdoebe, T.H.; Gorter, A.; Welters, M.J.; van Hall, T.; van der Burg, S.H. M2 macrophages induced by prostaglandin E2 and IL-6 from cervical carcinoma are switched to activated M1 macrophages by CD4+ Th1 cells. J. Immunol. 2011, 187, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.W.; Thorbecke, G.J.; Baer, R.L.; Gigli, I. Effect of indomethacin on alteration of ATPase-positive Langerhans cell density and cutaneous sunburn reaction induced by ultraviolet-B radiation. J. Investig. Dermatol. 1983, 81, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Ng, R.L.; Bisley, J.L.; Gorman, S.; Norval, M.; Hart, P.H. Ultraviolet irradiation of mice reduces the competency of bone marrow-derived CD11c+ cells via an indomethacin-inhibitable pathway. J. Immunol. 2010, 185, 7207–7215. [Google Scholar] [CrossRef] [PubMed]
- Baratelli, F.E.; Heuze-Vourc’h, N.; Krysan, K.; Dohadwala, M.; Riedl, K.; Sharma, S.; Dubinett, S.M. Prostaglandin E2-dependent enhancement of tissue inhibitors of metalloproteinases-1 production limits dendritic cell migration through extracellular matrix. J. Immunol. 2004, 173, 5458–5466. [Google Scholar] [CrossRef] [PubMed]
- Doyen, V.; Rubio, M.; Braun, D.; Nakajima, T.; Abe, J.; Saito, H.; Delespesse, G.; Sarfati, M. Thrombospondin 1 is an autocrine negative regulator of human dendritic cell activation. J. Exp. Med. 2003, 198, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.; Longman, R.S.; Albert, M.L. A two-step induction of indoleamine 2,3 dioxygenase (IDO) activity during dendritic-cell maturation. Blood 2005, 106, 2375–2381. [Google Scholar] [CrossRef]
- Muthuswamy, R.; Mueller-Berghaus, J.; Haberkorn, U.; Reinhart, T.A.; Schadendorf, D.; Kalinski, P. PGE(2) transiently enhances DC expression of CCR7 but inhibits the ability of DCs to produce CCL19 and attract naive T cells. Blood 2010, 116, 1454–1459. [Google Scholar] [CrossRef]
- Kalinski, P.; Schuitemaker, J.H.; Hilkens, C.M.; Kapsenberg, M.L. Prostaglandin E2 induces the final maturation of IL-12-deficient CD1a+CD83+ dendritic cells: The levels of IL-12 are determined during the final dendritic cell maturation and are resistant to further modulation. J. Immunol. 1998, 161, 2804–2809. [Google Scholar]
- Kalinski, P.; Hilkens, C.M.; Wierenga, E.A.; Kapsenberg, M.L. T-cell priming by type-1 and type-2 polarized dendritic cells: The concept of a third signal. Immunol. Today 1999, 20, 561–567. [Google Scholar] [CrossRef]
- Gustafsson, K.; Ingelsten, M.; Bergqvist, L.; Nystrom, J.; Andersson, B.; Karlsson-Parra, A. Recruitment and activation of natural killer cells in vitro by a human dendritic cell vaccine. Cancer Res. 2008, 68, 5965–5971. [Google Scholar] [CrossRef] [PubMed]
- Van der Pouw Kraan, T.C.; Boeije, L.C.; Smeenk, R.J.; Wijdenes, J.; Aarden, L.A. Prostaglandin-E2 is a potent inhibitor of human interleukin 12 production. J. Exp. Med. 1995, 181, 775–779. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Hirata, T.; Soontrapa, K.; Ma, X.; Takemori, H.; Narumiya, S. Prostaglandin E(2) promotes Th1 differentiation via synergistic amplification of IL-12 signalling by cAMP and PI3-kinase. Nat. Commun. 2013, 4, 1685. [Google Scholar] [CrossRef] [PubMed]
- Kalinski, P.; Vieira, P.L.; Schuitemaker, J.H.; de Jong, E.C.; Kapsenberg, M.L. Prostaglandin E(2) is a selective inducer of interleukin-12 p40 (IL-12p40) production and an inhibitor of bioactive IL-12p70 heterodimer. Blood 2001, 97, 3466–3469. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, D.S.; Bahjat, K.S.; Moldawer, L.L.; Clare-Salzler, M.J. Autoregulation of human monocyte-derived dendritic cell maturation and IL-12 production by cyclooxygenase-2-mediated prostanoid production. J. Immunol. 2000, 165, 4298–4304. [Google Scholar] [CrossRef] [PubMed]
- Gillessen, S.; Carvajal, D.; Ling, P.; Podlaski, F.J.; Stremlo, D.L.; Familletti, P.C.; Gubler, U.; Presky, D.H.; Stern, A.S.; Gately, M.K. Mouse interleukin-12 (IL-12) p40 homodimer: A potent IL-12 antagonist. Eur. J. Immunol. 1995, 25, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Wehbi, V.L.; Tasken, K. Molecular Mechanisms for cAMP-Mediated Immunoregulation in T cells-Role of Anchored Protein Kinase A Signaling Units. Front. Immunol. 2016, 7, 222. [Google Scholar] [CrossRef]
- Krause, P.; Bruckner, M.; Uermosi, C.; Singer, E.; Groettrup, M.; Legler, D.F. Prostaglandin E(2) enhances T-cell proliferation by inducing the costimulatory molecules OX40L, CD70, and 4-1BBL on dendritic cells. Blood 2009, 113, 2451–2460. [Google Scholar] [CrossRef]
- Luft, T.; Jefford, M.; Luetjens, P.; Toy, T.; Hochrein, H.; Masterman, K.A.; Maliszewski, C.; Shortman, K.; Cebon, J.; Maraskovsky, E. Functionally distinct dendritic cell (DC) populations induced by physiologic stimuli: Prostaglandin E(2) regulates the migratory capacity of specific DC subsets. Blood 2002, 100, 1362–1372. [Google Scholar] [CrossRef]
- Scandella, E.; Men, Y.; Gillessen, S.; Forster, R.; Groettrup, M. Prostaglandin E2 is a key factor for CCR7 surface expression and migration of monocyte-derived dendritic cells. Blood 2002, 100, 1354–1361. [Google Scholar] [CrossRef]
- Quillien, V.; Moisan, A.; Carsin, A.; Lesimple, T.; Lefeuvre, C.; Adamski, H.; Bertho, N.; Devillers, A.; Leberre, C.; Toujas, L. Biodistribution of radiolabelled human dendritic cells injected by various routes. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Jonuleit, H.; Kuhn, U.; Muller, G.; Steinbrink, K.; Paragnik, L.; Schmitt, E.; Knop, J.; Enk, A.H. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur. J. Immunol. 1997, 27, 3135–3142. [Google Scholar] [CrossRef] [PubMed]
- Chizzolini, C.; Chicheportiche, R.; Alvarez, M.; de Rham, C.; Roux-Lombard, P.; Ferrari-Lacraz, S.; Dayer, J.M. Prostaglandin E2 synergistically with interleukin-23 favors human Th17 expansion. Blood 2008, 112, 3696–3703. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, H.; Zhang, X.; Wen, D.; Yu, F.; Yang, S.; Jia, X.; Cong, B.; Ma, C. Prostaglandin I2-IP signalling regulates human Th17 and Treg cell differentiation. Prostaglandins Leukot. Essent. Fat. Acids 2013, 89, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Z.; Yao, B.; Wang, Y.; Yang, S.; Wang, S.; Fan, X.; Harris, R.C. Inhibition of cyclooxygenase-2 in hematopoietic cells results in salt-sensitive hypertension. J. Clin. Investig. 2015, 125, 4281–4294. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Packer, L. Antioxidant and redox regulation of gene transcription. FASEB J. 1996, 10, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Kirkby, N.S.; Chan, M.V.; Lundberg, M.H.; Massey, K.A.; Edmands, W.M.; MacKenzie, L.S.; Holmes, E.; Nicolaou, A.; Warner, T.D.; Mitchell, J.A. Aspirin-triggered 15-epi-lipoxin A4 predicts cyclooxygenase-2 in the lungs of LPS-treated mice but not in the circulation: Implications for a clinical test. FASEB J. 2013, 27, 3938–3946. [Google Scholar] [CrossRef]
- Sampson, A.K.; Andrews, K.L.; Graham, D.; McBride, M.W.; Head, G.A.; Thomas, M.C.; Chin-Dusting, J.P.; Dominiczak, A.F.; Jennings, G.L. Origin of the Y chromosome influences intrarenal vascular responsiveness to angiotensin I and angiotensin (1-7) in stroke-prone spontaneously hypertensive rats. Hypertension 2014, 64, 1376–1383. [Google Scholar] [CrossRef]
- Livio, M.; Benigni, A.; Zoja, C.; Begnis, R.; Morelli, C.; Rossini, M.; Garattini, S.; Remuzzi, G. Differential inhibition by aspirin of platelet thromboxane and renal prostaglandins in the rat. J. Pharmacol. Exp. Ther. 1989, 248, 334–341. [Google Scholar]
- Sousa, T.; Carvalho, J.; Jo?o Valente, M.; Vale, L.; Almeida, L.; Afonso, J.; Jo?o Martins, M.; Carvalho, F.; Albino-Teixeira, A. Low-dose aspirin improves renal function and antihypertensive efficacy in hypertensive rats treated with losartan. J. Nephrol. Ther. 2014, 4, 4. [Google Scholar]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, S.; Andrews, K.L.; Chin-Dusting, J.P.F. Cyclo-Oxygenase (COX) Inhibitors and Cardiovascular Risk: Are Non-Steroidal Anti-Inflammatory Drugs Really Anti-Inflammatory? Int. J. Mol. Sci. 2019, 20, 4262. https://doi.org/10.3390/ijms20174262
Khan S, Andrews KL, Chin-Dusting JPF. Cyclo-Oxygenase (COX) Inhibitors and Cardiovascular Risk: Are Non-Steroidal Anti-Inflammatory Drugs Really Anti-Inflammatory? International Journal of Molecular Sciences. 2019; 20(17):4262. https://doi.org/10.3390/ijms20174262
Chicago/Turabian StyleKhan, Shanzana, Karen L. Andrews, and Jaye P. F. Chin-Dusting. 2019. "Cyclo-Oxygenase (COX) Inhibitors and Cardiovascular Risk: Are Non-Steroidal Anti-Inflammatory Drugs Really Anti-Inflammatory?" International Journal of Molecular Sciences 20, no. 17: 4262. https://doi.org/10.3390/ijms20174262
APA StyleKhan, S., Andrews, K. L., & Chin-Dusting, J. P. F. (2019). Cyclo-Oxygenase (COX) Inhibitors and Cardiovascular Risk: Are Non-Steroidal Anti-Inflammatory Drugs Really Anti-Inflammatory? International Journal of Molecular Sciences, 20(17), 4262. https://doi.org/10.3390/ijms20174262